
Bacteriophage Tail Proteins as a Tool for Bacterial Pathogen Recognition—A Literature Review


Abstract
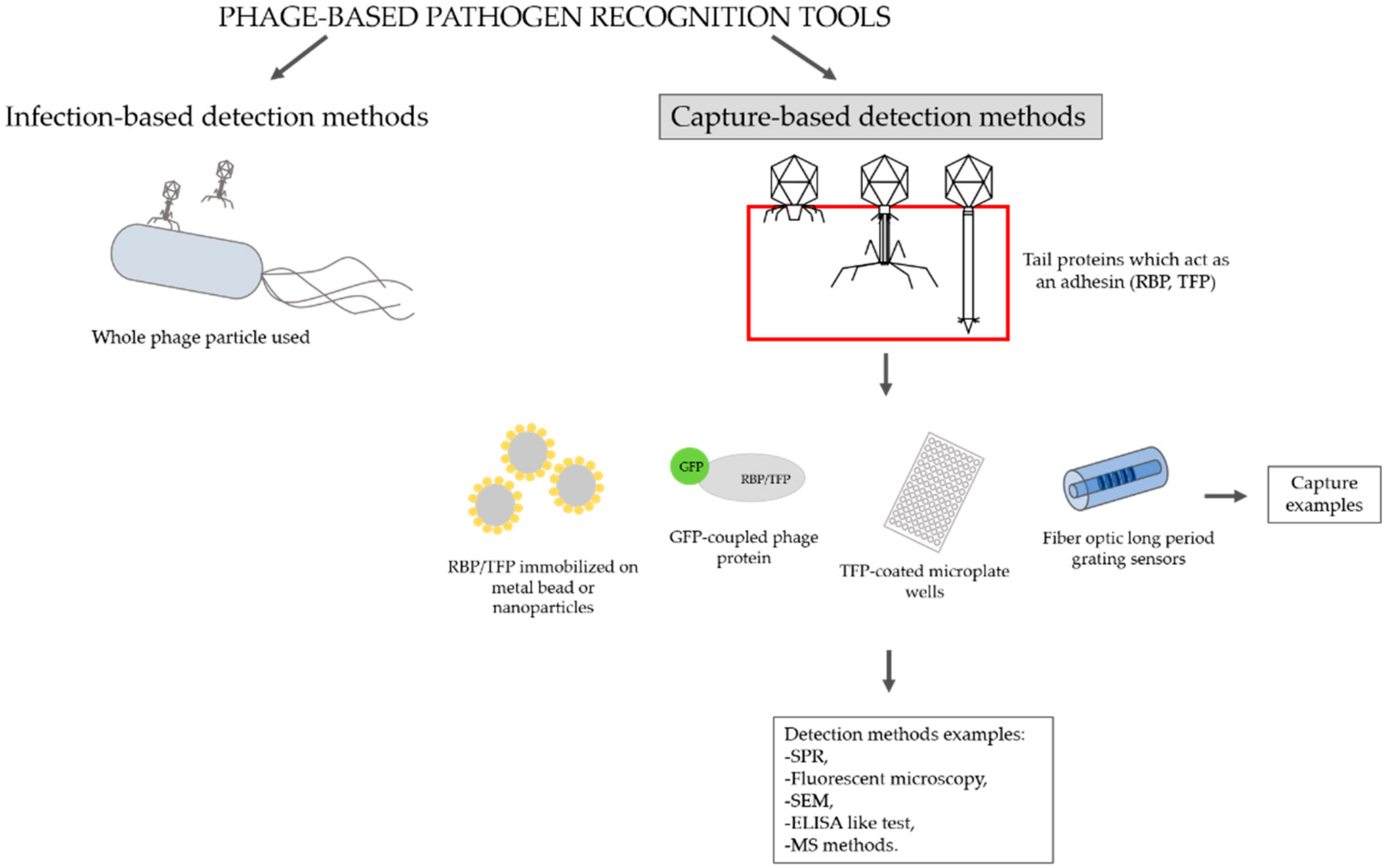
1. Introduction
2. Overview of Methodologies That Use Bacteriophage Tail Proteins for Detecting Pathogenic Bacteria
2.1. Acinetobacter baumannii Detection
2.2. Campylobacter spp. Detection
2.3. Listeria monocytogenes Detection
2.4. Yersinia pestis Detection
2.5. Pseudomonas aeruginosa Detection
2.6. Enterococcus spp. and Staphylococcus spp. Detection
2.7. Salmonella spp. Detection
2.8. Shigella Detection
2.9. Bacillus anthracis Detection
2.10. Klebsiella pneumoniae Detection
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ackerman, H.W. 5500 Phages examined in the electron microscope. Arch. Virol. 2007, 152, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Jończyk, E.; Kłak, M.; Międzybrodzki, R.; Górski, A. The influence of external factors on bacteriophages—Review. Folia Microbiol. 2011, 56, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef]
- Slopek, S.; Przondo-Hessek, H.; Milch, H.; Deák, S. A working scheme for bacteriophage typing of Klebsiella bacilli. Arch. Immunol. Ther. Exp. 1967, 15, 589–599. [Google Scholar]
- Abdelsattar, A.S.; Dawooud, A.; Rezk, N.; Makky, S.; Safwat, A.; Richards, P.J.; El-Shibiny, A. How to Train Your Phage: The Recent Efforts in Phage Training. Biologics 2021, 1, 70–88. [Google Scholar] [CrossRef]
- Stone, E.; Campbell, K.; Grant, I.; McAuli, O. Understanding and Exploiting Phage–Host Interactions. Viruses 2019, 11, 567. [Google Scholar] [CrossRef]
- Fernandes, S.; São-José, C. Enzymes and Mechanisms Employed by Tailed Bacteriophages to Breach the Bacterial Cell Barriers. Viruses 2018, 10, 396. [Google Scholar] [CrossRef]
- Pyra, A.; Brzozowska, E.; Pawlik, K.; Gamian, A.; Dauter, M.; Dauter, Z. Tail tubular protein A: A dual function tail protein of Klebsiella pneumoniae bacteriophage KP32. Sci. Rep. 2017, 7, 2223. [Google Scholar] [CrossRef]
- Pyra, A.; Filik, K.; Szermer-Olearnik, B.; Czarny, A.; Brzozowska, E. New Insights on the Feature and Function of Tail Tubular Protein B and Tail Fiber Protein of the Lytic Bacteriophage YeO3-12 Specific for Yersinia enterocolitica Serotype O:3. Molecules 2020, 25, 4392. [Google Scholar] [CrossRef]
- Kutter, E.; Sulakvelidze, A. Bacteriophages: Biology and Applications; CRC Press: Washington, DC, USA, 2004. [Google Scholar]
- Echeverría-Vega, A.; Morales-Vicencio, P.; Saez-Saavedra, C.; Alvarez, M.A.; Gordillo, F.; Del-Valle, R.; Solís, M.E.; Araya, R. Characterization of the Bacteriophage vB_VorS-PVo5 Infection on Vibrio ordalii: A Model for Phage-Bacteria Adsorption in Aquatic Environments. Front. Microbiol. 2020, 11, 2440. [Google Scholar] [CrossRef]
- Yin, J.; McCaskill, J.S. Replication of viruses in a growing plaque: A reaction-diffusion model. J. Biophys. Soc. 1992, 61, 1540–1549. [Google Scholar] [CrossRef]
- Brzozowska, E.; Bazan, J.; Gamian, A. The function of bacteriophage proteins. Postepy Hig. Med. Dosw. (Online) 2011, 65, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.H. Bacteriophages with Chapters; Anderson, E.S., Gots, J.S., Jacob, F., Wollman, E.L., Eds.; Interscience Publishers, Inc.: New York, NY, USA, 1959; Volume 5, p. xviii + 592. [Google Scholar]
- Szermer-Olearnik, B.; Drab, M.; Mąkosa, M.; Zembala, M.; Barbasz, J.; Dąbrowska, K.; Boratyński, J. Aggregation/dispersion transitions of T4 phage triggered by environmental ion availability. J. Nanobiotechnol. 2017, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Philippe, C.; Chaïb, A.; Jaomanjaka, F.; Cluzet, S.; Lagarde, A.; Ballestra, P.; Decendit, A.; Petrel, M.; Claisse, O.; Goulet, A.; et al. Wine Phenolic Compounds Dierently Affect the Host-Killing Activity of Two Lytic Bacteriophages Infecting the Lactic Acid Bacterium Oenococcus oeni. Viruses 2020, 12, 1316. [Google Scholar] [CrossRef] [PubMed]
- Delbrück, M. Biochemical mutants of bacterial viruses. J. Bacteriol. 1948, 56, 1–16. [Google Scholar] [CrossRef]
- Ackermann, H.W. Tailed bacteriophages: The order Caudovirales. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 1999; Volume 51, pp. 135–201. [Google Scholar] [CrossRef]
- Molineux, I.J. T7-LIKE PHAGES (PODOVIRIDAE). In Encyclopedia of Virology, 2nd ed.; Granoff, A., Webster, R.G., Eds.; Elsevier: Amsterdam, The Netherlands, 1999; pp. 1722–1729. [Google Scholar]
- Mosig, G. T4-LIKE PHAGES (MYOVIRIDAE). In Encyclopedia of Virology, 2nd ed.; Granoff, A., Webster, R.G., Eds.; Elsevier: Amsterdam, The Netherlands, 1999; pp. 1706–1716. [Google Scholar]
- Aksyuk, A.A.; Leiman, P.G.; Kurochkina, L.P.; Shneider, M.M.; Kostyuchenko, V.A.; Mesyanzhinov, V.V.; Rossmann, M.G. The tail sheath structure of bacteriophage T4: A molecular machine for infecting bacteria. EMBO J. 2009, 28, 821–829. [Google Scholar] [CrossRef]
- Leiman, P.G.; Arisaka, F.; Van Raaij, M.J.; Kostyuchenko, V.A.; Aksyuk, A.A.; Kanamaru, S.; Rossmann, M.G. Morphogenesis of the T4 tail and tail fibers. Virol. J. 2010, 7, 355. [Google Scholar] [CrossRef]
- Tao, Y.; Strelkov, S.V.; Mesyanzhinov, V.V.; Rossmann, M.G. Structure of bacteriophage T4 fibritin: A segmented coiled coil and the role of the C-terminal domain. Structure 1997, 5, 789–798. [Google Scholar] [CrossRef]
- Bartual, S.G.; Otero, J.M.; Garcia-Doval, C.; Llamas-Saiz, A.L.; Kahn, R.; Fox, G.C.; van Raaij, M.J. Structure of the bacteriophage T4 long tail fiber receptor-binding tip. Proc. Natl. Acad. Sci. USA 2010, 107, 20287–20292. [Google Scholar] [CrossRef]
- Campbell, A.M. Coliphage Lambda (Siphoviridae). In Encyclopedia of Virology, 2nd ed.; Granoff, A., Webster, R.G., Eds.; Elsevier: Amsterdam, The Netherlands, 1999; pp. 281–285. [Google Scholar]
- Simpson, D.J.; Sacher, J.C.; Szymanski, C.M. Development of an assay for the identification of receptor binding proteins from bacteriophages. Viruses 2016, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Meile, S.; Kilcher, S.; Loessner, M.J.; Dunne, M. Reporter Phage-Based Detection of Bacterial Pathogens: Design Guidelines and Recent Developments. Viruses 2020, 12, 944. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Arutyunov, D.; Szymanski, C.M.; Evoy, S. Bacteriophage based probes for pathogen detection. Analyst 2012, 137, 3405. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Poshtiban, S.; Evoy, S. Recent advances in bacteriophage based biosensors for food-borne pathogen detection. Sensors 2013, 13, 1763–1786. [Google Scholar] [CrossRef]
- Gao, J.; Jeffries, L.; Mach, K.E.; Craft, D.W.; Thomas, N.J.; Gau, V.; Liao, J.C.; Wong, P.K. A Multiplex Electrochemical Biosensor for Bloodstream Infection Diagnosis. SLAS Technol. 2017, 22, 466–474. [Google Scholar] [CrossRef]
- Vásquez, G.; Rey, A.; Rivera, C.; Iregui, C.; Orozco, J. Amperometric biosensor based on a single antibody of dual function for rapid detection of Streptococcus agalactiae. Biosens. Bioelectron. 2017, 87, 453–458. [Google Scholar] [CrossRef]
- Spehar-Délèze, A.M.; Julich, S.; Gransee, R.; Tomaso, H.; Dulay, S.B.; O’Sullivan, C.K. Electrochemiluminescence (ECL) immunosensor for detection of Francisella tularensis on screen-printed gold electrode array. Anal. Bioanal. Chem. 2016, 408, 7147–7153. [Google Scholar] [CrossRef]
- Farooq, U.; Ullah, M.W.; Yang, Q.; Aziz, A.; Xu, J.; Zhou, L.; Wang, S. High-density phage particles immobilization in surface-modified bacterial cellulose for ultra-sensitive and selective electrochemical detection of Staphylococcus aureus. Biosens. Bioelectron. 2020, 157, 112163. [Google Scholar] [CrossRef]
- Singh, A.; Arutyunov, D.; McDermott, M.T.; Szymanski, C.M.; Evoy, S. Specific detection of Campylobacter jejuni using the bacteriophage NCTC 12673 receptor binding protein as a probe. Analyst 2011, 136, 4780. [Google Scholar] [CrossRef]
- Peng, H.; Chen, I.A. Rapid Colorimetric Detection of Bacterial Species through the Capture of Gold Nanoparticles by Chimeric Phages. ACS Nano 2019, 13, 1244–1252. [Google Scholar] [CrossRef]
- Kong, M.; Sim, J.; Kang, T.; Nguyen, H.H.; Park, H.K.; Chung, B.H.; Ryu, S. A novel and highly specific phage endolysin cell wall binding domain for detection of Bacillus cereus. Eur. Biophys. J. 2015, 44, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Tolba, M.; Ahmed, M.U.; Tlili, C.; Eichenseher, F.; Loessner, M.J.; Zourob, M. A bacteriophage endolysin-based electrochemical impedance biosensor for the rapid detection of Listeria cells. Analyst 2012, 137, 5749–5756. [Google Scholar] [CrossRef] [PubMed]
- Le Brun, G.; Hauwaert, M.; Leprince, A.; Glinel, K.; Mahillon, J.; Raskin, J.P. Electrical Characterization of Cellulose-Based Membranes towards Pathogen Detection in Water. Biosensors 2021, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; McIvor, M.J.; Elliott, C.T.; Karoonuthaisiri, N.; Segatori, L.; Biswal, S.L. Rapid detection of pathogenic bacteria and screening of phage-derived peptides using microcantilevers. Anal. Chem. 2014, 86, 1671–1678. [Google Scholar] [CrossRef]
- Liu, P.; Han, L.; Wang, F.; Petrenko, V.A.; Liu, A. Gold nanoprobe functionalized with specific fusion protein selection from phage display and its application in rapid, selective and sensitive colorimetric biosensing of Staphylococcus aureus. Biosens. Bioelectron. 2016, 82, 195–203. [Google Scholar] [CrossRef]
- Farooq, U.; Yang, Q.; Ullah, M.W.; Wang, S. Bacterial biosensing: Recent advances in phage-based bioassays and biosensors. Biosens. Bioelectron. 2018, 118, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Anany, H.; Chou, Y.; Cucic, S.; Derda, R.; Evoy, S.; Griffiths, M.W. From Bits and Pieces to Whole Phage to Nanomachines: Pathogen Detection Using Bacteriophages. Annu. Rev. Food Sci. Technol. 2017, 8, 305–329. [Google Scholar] [CrossRef] [PubMed]
- Gerstmans, H.; Criel, B.; Briers, Y. Synthetic biology of modular endolysins. Biotechnol. Adv. 2018, 36, 624–640. [Google Scholar] [CrossRef]
- Loessner, M.J.; Kramer, K.; Ebel, F.; Scherer, S. C-terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high-affinity binding to bacterial cell wall carbohydrates. Mol. Microbiol. 2002, 44, 335–349. [Google Scholar] [CrossRef]
- Maciejewska, B.; Olszak, T.; Drulis-Kawa, Z. Applications of bacteriophages versus phage enzymes to combat and cure bacterial infections: An ambitious and also a realistic application? Appl. Microbiol. Biotechnol. 2018, 102, 2563–2581. [Google Scholar] [CrossRef]
- Oliveira, H.; Melo, L.D.; Santos, S.B.; Nóbrega, F.L.; Ferreira, E.C.; Cerca, N.; Azeredo, J.; Kluskens, L.D. Molecular aspects and comparative genomics of bacteriophage endolysins. J. Virol. 2013, 87, 4558–4570. [Google Scholar] [CrossRef] [PubMed]
- Mo, K.F.; Li, X.; Li, H.; Low, L.Y.; Quinn, C.P.; Boons, G.J. Endolysins of Bacillus anthracis bacteriophages recognize unique carbohydrate epitopes of vegetative cell wall polysaccharides with high affinity and selectivity. J. Am. Chem. Soc. 2012, 134, 15556–15562. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dams, D.; Briers, Y. Enzybiotics: Enzyme-Based Antibacterials as Therapeutics. In Therapeutic Enzymes: Function and Clinical Implications. Advances in Experimental Medicine and Biology; Labrou, N., Ed.; Springer: Singapore, 2019; Volume 1148. [Google Scholar] [CrossRef]
- Schmelcher, M.; Shabarova, T.; Eugster, M.R.; Eichenseher, F.; Tchang, V.S.; Banz, M.; Loessner, M.J. Rapid multiplex detection and differentiation of Listeria cells by use of fluorescent phage endolysin cell wall binding domains. Appl. Environ. Microbiol. 2010, 76, 5745–5756. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Kwon, J.G.; O’Sullivan, D.J.; Ryu, S.; Lee, J.H. Development of an endolysin enzyme and its cell wall-binding domain protein and their applications for biocontrol and rapid detection of Clostridium perfringens in food. Food Chem. 2021, 345, 128562. [Google Scholar] [CrossRef]
- Gómez-Torres, N.; Dunne, M.; Garde, S.; Meijers, R.; Narbad, A.; Ávila, M.; Mayer, M.J. Development of a specific fluorescent phage endolysin for in situ detection of Clostridium species associated with cheese spoilage. Microb. Biotechnol. 2018, 11, 332–345. [Google Scholar] [CrossRef]
- Chang, Y.; Ryu, S. Characterization of a novel cell wall binding domain-containing Staphylococcus aureus endolysin LysSA97. Appl. Microbiol. Biotechnol. 2016, 101, 147–158. [Google Scholar] [CrossRef]
- Walcher, G.; Stessl, B.; Wagner, M.; Eichenseher, F.; Loessner, M.J.; Hein, I. Evaluation of paramagnetic beads coated with recombinant Listeria phage endolysin-derived cell-wall-binding domain proteins for separation of Listeria monocytogenes from raw milk in combination with culture-based and real-time polymerase chain reaction-based quantification. Foodborne Pathog. Dis. 2010, 7, 1019–1024. [Google Scholar] [CrossRef]
- Kretzer, J.W.; Lehmann, R.; Schmelcher, M.; Banz, M.; Kim, K.P.; Korn, C.; Loessner, M.J. Use of high-affinity cell wall-binding domains of bacteriophage endolysins for immobilization and separation of bacterial cells. Appl. Environ. Microbiol. 2007, 73, 1992–2000. [Google Scholar] [CrossRef]
- Kretzer, J.W.; Schmelcher, M.; Loessner, M.J. Ultrasensitive and Fast Diagnostics of Viable Listeria Cells by CBD Magnetic Separation Combined with A511::luxAB Detection. Viruses 2018, 10, 626. [Google Scholar] [CrossRef]
- Park, C.; Kong, M.; Lee, J.H.; Ryu, S.; Park, S. Detection of bacillus cereus using bioluminescence assay with cell wall-binding domain conjugated magnetic nanoparticles. BioChip J. 2018, 12, 287–293. [Google Scholar] [CrossRef]
- Kong, M.; Shin, J.H.; Heu, S.; Park, J.K.; Ryu, S. Lateral flow assay-based bacterial detection using engineered cell wall binding domains of a phage endolysin. Biosens. Bioelectron. 2017, 96, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, X.; Kang, G.; Bai, L.; Wang, P.; Huang, H. Isolation and Characterization of AbTJ, an Acinetobacter baumannii Phage, and Functional Identification of Its Receptor-Binding Modules. Viruses 2020, 12, 205. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.L.; Shahed-Al-Mahmud, M.; Selvaprakash, K.; Lin, N.T.; Chen, Y.C. Tail Fiber Protein-Immobilized Magnetic Nanoparticle-Based Affinity Approaches for Detection of Acinetobacter baumannii. Anal. Chem. 2019, 91, 10335–10342. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.M.; Tu, I.F.; Yang, F.L.; Wu, S.H. Bacteriophage Tail-Spike Proteins Enable Detection of Pseudaminic-Acid-Coated Pathogenic Bacteria and Guide the Development of Antiglycan Antibodies with Cross-Species Antibacterial Activity. J. Am. Chem. Soc. 2020, 142, 19446–19450. [Google Scholar] [CrossRef]
- Murdoch, D.R.; Corey, G.R.; Hoen, B.; Miró, J.M.; Fowler, V.G., Jr.; Bayer, A.S.; Karchmer, A.W.; Olaison, L.; Pappas, P.A.; Moreillon, P.; et al. International Collaboration on Endocarditis-Prospective Cohort Study (ICE-PCS) Investigators. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: The International Collaboration on Endocarditis-Prospective Cohor Study. Arch. Intern. Med. 2009, 169, 463–473. [Google Scholar] [CrossRef]
- Vidal, F.; Mensa, J.; Almela, M.; Martinez, J.A.; Marco, F.; Casals, C.; Gatell, J.M.; Soriano, E.; de Jimenez Anta, M.T. Epidemiology and outcome of Pseudomonas aeruginosa bacteremia, with special emphasis on the influence of antibiotic treatment. Analysis of 189 episodes. Arch. Intern. Med. 1996, 156, 2121–2126. [Google Scholar] [CrossRef]
- Perry, R.D.; Fetherston, J.D. Yersinia pestis--etiologic agent of plague. Clin. Microbiol. Rev. 1997, 10, 35–66. [Google Scholar] [CrossRef]
- Poshtiban, S.; Singh, A.; Fitzpatrick, G.; Evoy, S. Bacteriophage tail-spike protein derivitized microresonator arrays for specific detection of pathogenic bacteria. Sens. Actuators B Chem. 2013, 181, 410–416. [Google Scholar] [CrossRef]
- Javed, M.A.; Poshtiban, S.; Arutyunov, D.; Evoy, S.; Szymanski, C.M. Bacteriophage receptor binding protein based assays for the simultaneous detection of Campylobacter jejuni and Campylobacter coli. PLoS ONE 2013, 8, e69770. [Google Scholar] [CrossRef]
- Denyes, J.M.; Dunne, M.; Steiner, S.; Mittelviefhaus, M.; Weiss, A.; Schmidt, H.; Klumpp, J.; Loessner, M.J. Modified bacteriophage S16 long tail fiber proteins for rapid and specific immobilization and detection of Salmonella cells. Appl. Environ. Microbiol. 2017, 83, e00277-17. [Google Scholar] [CrossRef]
- Andrews, W.H.; Jacobson, A.; Hammack, T. “Bacteriological Analytical Manual (BAM) Chapter 5: Salmonella”. Bacteriological Analytical Manual (2018).* Bacteriological Analytical Manual Chepter 5: Salmonella. Available online: https://www.fda.gov/food/laboratory-methods-food/bam-chapter-5-salmonella (accessed on 26 September 2021).
- Coimbra, R.S.; Grimont, F.; Grimont, P.A. Identification of Shigella serotypes by restriction of amplified O-antigen gene cluster. Res. Microbiol. 1999, 150, 543–553. [Google Scholar] [CrossRef]
- Braun, P.; Rupprich, N.; Neif, D.; Grass, G. Enzyme-Linked Phage Receptor Binding Protein Assays (ELPRA) Enable Identification of Bacillus anthracis Colonies. Viruses 2021, 13, 1462. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, C.L.; Pires, D.P.; Monteiro, R.; Santos, S.B.; Carvalho, C.M. Exploitation of a Klebsiella Bacteriophage Receptor-Binding Protein as a Superior Biorecognition Molecule. ACS Infect. Dis. 2021, 7, 3077–3087. [Google Scholar] [CrossRef] [PubMed]
- Garnacho-Montero, J.; Timsit, J.F. Managing Acinetobacter baumannii infections. Curr. Opin. Infect. Dis. 2019, 32, 69–76. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- Harding, C.M.; Hennon, S.W.; Feldman, M.F. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 2018, 16, 91–102. [Google Scholar] [CrossRef]
- Shashkov, A.S.; Kenyon, J.J.; Arbatsky, N.P.; Shneider, M.M.; Popova, A.V.; Miroshnikov, K.A.; Volozhantsev, N.V.; Knirel, Y.A. Structures of three different neutral polysaccharides of Acinetobacter baumannii, NIPH190, NIPH201, and NIPH615, assigned to K30, K45, and K48 capsule types, respectively, based on capsule biosynthesis gene clusters. Carbohydr. Res. 2015, 417, 81–88. [Google Scholar] [CrossRef]
- Wyres, K.L.; Cahill, S.M.; Holt, K.E.; Hall, R.M.; Kenyon, J.J. Identification of Acinetobacter baumannii loci for capsular polysaccharide (KL) and lipooligosaccharide outer core (OCL) synthesis in genome assemblies using curated reference databases compatible with Kaptive. Microb. Genom. 2020, 6, e000339. [Google Scholar] [CrossRef]
- Gordillo Altamirano, F.; Forsyth, J.H.; Patwa, R.; Kostoulias, X.; Trim, M.; Subedi, D.; Archer, S.K.; Morris, F.C.; Oliveira, C.; Kielty, L.; et al. Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 2021, 6, 157–161. [Google Scholar] [CrossRef]
- Shchurova, A.S.; Shneider, M.M.; Arbatsky, N.P.; Shashkov, A.S.; Chizhov, A.O.; Skryabin, Y.P.; Mikhaylova, Y.V.; Sokolova, O.S.; Shelenkov, A.A.; Miroshnikov, K.A.; et al. Novel Acinetobacter baumannii Myovirus TaPaz Encoding Two Tailspike Depolymerases: Characterization and Host-Recognition Strategy. Viruses 2021, 13, 978. [Google Scholar] [CrossRef] [PubMed]
- Popova, A.V.; Lavysh, D.G.; Klimuk, E.I.; Edelstein, M.V.; Bogun, A.G.; Shneider, M.M.; Goncharov, A.E.; Leonov, S.V.; Severinov, K.V. Novel Fri1-like Viruses Infecting Acinetobacter baumannii-vB_AbaP_AS11 and vB_AbaP_AS12-Characterization, Comparative Genomic Analysis, and Host-Recognition Strategy. Viruses 2017, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Popova, A.V.; Shneider, M.M.; Myakinina, V.P.; Bannov, V.A.; Edelstein, M.V.; Rubalskii, E.O.; Aleshkin, A.V.; Fursova, N.K.; Volozhantsev, N.V. Characterization of myophage AM24 infecting Acinetobacter baumannii of the K9 capsular type. Arch. Virol. 2019, 164, 1493–1497. [Google Scholar] [CrossRef]
- Popova, A.V.; Shneider, M.M.; Arbatsky, N.P.; Kasimova, A.A.; Senchenkova, S.N.; Shashkov, A.S.; Dmitrenok, A.S.; Chizhov, A.O.; Mikhailova, Y.V.; Shagin, D.A.; et al. Specific Interaction of Novel Friunavirus Phages Encoding Tailspike Depolymerases with Corresponding Acinetobacter baumannii Capsular Types. J. Virol. 2020, 95, e01714-20. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.; Costa, A.R.; Konstantinides, N.; Ferreira, A.; Akturk, E.; Sillankorva, S.; Nemec, A.; Shneider, M.; Dötsch, A.; Azeredo, J. Ability of phages to infect Acinetobacter calcoaceticus-Acinetobacter baumannii complex species through acquisition of different pectate lyase depolymerase domains. Environ. Microbiol. 2017, 19, 5060–5077. [Google Scholar] [CrossRef]
- Hernandez-Morales, A.C.; Lessor, L.L.; Wood, T.L.; Migl, D.; Mijalis, E.M.; Cahill, J.; Russell, W.K.; Young, R.F.; Gill, J.J. Genomic and Biochemical Characterization of Acinetobacter Podophage Petty Reveals a Novel Lysis Mechanism and Tail-Associated Depolymerase Activity. J. Virol. 2018, 92, e01064-17. [Google Scholar] [CrossRef]
- Liu, Y.; Mi, Z.; Mi, L.; Huang, Y.; Li, P.; Liu, H.; Yuan, X.; Niu, W.; Jiang, N.; Bai, C.; et al. Identification and characterization of capsule depolymerase Dpo48 from Acinetobacter baumannii phage IME200. PeerJ 2019, 7, e6173. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Leung, S.; Guo, Y.; Zhao, L.; Jiang, N.; Mi, L.; Li, P.; Wang, C.; Qin, Y.; Mi, Z.; et al. The Capsule Depolymerase Dpo48 Rescues Galleria mellonella and Mice from Acinetobacter baumannii Systemic Infections. Front. Microbiol. 2019, 10, 545. [Google Scholar] [CrossRef]
- Shahed-Al-Mahmud, M.; Roy, R.; Sugiokto, F.G.; Islam, M.N.; Lin, M.D.; Lin, L.C.; Lin, N.T. Phage φAB6-Borne Depolymerase Combats Acinetobacter baumannii Biofilm Formation and Infection. Antibiotics 2021, 10, 279. [Google Scholar] [CrossRef]
- Wang, C.; Li, P.; Zhu, Y.; Huang, Y.; Gao, M.; Yuan, X.; Niu, W.; Liu, H.; Fan, H.; Qin, Y.; et al. Identification of a Novel Acinetobacter baumannii Phage-Derived Depolymerase and Its Therapeutic Application in Mice. Front. Microbiol. 2020, 11, 1407. [Google Scholar] [CrossRef]
- Oliveira, H.; Costa, A.R.; Ferreira, A.; Konstantinides, N.; Santos, S.B.; Boon, M.; Noben, J.P.; Lavigne, R.; Azeredo, J. Functional Analysis and Antivirulence Properties of a New Depolymerase from a Myovirus That Infects Acinetobacter baumannii Capsule K45. J. Virol. 2019, 93, e01163-18. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.M.; Tu, I.F.; Yang, F.L.; Ko, T.P.; Liao, J.H.; Lin, N.T.; Wu, C.Y.; Ren, C.T.; Wang, A.H.; Chang, C.M.; et al. Structural basis for fragmenting the exopolysaccharide of Acinetobacter baumannii by bacteriophage ΦAB6 tailspike protein. Sci. Rep. 2017, 7, 42711. [Google Scholar] [CrossRef] [PubMed]
- Zunk, M.; Kiefel, M. The occurrence and biological significance of the α-keto-sugars pseudaminic acid and legionaminic acid within pathogenic bacteria. RSC Adv. 2014, 4, 3413–3421. [Google Scholar] [CrossRef]
- Kenyon, J.J.; Marzaioli, A.M.; De Castro, C.; Hall, R.M. 5,7-di-N-acetyl-acinetaminic acid: A novel non-2-ulosonic acid found in the capsule of an Acinetobacter baumannii isolate. Glycobiology 2015, 25, 644–654. [Google Scholar] [CrossRef]
- Skirrow, M.B. Campylobacter enteritis: A “new” disease. Br. Med. J. 1997, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Leite, D.; Fernandes, M.; Mena, C.; Gibbs, P.A.; Teixeira, P. Campylobacter spp. as a foodborne pathogen: A review. Front. Microbiol. 2011, 2, 200. [Google Scholar] [CrossRef]
- Tauxe, R.V. “Epidemiology of Campylobacter jejuni infection in the United States and other industrialized nations,” Chapter 2. In Campylobacter jejuni: Current Status and Future Trends; Nachamkin, I., Blaser, M.J., Tompkins, L.S., Eds.; American Association of Microbiologists: Washington, DC, USA, 1992; pp. 9–19. [Google Scholar]
- Skirrow, M.B. Campylobacteriosis. In Zoonoses; Palmer, S.R., Lord Soulsby, S.R., Simpson, D.I.H., Eds.; Oxford University Press: New York, NY, USA, 1998; pp. 37–46. [Google Scholar]
- Corry, J.E.L.; Atabay, H.I. Poultry as a source of Campylobacter and related organisms. J. Appl. Microbiol. 2001, 90, 96S–114S. [Google Scholar] [CrossRef]
- Humphrey, T.; O’Brien, S.; Madsen, M. Campylobacters as zoonotic pathogens: A food production perspective. Int. J. Food Microbiol. 2007, 117, 237–257. [Google Scholar] [CrossRef]
- Eberhart-Phillips, J.; Walker, N.; Garrett, N.; Bell, D.; Sinclair, D.; Rainger, W.; Bates, M. Campylobacteriosis in New Zealand: Results of a case-control study. J. Epidemiol. Community Health 1997, 51, 686–691. [Google Scholar] [CrossRef]
- Portner, D.C.; Leuschner, R.G.K.; Murray, B.S. Optimising the viability during storage of freeze-dried cell preparations of Campylobacter jejuni. Cryobiology 2007, 54, 265–270. [Google Scholar] [CrossRef]
- Montagut, Y.; Garcia, J.; Jiménez, Y.; March, C.; Montoya, Á.; Arnau, A. QCM technology in biosensors. In Biosensors-Emerging Materials and Application; Serra, P.A., Ed.; IntechOpen: London, UK, 2011; pp. 153–178. [Google Scholar] [CrossRef]
- Law, J.W.; Ab Mutalib, N.S.; Chan, K.G.; Lee, L.H. An insight into the isolation, enumeration, and molecular detection of Listeria monocytogenes in food. Front. Microbiol. 2015, 6, 1227. [Google Scholar] [CrossRef] [PubMed]
- Schlech, W.F. Epidemiology and Clinical Manifestations of Listeria monocytogenes Infection. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Radoshevich, L.; Cossart, P. Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Archer, D.L. The evolution of FDA’s policy on Listeria monocytogenes in ready-to-eat foods in the United States. Curr. Opin. Food Sci. 2018, 20, 64–68. [Google Scholar] [CrossRef]
- COMMISSION REGULATION (EC) No 2073/2005 of 15 November 2005 on Microbiological Criteria for Foodstuffs. Available online: https://eur-lex.europa.eu/eli/reg/2005/2073 (accessed on 9 February 2022).
- Jadhav, S.; Bhave, M.; Palombo, E.A. Methods used for the detection and subtyping of Listeria monocytogenes. J. Microbiol. Methods 2012, 88, 327–341. [Google Scholar] [CrossRef]
- Bernardo, R.; Duarte, A.; Tavares, L.; Barreto, A.S.; Henriques, A.R. Listeria monocytogenes Assessment in a Ready-to-Eat Salad Shelf-Life Study Using Conventional Culture-Based Methods, Genetic Profiling, and Propidium Monoazide Quantitative PCR. Foods 2021, 10, 235. [Google Scholar] [CrossRef]
- Datta, A.R.; Burall, L.S. Serotype to genotype: The changing landscape of listeriosis outbreak investigations. Food Microbiol. 2018, 75, 18–27. [Google Scholar] [CrossRef]
- Sumrall, E.T.; Röhrig, C.; Hupfeld, M.; Selvakumar, L.; Du, J.; Dunne, M.; Schmelcher, M.; Shen, Y.; Loessner, M.J. Glycotyping and Specific Separation of Listeria monocytogenes with a Novel Bacteriophage Protein Tool Kit. Appl. Environ. Microbiol. 2020, 86, e00612-20. [Google Scholar] [CrossRef]
- Bhagwat, A.; Mixon, M.; Collins, C.H.; Dordick, J.S. Opportunities for broadening the application of cell wall lytic enzymes. Appl. Microbiol. Biotechnol. 2020, 104, 9019–9040. [Google Scholar] [CrossRef]
- Hagens, S.; de Wouters, T.; Vollenweider, P.; Loessner, M.J. Reporter bacteriophage A511::celB transduces a hyperthermostable glycosidase from Pyrococcus furiosus for rapid and simple detection of viable Listeria cells. Bacteriophage 2011, 1, 143–151. [Google Scholar] [CrossRef][Green Version]
- Crowley, E.; Bird, P.; Flannery, J.; Benzinger, M.J., Jr.; Fisher, K.; Boyle, M.; Huffman, T.; Bastin, B.; Bedinghaus, P.; Judd, W.; et al. Evaluation of VIDAS® UP Listeria assay (LPT) for the detection of Listeria in a variety of foods and environmental surfaces: First Action 2013.10. J. AOAC Int. 2014, 97, 431–441. [Google Scholar] [CrossRef] [PubMed]
- VIDAS®, L. Monocytogenes Xpress (LMX) Ultra Performance Summary. Available online: https://www.biomerieux-usa.com/sites/subsidiary_us/files/vidas_l._monocytogenes_xpress_8.5x11_v4.pdf (accessed on 9 February 2022).
- Junillon, T.; Vimont, A.; Mosticone, D.; Mallen, B.; Baril, F.; Rozand, C.; Flandrois, J.P. Simplified detection of food-borne pathogens: An in situ high affinity capture and staining concept. J. Microbiol. Methods 2012, 91, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Born, F.; Braun, P.; Scholz, H.C.; Grass, G. Specific detection of yersinia pestis based on receptor binding proteins of phages. Pathogens 2020, 9, 611. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Martín, I.; Sainz-Mejías, M.; McClean, S. Pseudomonas aeruginosa: An Audacious Pathogen with Adaptable Arsenal of Virulence Factors. Int. J. Mol. Sci. 2021, 22, 3128. [Google Scholar] [CrossRef] [PubMed]
- Blanc, D.S.; Petignat, C.; Janin, B.; Bille, J.; Francioli, P. Frequency and molecular diversity of Pseudomonas aeruginosa upon admission and during hospitalization: A prospective epidemiologic study. Clin. Microbiol. Infect. 1998, 4, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Strateva, T.; Yordanov, D. Pseudomonas aeruginosa—A phenomenon of bacterial resistance. J. Med. Microbiol. 2009, 58, 1133–1148. [Google Scholar] [CrossRef]
- He, Y.; Shi, Y.; Liu, M.; Wang, Y.; Wang, L.; Lu, S.; Fu, Z. Nonlytic Recombinant Phage Tail Fiber Protein for Specific Recognition of Pseudomonas aeruginosa. Anal. Chem. 2018, 90, 14462–14468. [Google Scholar] [CrossRef]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Kluytmans, J.; van Belkum, A.; Verbrugh, H. Nasal carriage of Staphylococcus aureus: Epidemiology underlying mechanisms, and associated risks. Clin. Microbiol. Rev. 1997, 10, 505–520. [Google Scholar] [CrossRef]
- Chambers, H.F.; DeLeo, F.R. Waves of Resistance: Staphylococcus aureus in the Antibiotic Era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef]
- Laupland, K.B.; Lyytikäinen, O.; Søgaard, M.; Kennedy, K.J.; Knudsen, J.D.; Ostergaard, C.; Galbraith, J.C.; Valiquette, L.; Jacobsson, G.; Collignon, P.; et al. The changing epidemiology of Staphylococcus aureus bloodstream infection: A multinational population—Based surveillance study. Clin. Microbiol. Infect. 2013, 19, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, S.L.; Hulten, K.G.; Gonzalez, B.E.; Hammerman, W.A.; Lamberth, L.; Versalovic, J.; Mason, E.O., Jr. Three-year surveillance of community-acquired Staphylococcus aureus infections in children. Clin. Infect. Dis. 2005, 40, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Han, L.; Cai, Y.; Ren, H.; Ma, T.; Li, X.; Petrenko, V.A.; Liu, A. Colorimetric Assay of Bacterial Pathogens Based on Co3O4 Magnetic Nanozymes Conjugated with Specific Fusion Phage Proteins and Magnetophoretic Chromatography. ACS Appl. Mater. Interfaces 2020, 12, 9090–9097. [Google Scholar] [CrossRef]
- Agudelo Higuita, N.I.; Huycke, M.M. Enterococcal Disease, Epidemiology, and Implications for Treatment. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Massachusetts Eye and Ear Infirmary: Boston, MD, USA, 2014; NBK190429. [Google Scholar]
- Hall, L.M.; Duke, B.; Urwin, G.; Guiney, M. Epidemiology of Enterococcus faecalis urinary track infection in a teaching hospital in London, United Kingdom. J. Clin. Microbiol. 1992, 30, 1953–1957. [Google Scholar] [CrossRef] [PubMed]
- Noskin, G.A.; Peterson, L.R.; Warren, J.R. Enterococcus faecium and Enterococcus faecalis bacteremia: Acquisition and outcome. Clin. Infect. Dis. 1995, 20, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Hidron, A.I.; Edwards, J.R.; Patel, J.; Horan, T.C.; Sievert, D.M.; Pollock, D.A.; Fridkin, S.K. National Healthcare Safety Network Team. NHSN annual update: Antimicrobial-resistant pathogens associated with healthcare-associated infections: Annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control Hosp. Epidemiol. 2008, 29, 996–1011. [Google Scholar] [CrossRef]
- Foudeh, A.M.; Didar, T.F.; Veres, T.; Tabrizian, M. Microfluidic designs and techniques using lab-on-a-chip devices for pathogen detection for point-of-care diagnostics. Lab Chip 2012, 12, 3249–3266. [Google Scholar] [CrossRef]
- Santos, S.B.; Cunha, A.P.; Macedo, M.; Nogueira, C.L.; Brandão, A.; Costa, S.P.; Melo, L.D.R.; Azeredo, J.; Carvalho, C.M. Bacteriophage-receptor binding proteins for multiplex detection of Staphylococcus and Enterecoccus in blood. Biotechnol. Bioeng. 2020, 117, 3286–3298. [Google Scholar] [CrossRef]
- Patel, J.D.; O’Carra, R.; Jones, J.; Woodward, J.G.; Mumper, R.J. Preparation and characterization of nickel nanoparticles for binding to his-tag proteins and antigens. Pharm. Res. 2007, 24, 343–352. [Google Scholar] [CrossRef]
- Cunha, A.P.; Henriques, R.; Cardoso, S.; Freitas, P.P.; Carvalho, C.M. Rapid and multiplex detection of nosocomial pathogens on a phage-based magnetoresistive lab-on-chip platform. Biotechnol. Bioeng. 2021, 118, 3164–3174. [Google Scholar] [CrossRef]
- Majowicz, S.E.; Musto, J.; Scallan, E.; Angulo, F.J.; Kirk, M.; O’Brien, S.J.; Jones, T.F.; Fazil, A.; Hoekstra, R.M.; for the International Collaboration on Enteric Disease “Burden of Illness” Studies. The global burden of nontyphoidal Salmonella gastroenteritis. Clin. Infect. Dis. 2010, 50, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, A.; Losasso, C.; Barco, L.; Eckert, E.M.; Conficoni, D.; Sarasini, G.; Corno, G.; Ricci, A. Diverse distribution of Toxin-Antitoxin II systems in Salmonella enterica serovars. Sci. Rep. 2016, 6, 28759. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.L.; Jarvis, K.G.; Ottesen, A.R.; McFarland, M.A.; Brown, E.W. Recent and emerging innovations in Salmonella detection: A food and environmental perspective. Microb. Biotechnol. 2016, 9, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.P.; Frank, K.M. Salmonella, Shigella, and yersinia. Clin. Lab. Med. 2015, 35, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Priyanka, B.; Patil, R.K.; Dwarakanath, S. A review on detection methods used for foodborne pathogens. Indian J. Med. Res. 2016, 144, 327. [Google Scholar] [CrossRef]
- Singh, A.; Arya, S.K.; Glass, N.; Hanifi-Moghaddam, P.; Naidoo, R.; Szymanski, C.M.; Tanha, J.; Evoy, S. Bacteriophage tailspike proteins as molecular probes for sensitive and selective bacterial detection. Biosens. Bioelectron. 2010, 26, 131–138. [Google Scholar] [CrossRef]
- Walter, M.; Fiedler, C.; Grassl, R.; Biebl, M.; Rachel, R.; Hermo-Parrado, X.L.; Llamas-Saiz, A.L.; Seckler, R.; Miller, S.; van Raaij, M.J. Structure of the receptor-binding protein of bacteriophage det7: A podoviral tail spike in a myovirus. J. Virol. 2008, 82, 2265–2273. [Google Scholar] [CrossRef]
- Lazcka, O.; Del Campo, F.J.; Munoz, F.X. Pathogen detection: A perspective of traditional methods and biosensors. Biosens. Bioelectron. 2007, 22, 1205–1217. [Google Scholar] [CrossRef]
- Hyeon, S.H.; Lim, W.K.; Shin, H.J. Novel surface plasmon resonance biosensor that uses full-length Det7 phage tail protein for rapid and selective detection of Salmonella enterica serovar Typhimurium. Biotechnol. Appl. Biochem. 2021, 68, 5–12. [Google Scholar] [CrossRef]
- Edgar, R.H. Evolution of Bacteriophage Host Attachment Using Det7 as a Model. Master’s Thesis, University of Pittsburgh, Pittsburgh, PA, USA, 4 February 2014. [Google Scholar]
- Odumeru, J.A.; León-Velarde, C.G. Salmonella detection methods for food and food ingredients. In Salmonella-A Dangerous Foodborne Pathogen; In Tec: Rijeka, Croatia, 2012; pp. 373–392. [Google Scholar]
- Liang, B.; Roberts, A.P.; Xu, X.; Yang, C.; Yang, X.; Wang, J.; Yi, S.; Li, Y.; Ma, Q.; Wu, F.; et al. Transferable plasmid-borne mcr-1 in a colistin-resistant Shigella flexneri isolate. Appl. Environ. Microbiol. 2018, 84, e02655-17. [Google Scholar] [CrossRef]
- Warren, B.R.; Parish, M.E.; Schneider, K.R. Shigella as a foodborne pathogen and current methods for detection in food. Crit. Rev. Food Sci. Nutr. 2006, 46, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Muthuirulandi Sethuvel, D.P.; Devanga Ragupathi, N.K.; Anandan, S.; Veeraraghavan, B. Update on: Shigella new serogroups/serotypes and their antimicrobial resistance. Lett. Appl. Microbiol. 2017, 64, 8–18. [Google Scholar] [CrossRef] [PubMed]
- DuPont, H.L.; Levine, M.M.; Hornick, R.B.; Formal, S.B. Inoculum size in shigellosis and implications for expected mode of transmission. J. Infect. Dis. 1989, 159, 1126–1128. [Google Scholar] [CrossRef]
- Kunstmann, S.; Scheidt, T.; Buchwald, S.; Helm, A.; Mulard, L.A.; Fruth, A.; Barbirz, S. Bacteriophage Sf6 tailspike protein for detection of Shigella flexneri pathogens. Viruses 2018, 10, 431. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Rabsch, W.; Broeker, N.K.; Barbirz, S. Bacteriophage tailspike protein based assay to monitor phase variable glucosylations in Salmonella O-antigens. BMC Microbiol. 2016, 16, 207. [Google Scholar] [CrossRef]
- Spencer, R.C. Bacillus anthracis. J. Clin. Pathol. 2003, 56, 182–187. [Google Scholar] [CrossRef]
- Swartz, M.N. Recognition and management of anthrax—An update. N. Engl. J. Med. 2001, 345, 1607–1610. [Google Scholar] [CrossRef]
- Salmanov, A.; Litus, V.; Vdovychenko, S.; Litus, O.; Davtian, L.; Ubogov, S.; Bisyuk, Y.; Drozdova, A.; Vlasenko, I. Healthcare-Associated Infections in Intensive Care Units. Wiad. Lek. 2019, 72, 963–969. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. Surveillance of Antimicrobial Resistance in Europe: Annual Report of the European Antimicrobial Resistance Surveillance Network (EARS-Net) 2017; ECDC: Solna, Sweden, 2018.
- World Health Organization. Central Asian and European Surveillance of Antimicrobial Resistance: Annual Report 2019; World Health Organization: Geneva, Switzerland, 2019.
- Suetens, C.; Latour, K.; Kärki, T.; Ricchizzi, E.; Kinross, P.; Moro, M.L.; Jans, B.; Hopkins, S.; Hansen, S.; Lyytikäinen, O.; et al. Prevalence of Healthcare-Associated Infections, Estimated Incidence and Composite Antimicrobial Resistance Index in Acute Care Hospitals and Long-Term Care Facilities: Results from Two European Point Prevalence Surveys, 2016 to 2017. Eurosurveillance 2018, 23, 1800516. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target Species | Capture Method | Detection (Visualization) Method | Limit of Detection | Reference |
|---|---|---|---|---|
| Acinetobacter baumannii | Sandwich fluorescence assay | Fluorescence (FITC-labeled probes) | 6.2 × 102 CFU/mL | [58] |
| Magnetic beads coated with TFP | Bioluminescence (ATP release with luciferin/luciferase detection) | |||
| Magnetic nanoparticles coated with TFP | MALDI-TOF MS | ∼2.34 × 105 and ∼4.48 × 104 CFU/mL, depending on the strain | [59] | |
| Acinetobacter baumannii Pse + other Pse bacteria (Pse–Ppseudoaminic acid) | Incubation in solution | Fluorescently labeled probe | - | [60] |
| TFP-coated microplate wells | FITC labeling of bacteria | |||
| Enterococcus faecalis, Enterococcus faecium | Magnetic nanoparticles coated with His-tagged TFP | Array of spin-valve sensors on the biochip | 10 CFU/mL | [61] |
| Staphylococcus aureus | ||||
| Pseudomonas aeruginosa | Magnetic particles | Magnetic separation | 6.7 × 102 CFU/mL and 1.7 × 102 CFU/mL | [62] |
| Fluorescent labeling by TRITC | Fluorescent microscopy | |||
| Yersinia pestis | Fluorescent probe (RBP proteins from Y. pestis phages φA1122 and L-413C) | Fluorescent microscopy | - | [63] |
| Campylobacter jejuni, Campylobacter coli | Microresonator functionalized with the GST-Gp48 tailspike | Biosensors | - | [64] |
| RBP and GFP-coupled RBP (Gp047) | Agglutination assay on glass slide and fluorescent microscopy | - | [65] | |
| Salmonella spp. | TSP-coated gold/incubation in solution | SEM or SPR | 103 CFU/mL in case of SPR | [66] |
| Det7T loaded to the gold-coated surfaces of a CM5 chip | SPR | 5 × 107 CFU/mL | [67] | |
| Metal beads conjugated with HRP-LTF; incubation in solution | enzyme-linked LTF assay (ELLTA) | 102 CFU/mL | ||
| Shigella spp. | Sf6TSP cloned with Strep-Tag coated microplate wells | ELISA-like tailspike adsorption assay (ELITA) | 103 CFU/mL | [68] |
| Bacillus anthracis | NanoLuc-RBPλ03Δ1-120, RBP conjugated with commercial luciferase | Enzyme-linked phage receptor binding protein assay (ELPRA)-luminescence | - | [69] |
| HRP moiety directly conjugated to the RBPλ03Δ1-120 | ELPRA-colorimetric assay | |||
| Klebsiella pneumoniae | Gp86 RBP fused with mCherry | Fluorescence microscopy and RBP-based fluorescent spectroscopy | - | [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filik, K.; Szermer-Olearnik, B.; Oleksy, S.; Brykała, J.; Brzozowska, E. Bacteriophage Tail Proteins as a Tool for Bacterial Pathogen Recognition—A Literature Review. Antibiotics 2022, 11, 555. https://doi.org/10.3390/antibiotics11050555
Filik K, Szermer-Olearnik B, Oleksy S, Brykała J, Brzozowska E. Bacteriophage Tail Proteins as a Tool for Bacterial Pathogen Recognition—A Literature Review. Antibiotics. 2022; 11(5):555. https://doi.org/10.3390/antibiotics11050555
Chicago/Turabian StyleFilik, Karolina, Bożena Szermer-Olearnik, Sabina Oleksy, Jan Brykała, and Ewa Brzozowska. 2022. "Bacteriophage Tail Proteins as a Tool for Bacterial Pathogen Recognition—A Literature Review" Antibiotics 11, no. 5: 555. https://doi.org/10.3390/antibiotics11050555
APA StyleFilik, K., Szermer-Olearnik, B., Oleksy, S., Brykała, J., & Brzozowska, E. (2022). Bacteriophage Tail Proteins as a Tool for Bacterial Pathogen Recognition—A Literature Review. Antibiotics, 11(5), 555. https://doi.org/10.3390/antibiotics11050555