
Phage-Host Interaction Analysis by Flow Cytometry Allows for Rapid and Efficient Screening of Phages



Abstract
:1. Introduction
2. Results
2.1. Real-Time Monitoring of P. aeruginosa PAO1 Infection with Two Different Phages Using Flow Cytometry
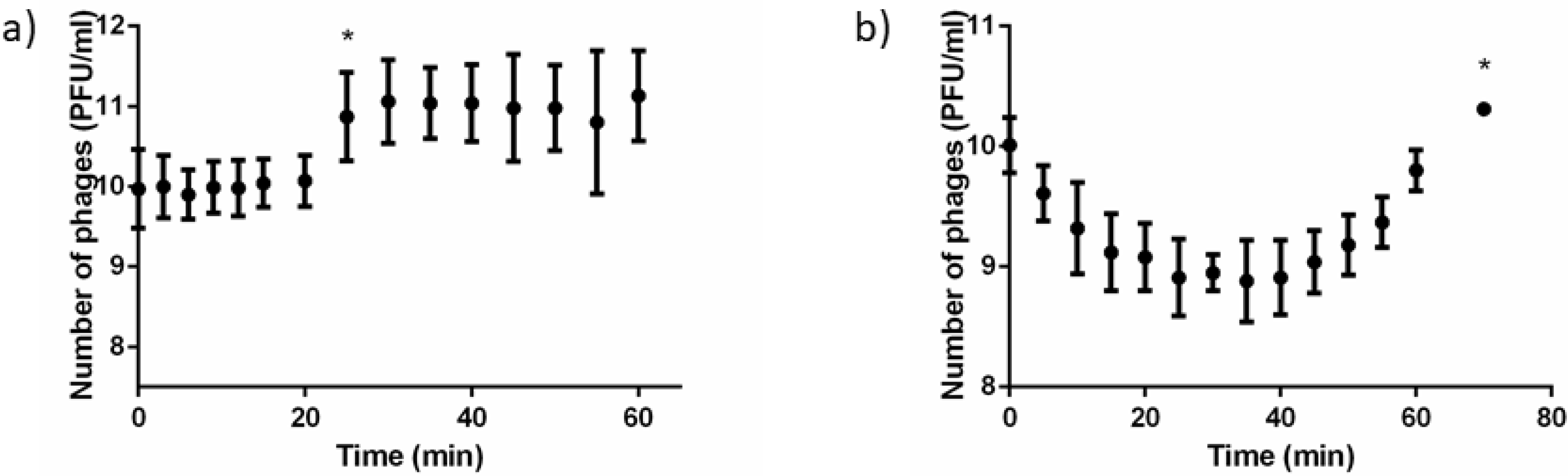
2.2. Phage Replication Assessed through Plaque Forming Units Counting
2.3. Gene Expression
3. Discussion
4. Materials and Methods
4.1. Bacterial Strain and Phages
4.2. Phage Production and Titration
4.3. Synchronized Infection Assays
4.4. Phage Quantification
4.5. Flow Cytometry
4.6. RNA Extraction
4.7. Gene Expression Quantification
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 31 December 2021).
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G., Jr. Bacteriophage Therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Ma, Y.; Wang, Y.; Yang, H.; Shen, W.; Chen, X. Transcription regulation mechanisms of bacteriophages: Recent advances and future prospects. Bioengineered 2014, 5, 300–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roucourt, B.; Lavigne, R. The role of interactions between phage and bacterial proteins within the infected cell: A diverse and puzzling interactome. Environ. Microbiol. 2009, 11, 2789–2805. [Google Scholar] [CrossRef]
- Leskinen, K.; Blasdel, B.G.; Lavigne, R.; Skurnik, M. RNA-Sequencing Reveals the Progression of Phage-Host Interactions between φR1-37 and Yersinia enterocolitica. Viruses 2016, 8, 111. [Google Scholar] [CrossRef] [Green Version]
- Tadmor, A.D.; Ottesen, E.A.; Leadbetter, J.R.; Phillips, R. Probing Individual Environmental Bacteria for Viruses by Using Microfluidic Digital PCR. Science 2011, 333, 58–62. [Google Scholar] [CrossRef] [Green Version]
- Barrero-Canosa, J.; Moraru, C. PhageFISH for Monitoring Phage Infections at Single Cell Level. Methods Mol. Biol. 2019, 1898, 1–26. [Google Scholar] [CrossRef]
- Álvarez-Barrientos, A.; Arroyo, J.; Cantón, R.; Nombela, C.; Sánchez-Pérez, M. Applications of Flow Cytometry to Clinical Microbiology. Clin. Microbiol. Rev. 2000, 13, 167–195. [Google Scholar] [CrossRef] [Green Version]
- Grainha, T.; Magalhães, A.P.; Melo, L.D.R.; Pereira, M.O. Pitfalls Associated with Discriminating Mixed-Species Biofilms by Flow Cytometry. Antibiotics 2020, 9, 741. [Google Scholar] [CrossRef]
- Teixeira, P.; Fernandes, B.; Silva, A.M.; Dias, N.; Azeredo, J. Evaluation by Flow Cytometry of Escherichia coli Viability in Lettuce after Disinfection. Antibiotics 2019, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berney, M.; Hammes, F.; Bosshard, F.; Weilenmann, H.-U.; Egli, T. Assessment and Interpretation of Bacterial Viability by Using the LIVE/DEAD BacLight Kit in Combination with Flow Cytometry. Appl. Environ. Microbiol. 2007, 73, 3283–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerca, F.; Trigo, G.; Correia, A.; Cerca, N.; Azeredo, J.; Vilanova, M. SYBR green as a fluorescent probe to evaluate the biofilm physiological state of Staphylococcus epidermidis, using flow cytometry. Can. J. Microbiol. 2011, 57, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Melo, L.D.R.; França, A.; Brandão, A.; Sillankorva, S.; Cerca, N.; Azeredo, J. Assessment of Sep1virus interaction with stationary cultures by transcriptional and flow cytometry studies. FEMS Microbiol. Ecol. 2018, 94, fiy143. [Google Scholar] [CrossRef]
- Oliveira, A.; Ribeiro, H.G.; Silva, A.C.; Silva, M.D.; Sousa, J.C.; Rodrigues, C.F.; Melo, L.D.R.; Henriques, A.F.; Sillankorva, S. Synergistic Antimicrobial Interaction between Honey and Phage against Escherichia coli Biofilms. Front. Microbiol. 2017, 8, 2407. [Google Scholar] [CrossRef]
- Brandão, A.; Pires, D.P.; Coppens, L.; Voet, M.; Lavigne, R.; Azeredo, J. Differential transcription profiling of the phage LUZ19 infection process in different growth media. RNA Biol. 2021, 18, 1778–1790. [Google Scholar] [CrossRef]
- Brussaard, C.P. Enumeration of Bacteriophages Using Flow Cytometry. Methods Mol. Biol. 2009, 501, 97–111. [Google Scholar] [CrossRef]
- Verthé, K.; Verstraete, W. Use of flow cytometry for analysis of phage-mediated killing of Enterobacter aerogenes. Res. Microbiol. 2006, 157, 613–618. [Google Scholar] [CrossRef]
- Low, H.Z.; Böhnlein, C.; Sprotte, S.; Wagner, N.; Fiedler, G.; Kabisch, J.; Franz, C. Fast and Easy Phage-Tagging and Live/Dead Analysis for the Rapid Monitoring of Bacteriophage Infection. Front. Microbiol. 2020, 11, 602444. [Google Scholar] [CrossRef]
- Parcey, M.; Gayder, S.; Castle, A.J.; Svircev, A.M. Molecular Profile of Phage Infection: A Novel Approach for the Characterization of Erwinia Phages through qPCR. Int. J. Mol. Sci. 2020, 21, 553. [Google Scholar] [CrossRef] [Green Version]
- Wicke, L.; Ponath, F.; Coppens, L.; Gerovac, M.; Lavigne, R.; Vogel, J. Introducing differential RNA-seq mapping to track the early infection phase for Pseudomonas phage ФKZ. RNA Biol. 2021, 18, 1099–1110. [Google Scholar] [CrossRef]
- Fernández, L.; Gutiérrez, D.; García, P.; Rodríguez, A. The Perfect Bacteriophage for Therapeutic Applications—A Quick Guide. Antibiotics 2019, 8, 126. [Google Scholar] [CrossRef] [Green Version]
- Lood, C.; Danis-Wlodarczyk, K.; Blasdel, B.G.; Jang, H.B.; Vandenheuvel, D.; Briers, Y.; Noben, J.P.; van Noort, V.; Drulis-Kawa, Z.; Lavigne, R. Integrative omics analysis of Pseudomonas aeruginosa virus PA5oct highlights the molecular complexity of jumbo phages. Environ. Microbiol. 2020, 22, 2165–2181. [Google Scholar] [CrossRef] [Green Version]
- Cerca, F.; Andrade, F.; França, A.; Andrade, E.B.; Ribeiro, A.; Almeida, A.A.; Cerca, N.; Pier, G.; Azeredo, J.; Vilanova, M. Staphylococcus epidermidis biofilms with higher proportions of dormant bacteria induce a lower activation of murine macrophages. J. Med. Microbiol. 2011, 60, 1717–1724. [Google Scholar] [CrossRef] [Green Version]
- Silveira, M.G.; Romão, M.V.S.; Loureiro-Dias, M.C.; Rombouts, F.M.; Abee, T. Flow Cytometric Assessment of Membrane Integrity of Ethanol-Stressed Oenococcus oeni Cells. Appl. Environ. Microbiol. 2002, 68, 6087–6093. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, O.; Lavysh, D.; Klimuk, E.; Djordjevic, M.; Ravcheev, D.A.; Gelfand, M.S.; Severinov, K.; Akulenko, N. Temporal Regulation of Gene Expression of the Escherichia coli Bacteriophage phiEco32. J. Mol. Biol. 2012, 416, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Duffy, C.; Feiss, M. The large subunit of bacteriophage λ’s terminase plays a role in DNA translocation and packaging termination. J. Mol. Biol. 2002, 316, 547–561. [Google Scholar] [CrossRef]
- Bull, J.J.; Gill, J.J. The habits of highly effective phages: Population dynamics as a framework for identifying therapeutic phages. Front. Microbiol. 2014, 5, 618. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.P.; Sillankorva, S.; Kropinski, A.M.; Lu, T.K.; Azeredo, J. Complete Genome Sequence of Pseudomonas aeruginosa Phage vB_PaeM_CEB_DP1. Genome Announc. 2015, 3, e00918-15. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.P.; Monteiro, R.; Mil-Homens, D.; Fialho, A.; Lu, T.K.; Azeredo, J. Designing P. aeruginosa synthetic phages with reduced genomes. Sci. Rep. 2021, 11, 2164. [Google Scholar] [CrossRef]
- Pires, D.P.; Dötsch, A.; Anderson, E.M.; Hao, Y.; Khursigara, C.M.; Lam, J.S.; Sillankorva, S.; Azeredo, J. A Genotypic Analysis of Five, P. aeruginosa Strains after Biofilm Infection by Phages Targeting Different Cell Surface Receptors. Front. Microbiol. 2017, 8, 1229. [Google Scholar] [CrossRef] [Green Version]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages by double agar overlay plaque assay. Methods Mol. Biol. 2009, 501, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Melo, L.D.R. In Vitro Activity of Bacteriophages against Planktonic and Biofilm Populations Assessed by Flow Cytometry. Methods Mol. Biol. 2018, 1693, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5’ → 3’) | Amplicon Size (bp) | Description |
|---|---|---|---|
| PE3_TerL_Fwd | GCAATGAGCGTTCCGTGTTCC | 176 | Amplify phage PE3 terminase, large subunit |
| PE3_TerL_Rev | CCATTCCTTCTTGGCAGCCTC | ||
| PE3_hel_Fwd | GCGCATCAGAAGGTAGACC | 204 | Amplify phage PE3 DNA helicase |
| PE3_hel_Rev | GGTTGTACTGCGCCAGGAG | ||
| PE3_gp3_Fwd | CGTGGTACAGCTTCAAGCC | 139 | Amplify phage PE3 gp3 |
| PE3_gp3_Rev | AGGTCACCCAGCAGTTCC | ||
| DP1_TerL_Fwd | GAAGCTTATGAGCGCGACC | 159 | Amplify phage DP1 terminase, large subunit |
| DP1_TerL_Rev | CCGATGCGCTTCGATCC | ||
| DP1_hel_Fwd | CAGGTTGCGCTTCCACTC | 171 | Amplify phage DP1 DNA helicase |
| DP1_hel_Rev | GCAGACGTGGCCATCTAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melo, L.D.R.; Monteiro, R.; Pires, D.P.; Azeredo, J. Phage-Host Interaction Analysis by Flow Cytometry Allows for Rapid and Efficient Screening of Phages. Antibiotics 2022, 11, 164. https://doi.org/10.3390/antibiotics11020164
Melo LDR, Monteiro R, Pires DP, Azeredo J. Phage-Host Interaction Analysis by Flow Cytometry Allows for Rapid and Efficient Screening of Phages. Antibiotics. 2022; 11(2):164. https://doi.org/10.3390/antibiotics11020164
Chicago/Turabian StyleMelo, Luís D. R., Rodrigo Monteiro, Diana P. Pires, and Joana Azeredo. 2022. "Phage-Host Interaction Analysis by Flow Cytometry Allows for Rapid and Efficient Screening of Phages" Antibiotics 11, no. 2: 164. https://doi.org/10.3390/antibiotics11020164
APA StyleMelo, L. D. R., Monteiro, R., Pires, D. P., & Azeredo, J. (2022). Phage-Host Interaction Analysis by Flow Cytometry Allows for Rapid and Efficient Screening of Phages. Antibiotics, 11(2), 164. https://doi.org/10.3390/antibiotics11020164