
Antimicrobial Activity and 70S Ribosome Binding of Apidaecin-Derived Api805 with Increased Bacterial Uptake Rate


Abstract
:1. Introduction
2. Results
2.1. BW25113 Is Not Susceptible to Api805
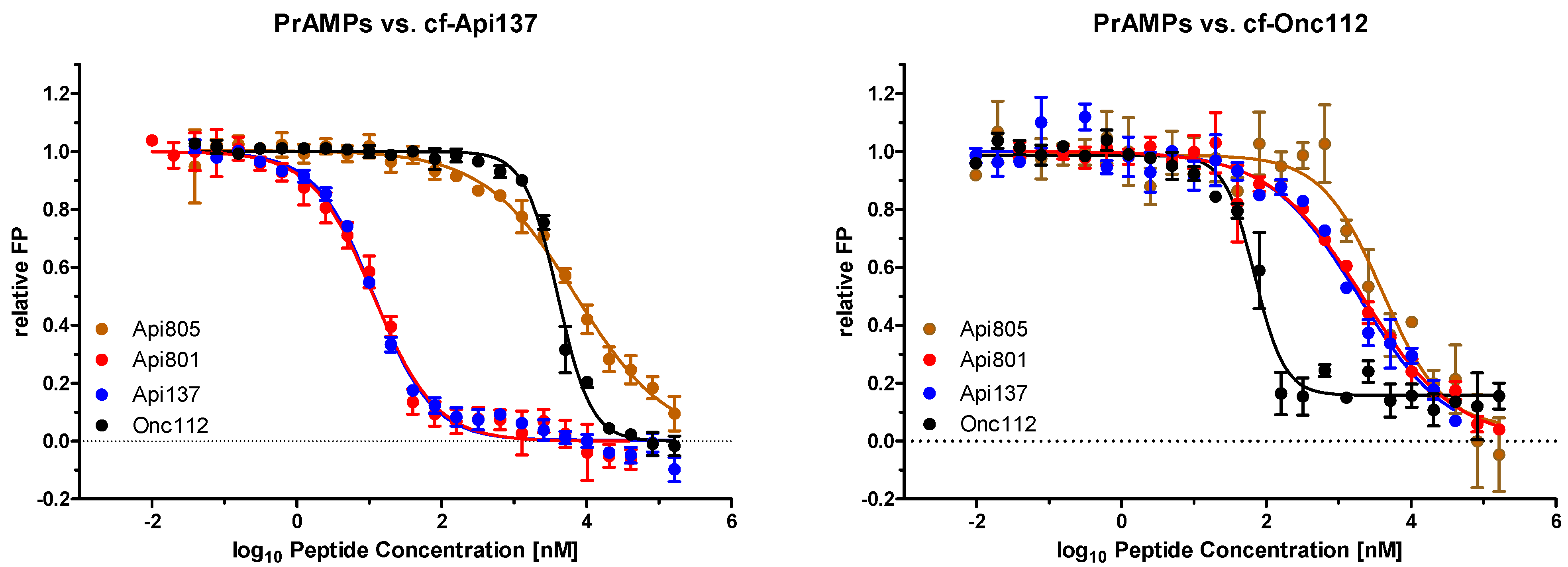
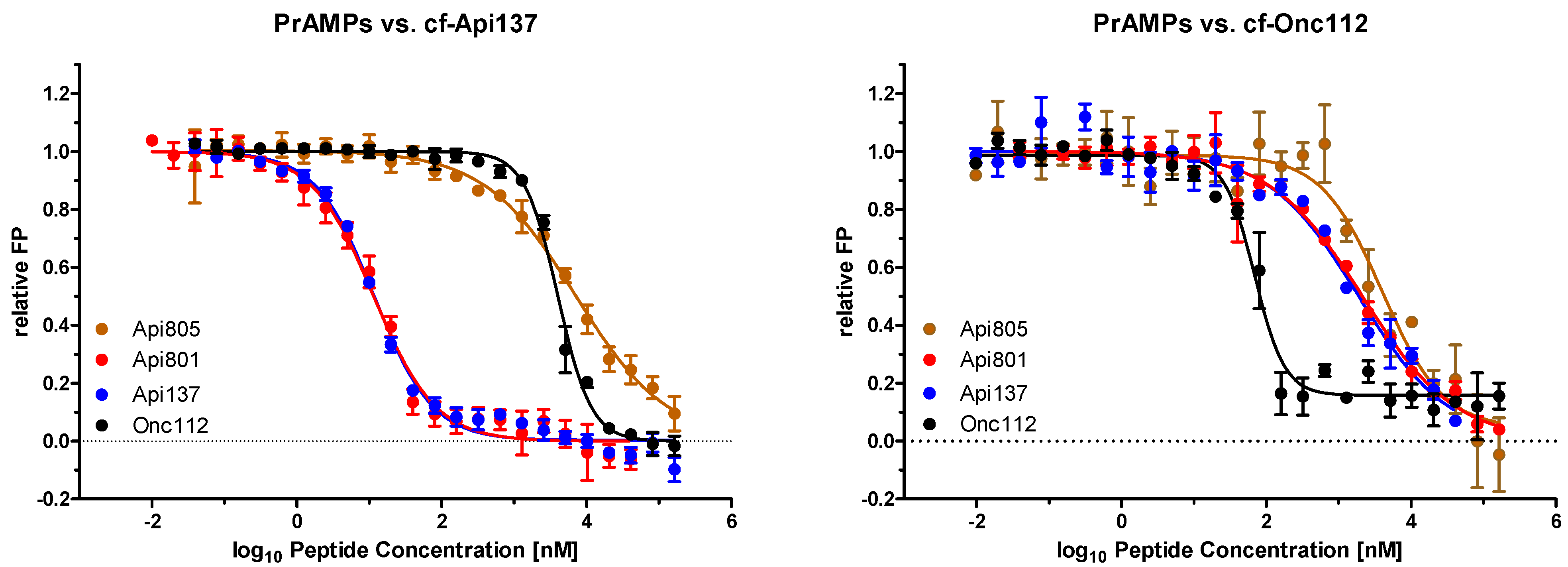
2.2. Api805 Binding to Riboome Extracts of E. coli BW25113 and Rosetta
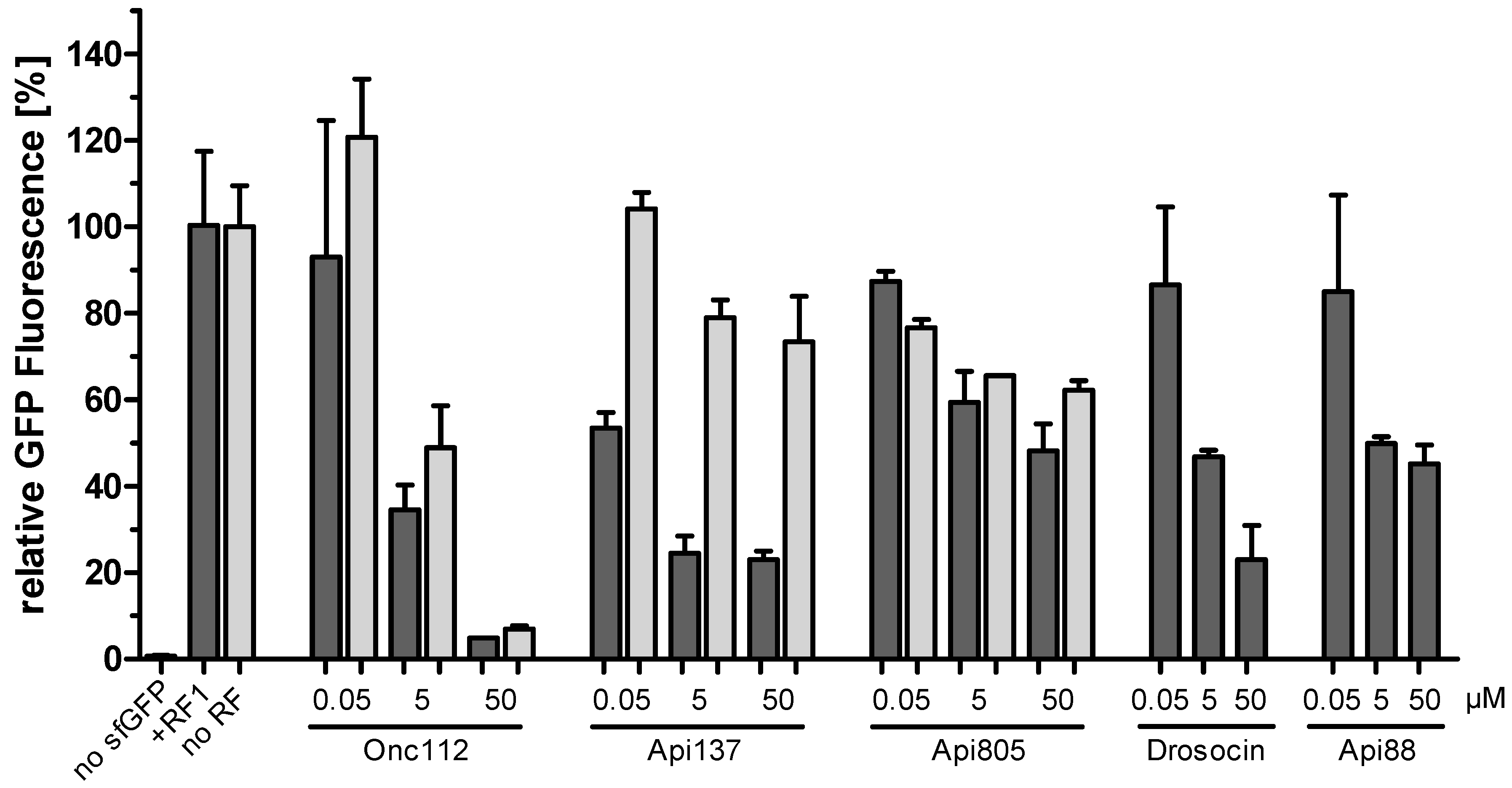
2.3. Api805 Inhibits Protein Translation
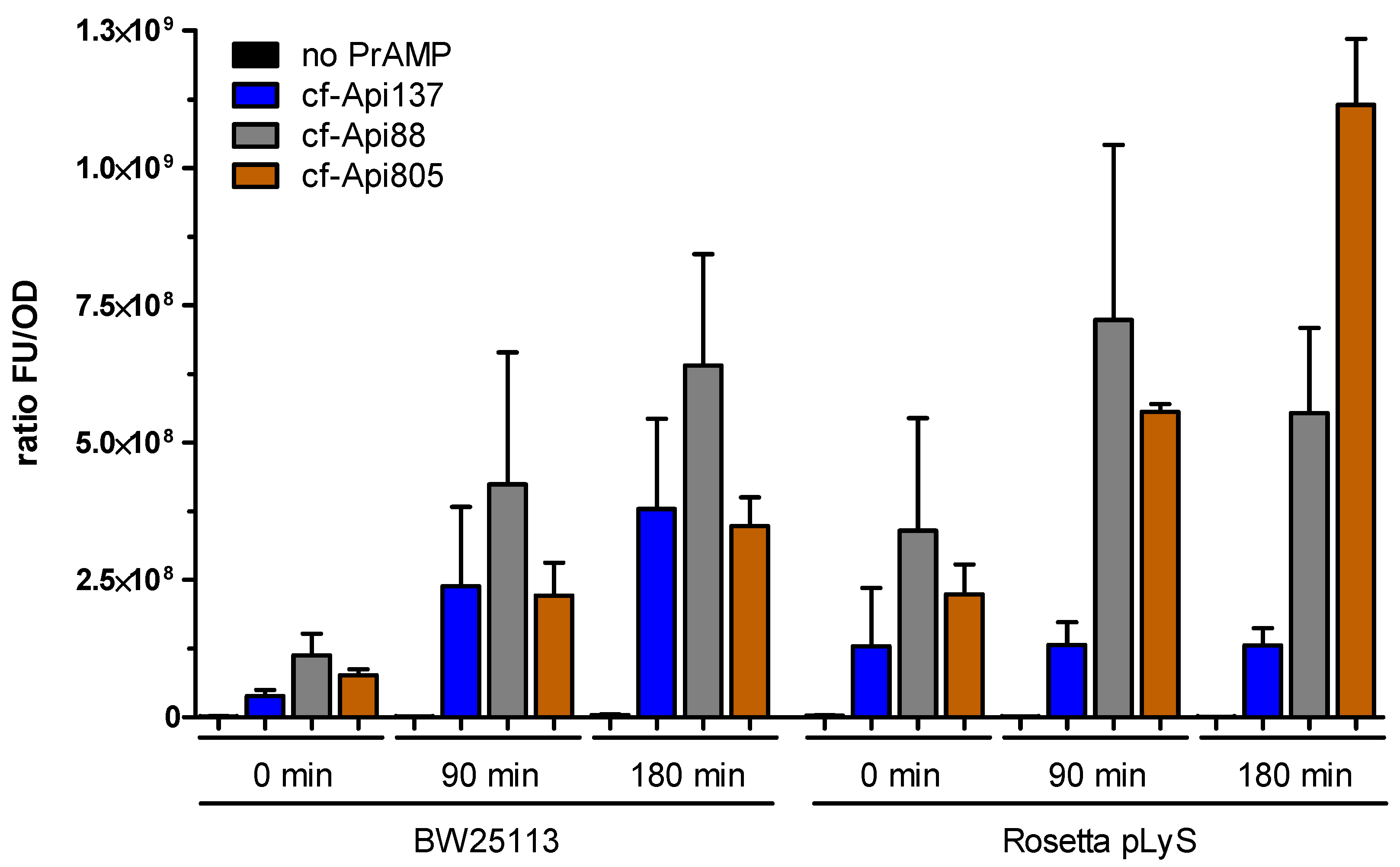
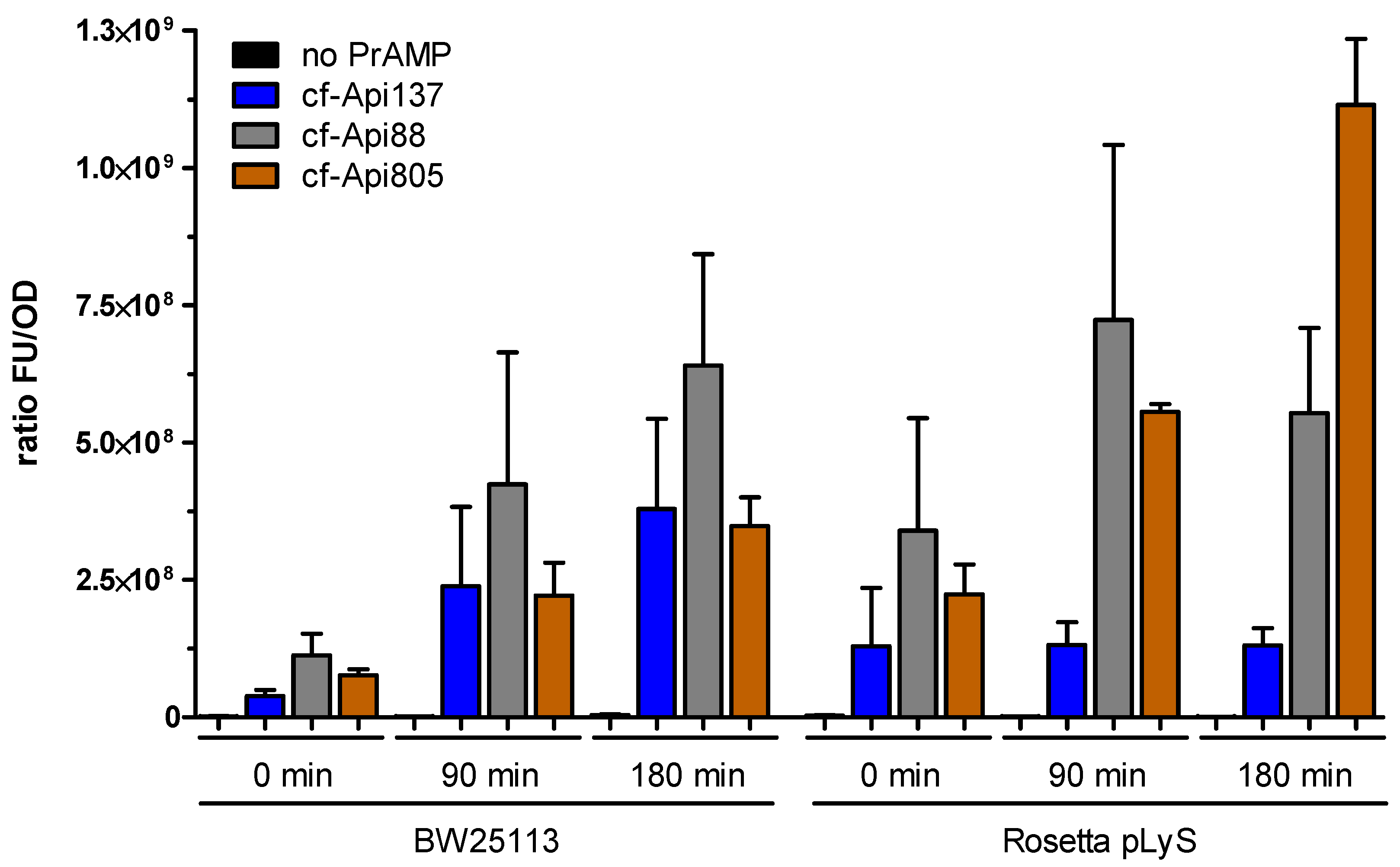
2.4. Bacterial Uptake of Api805
2.5. Api805 Uptake in E. coli Cell Lines with a Disturbed SbmA Transporter
3. Discussion
4. Materials and Methods
4.1. Materials and Chemicals
4.2. Peptide Synthesis
4.3. Antibacterial Activity
4.4. Extraction of E. coli Ribosomes
4.5. Fluorescence Polarization
4.6. In Vitro Translation Assay
4.7. Uptake of cf-Labeled PrAMPs
4.8. Transposon Mutagenesis
4.9. Peptide Stability in E. coli Lysates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations the Review on Antimicrobial Resistance Chaired; Review on Antimicrobial Resistance: London, UK, 2014. [Google Scholar]
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [Green Version]
- Otvos, L. Antibacterial peptides isolated from insects. J. Pept. Sci. 2000, 6, 497–511. [Google Scholar] [CrossRef]
- Krizsan, A.; Volke, D.; Weinert, S.; Sträter, N.; Knappe, D.; Hoffmann, R. Insect-Derived Proline-Rich Antimicrobial Peptides Kill Bacteria by Inhibiting Bacterial Protein Translation at the 70 S Ribosome. Angew. Chemie Int. Ed. 2014, 53, 12236–12239. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L. The short proline-rich antibacterial peptide family. Cell Mol. Life Sci. 2002, 59, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.; Schäfer, I.; Knappe, D.; Seibel, P.; Hoffmann, R. Intracellular toxicity of proline-rich antimicrobial peptides shuttled into mammalian cells by the cell-penetrating peptide penetratin. Antimicrob. Agents Chemother. 2012, 56, 5194–5201. [Google Scholar] [CrossRef] [Green Version]
- Knappe, D.; Piantavigna, S.; Hansen, A.; Mechler, A.; Binas, A.; Nolte, O.; Martin, L.L.; Hoffmann, R. Oncocin (VDKPPYLPRPRPPRRIYNR-NH2): A novel antibacterial peptide optimized against gram-negative human pathogens. J. Med. Chem. 2010, 53, 5240–5247. [Google Scholar] [CrossRef]
- Mattiuzzo, M.; Bandiera, A.; Gennaro, R.; Benincasa, M.; Pacor, S.; Antcheva, N.; Scocchi, M. Role of the Escherichia coli SbmA in the antimicrobial activity of proline-rich peptides. Mol. Microbiol. 2007, 66, 151–163. [Google Scholar] [CrossRef]
- Krizsan, A.; Knappe, D.; Hoffmann, R. Influence of the yjiL-mdtM gene cluster on the antibacterial activity of proline-rich antimicrobial peptides overcoming Escherichia coli resistance induced by the missing SbmA transporter system. Antimicrob. Agents Chemother. 2015, 59, 5992–5998. [Google Scholar] [CrossRef] [Green Version]
- Graf, M.; Mardirossian, M.; Nguyen, F.; Seefeldt, A.C.; Guichard, G.; Scocchi, M.; Innis, C.A.; Wilson, D.N. Proline-rich antimicrobial peptides targeting protein synthesis. Nat. Prod. Rep. 2017, 34, 702–711. [Google Scholar] [CrossRef]
- Krizsan, A.; Prahl, C.; Goldbach, T.; Knappe, D.; Hoffmann, R. Short Proline-Rich Antimicrobial Peptides Inhibit Either the Bacterial 70S Ribosome or the Assembly of its Large 50S Subunit. ChemBioChem 2015, 16, 2304–2308. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Wilson, D.N. Intracellular Antimicrobial Peptides Targeting the Protein Synthesis Machinery. Antimicrob. Pept. 2019, 117, 73–89. [Google Scholar] [CrossRef]
- Berthold, N.; Czihal, P.; Fritsche, S.; Sauer, U.; Schiffer, G.; Knappe, D.; Alber, G.; Hoffmann, R. Novel apidaecin 1b analogs with superior serum stabilities for treatment of infections by Gram-negative pathogens. Antimicrob. Agents Chemother. 2013, 57, 402–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florin, T.; Maracci, C.; Graf, M.; Karki, P.; Klepacki, D.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Wilson, D.N.; Rodnina, M.V.; et al. An antimicrobial peptide that inhibits translation by trapping release factors on the ribosome. Nat. Struct. Mol. Biol. 2017, 24, 752–757. [Google Scholar] [CrossRef]
- Schmidt, A.; Kochanowski, K.; Vedelaar, S.; Ahrné, E.; Volkmer, B.; Callipo, L.; Knoops, K.; Bauer, M.; Aebersold, R.; Heinemann, M. The quantitative and condition-dependent Escherichia coli proteome. Nat. Biotechnol. 2016, 34, 104–110. [Google Scholar] [CrossRef]
- Seefeldt, A.C.; Nguyen, F.; Antunes, S.; Pérébaskine, N.; Graf, M.; Arenz, S.; Inampudi, K.K.; Douat, C.; Guichard, G.; Wilson, D.N.; et al. The proline-rich antimicrobial peptide Onc112 inhibits translation by blocking and destabilizing the initiation complex. Nat. Struct. Mol. Biol. 2015, 22, 470–475. [Google Scholar] [CrossRef]
- Czihal, P.; Knappe, D.; Fritsche, S.; Zahn, M.; Berthold, N.; Piantavigna, S.; Müller, U.; Van Dorpe, S.; Herth, N.; Binas, A.; et al. Api88 Is a Novel Antibacterial Designer Peptide To Treat Systemic Infections with Multidrug-Resistant Gram-Negative Pathogens. ACS Chem. Biol. 2012, 7, 1281–1291. [Google Scholar] [CrossRef]
- Bulet, P.; Dimarcq, J.L.; Hetru, C.; Lagueux, M.; Charlet, M.; Hegy, G.; Van Dorsselaer, A.; Hoffmann, J.A. A novel inducible antibacterial peptide of Drosophila carries an O- glycosylated substitution. J. Biol. Chem. 1993, 268, 14893–14897. [Google Scholar] [CrossRef]
- Knappe, D.; Kabankov, N.; Hoffmann, R. Bactericidal oncocin derivatives with superior serum stabilities. Int. J. Antimicrob. Agents 2011, 37, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Bremer, H.; Dennis, P.P. Modulation of Chemical Composition and Other Parameters of the Cell at Different Exponential Growth Rates. EcoSal Plus 2008, 3, ecosal.5.2.3. [Google Scholar] [CrossRef]
- Adio, S.; Sharma, H.; Senyushkina, T.; Karki, P.; Maracci, C.; Wohlgemuth, I.; Holtkamp, W.; Peske, F.; Rodnina, M.V. Dynamics of ribosomes and release factors during translation termination in E. coli. Elife 2018, 7, e34252. [Google Scholar] [CrossRef] [PubMed]
- Dreyfus, M.; Heurgué-Hamard, V. Termination troubles in Escherichia coli K12. Mol. Microbiol. 2011, 79, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Florin, M.; Mangano, K.; Florin, T.; Shao, X.; Klepacki, D.; Chelysheva, I. Genome-wide effects of the antimicrobial peptide apidaecin on translation termination. Elife 2020, 9, e62655. [Google Scholar]
- Schavemaker, P.E.; Śmigiel, W.M.; Poolman, B. Ribosome surface properties may impose limits on the nature of the cytoplasmic proteome. Elife 2017, 6, e30084. [Google Scholar] [CrossRef]
- Volkmer, B.; Heinemann, M. Condition-Dependent cell volume and concentration of Escherichia coli to facilitate data conversion for systems biology modeling. PLoS ONE 2011, 6, e23126. [Google Scholar] [CrossRef] [Green Version]
- Holfeld, L.; Hoffmann, R.; Knappe, D. Correlating uptake and activity of proline-rich antimicrobial peptides in Escherichia coli. Anal. Bioanal. Chem. 2017, 409, 5581–5592. [Google Scholar] [CrossRef]
- Ghilarov, D.; Inaba-inoue, S.; Stepien, P.; Qu, F.; Michalczyk, E.; Pakosz, Z.; Nomura, N.; Ogasawara, S.; Walker, G.C.; Rebuffat, S.; et al. Molecular mechanism of SbmA, a promiscuous transporter exploited by antimicrobial peptides. Sci. Adv. 2021, 7, eabj5363. [Google Scholar] [CrossRef]
- Knappe, D.; Kabankov, N.; Herth, N.; Hoffmann, R. Insect-derived short proline-rich and murine cathelicidin-related antimicrobial peptides act synergistically on Gram-negative bacteria in vitro. Future Med. Chem. 2016, 8, 1035–1045. [Google Scholar] [CrossRef]
- Schmidt, R.; Krizsan, A.; Volke, D.; Knappe, D.; Hoffmann, R. Identification of new resistance mechanisms in Escherichia coli against apidaecin 1b using quantitative gel- and LC-MS-based proteomics. J. Proteome Res. 2016, 15, 2607–2617. [Google Scholar] [CrossRef]
- Schneider, D.; Duperchy, E.; Depeyrot, J.; Coursange, E.; Lenski, R.E.; Blot, M. Genomic comparisons among Escherichia coli strains B, K-12, and O157:H7using IS elements as molecular markers. BMC Microbiol. 2002, 2, 18. [Google Scholar] [CrossRef] [Green Version]
- Jansson, P.-E.; Lindberg, A.A.; Lindberg, B.; Wollin, R. Structural Studies on the Hexose Region of the Core in Lipopolysaccharides from Enterobacteriaceae. Eur. J. Biochem. 1981, 115, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Quillardet, P.; de Bellecombe, C.; Hofnung, M. The SOS Chromotest, a colorimetric bacterial assay for genotoxins: Validation study with 83 compounds. Mutat. Res. Mutagen. Relat. Subj. 1985, 147, 79–95. [Google Scholar] [CrossRef]
- Herrera, G.; MArtinez, A.; Blanco, M.; O’Connor, J.-E. Assessment of Escherichia coli B With Enhanced Permeability to Fluorochromes for Flow Cytometric Assays of Bacterial Cell Function. Clin. Cytom. 2002, 50, 239–242. [Google Scholar] [CrossRef]
- Ebbensgaard, A.; Mordhorst, H.; Aarestrup, F.M.; Hansen, E.B. The role of outer membrane proteins and lipopolysaccharides for the sensitivity of escherichia coli to antimicrobial peptides. Front. Microbiol. 2018, 9, 2153. [Google Scholar] [CrossRef] [Green Version]
- Mathias, U.; Jung, M. Determination of drug-serum protein interactions via fluorescence polarization measurements. Anal. Bioanal. Chem. 2007, 388, 1147–1156. [Google Scholar] [CrossRef]
- Kleckner, N.; Barker, D.F.; Ross, D.G.; Botstein, D. Properties of the translocatable tetracycline-resistance element Tn10 in Escherichia coli and bacteriophage lambda. Genetics 1978, 90, 427–461. [Google Scholar] [CrossRef]
- Kleckner, N.; Bender, J.; Gottesman, S. Uses of transposons with emphasis on Tn10. Methods Enzymol. 1991, 204, 139–180. [Google Scholar]
- Wilson, K. Preparation of genomic DNA from bacteria. Curr. Protoc. Mol. Biol. 2001, 56, 2–4. [Google Scholar] [CrossRef]
- Das, S.; Noe, J.C.; Paik, S.; Kitten, T. An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J. Microbiol. Methods 2005, 63, 89–94. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Peptides | Sequence | Reference |
|---|---|---|
| Api137 | gu-ONNRPVYIPRPRPPHPRL-OH | [14] |
| Api88 | gu-ONNRPVYIPRPRPPHPRL-NH2 | [18] |
| Api801 | GNNRPIYIPRPRPPHPRL-OH | This publication |
| Api805 | GNNRPIYIPRPRPPHPRPIRV-OH | |
| Drosocin | GKPRPYSPRPTSHPRPIRV-OH | [19] |
| Onc112 | VDKPPYLPRPRPPRrIYNr-NH2 | [20] |
| PrAMP | MIC [µg/mL] * | Kd [µmol/L] ** | Ki [µmol/L] *** | |||
|---|---|---|---|---|---|---|
| BW | Ros | BW | Ros | cf-Api137 | cf-Onc112 | |
| Api137 | 2 | 2 | 0.382 [0.337–0.432] | 0.198 [0.179–0.217] | 0.002 [0.0015–0.0020] | 0.219 [0.105–0.459] |
| Api801 | 16 | 4 | n.d. | n.d. | 0.002 [0.0015–0.0020] | 0.247 [0.172–0.354] |
| Api805 | 128 | 4 | 0.557 [0.475–0.652] | 0.515 [0.442–0.599] | 1.070 [0.769–1.489] | 0.489 [0.316–0.759] |
| Drosocin | 128 | 8 | n.d. | 1.08 [a] | 0.009 [a] | 1.78 [a] |
| Api88 | 2 | 1 | 0.906 [0.740–1.108] | 1.22 [a] | 0.18 [a] | 0.11 [a] |
| Onc112 | 4 | 4 | 0.027 [0.023–0.030] | 0.051 [0.044–0.061] | 0.606 [0.552–0.666] | 0.008 [0.007–0.011] |
| Strain | MIC (µg/mL) | ||||
|---|---|---|---|---|---|
| 25% MHBII | 33% TSB | ||||
| Api137 | Api805 | Api137 | Api805 | ||
| E. coli BL21 (DE3) | 4 | 4 | 2 | 2 | |
| sbmA::Tn10Cm | 16 | 128 | 16 | 128 | |
| sbmA::Tn10Cm + psbmA | 8 | 16 | 8 | 32 | |
| sbmA::Tn10Cm + psbmA_A31G/G219T | 4 | 16 | 8 | 32 | |
| BW25113 ΔsbmA | 8 | >128 | 64 | >128 | |
| +psbmA | 4 | >128 | 8 | >128 | |
| +psbmA_A31G/G219T | 2 | >128 | 8 | >128 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ludwig, T.; Krizsan, A.; Mohammed, G.K.; Hoffmann, R. Antimicrobial Activity and 70S Ribosome Binding of Apidaecin-Derived Api805 with Increased Bacterial Uptake Rate. Antibiotics 2022, 11, 430. https://doi.org/10.3390/antibiotics11040430
Ludwig T, Krizsan A, Mohammed GK, Hoffmann R. Antimicrobial Activity and 70S Ribosome Binding of Apidaecin-Derived Api805 with Increased Bacterial Uptake Rate. Antibiotics. 2022; 11(4):430. https://doi.org/10.3390/antibiotics11040430
Chicago/Turabian StyleLudwig, Tobias, Andor Krizsan, Gubran Khalil Mohammed, and Ralf Hoffmann. 2022. "Antimicrobial Activity and 70S Ribosome Binding of Apidaecin-Derived Api805 with Increased Bacterial Uptake Rate" Antibiotics 11, no. 4: 430. https://doi.org/10.3390/antibiotics11040430
APA StyleLudwig, T., Krizsan, A., Mohammed, G. K., & Hoffmann, R. (2022). Antimicrobial Activity and 70S Ribosome Binding of Apidaecin-Derived Api805 with Increased Bacterial Uptake Rate. Antibiotics, 11(4), 430. https://doi.org/10.3390/antibiotics11040430