
Theoretical Study at the Molecular Mechanics Level of the Interaction of Tetracycline and Chloramphenicol with the Antibiotic Receptors Present in Enterococcus faecalis (Q839F7) and Streptococcus mutans (Q8DS20)

 , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Computational Details
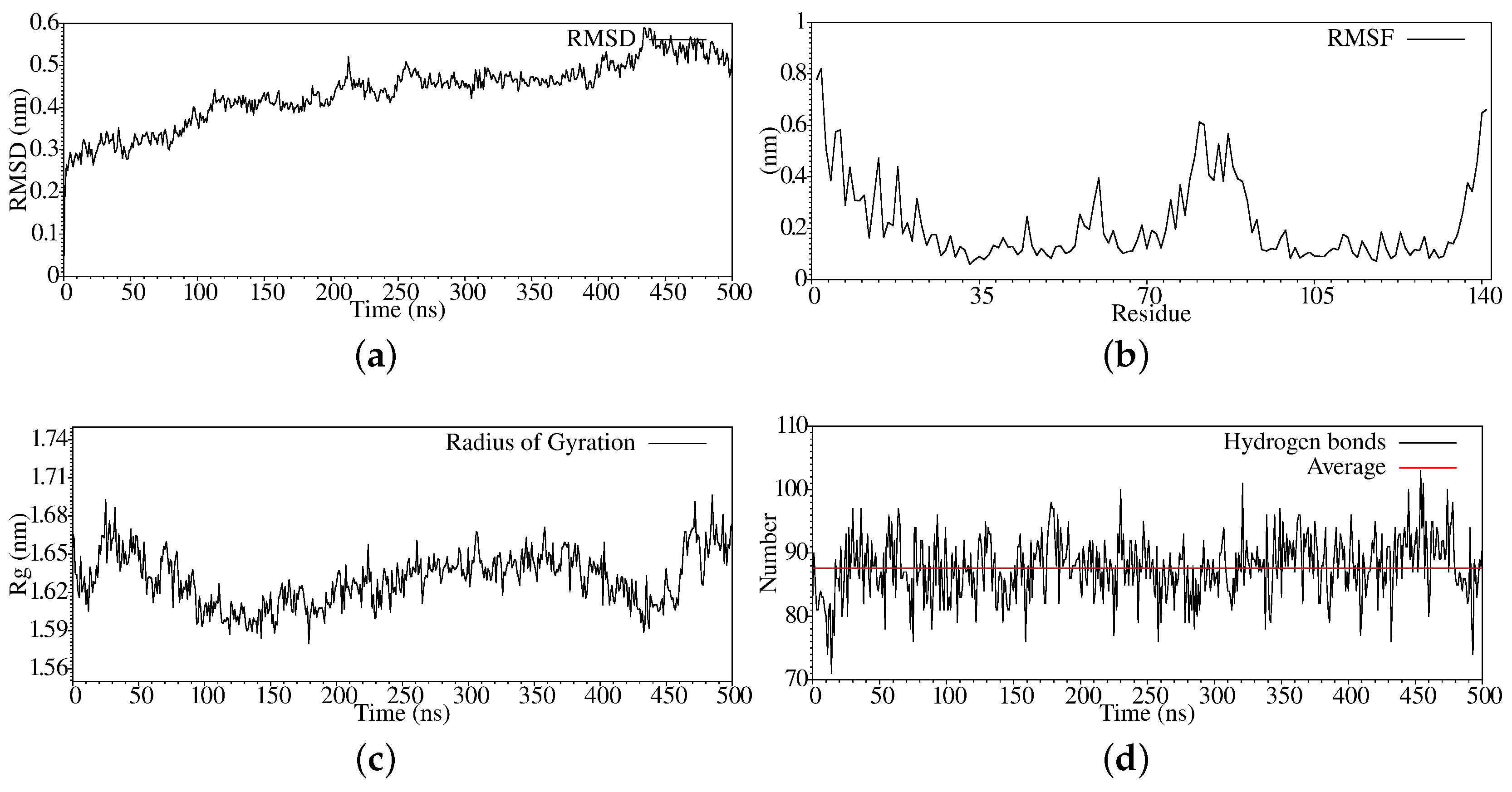
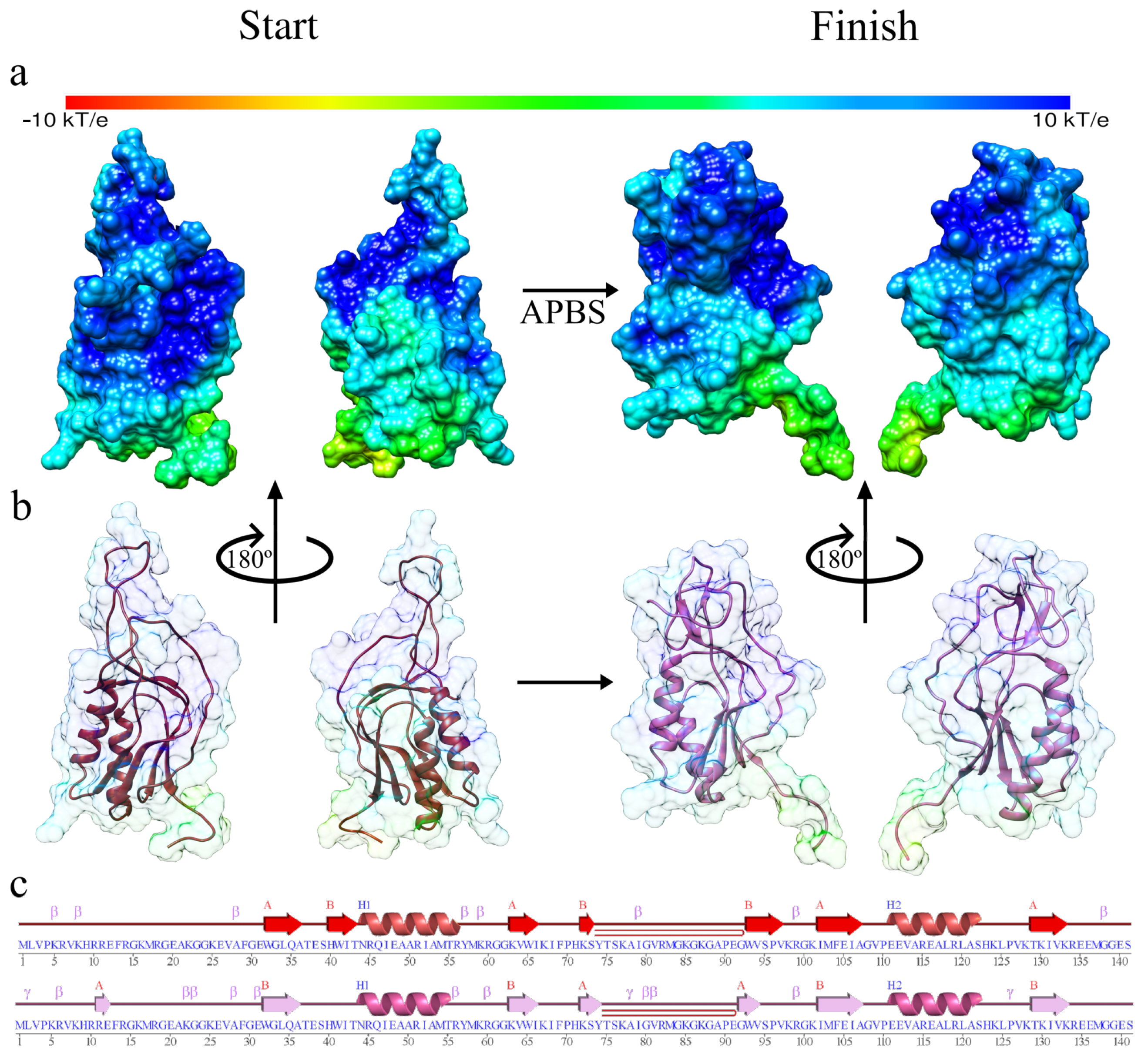
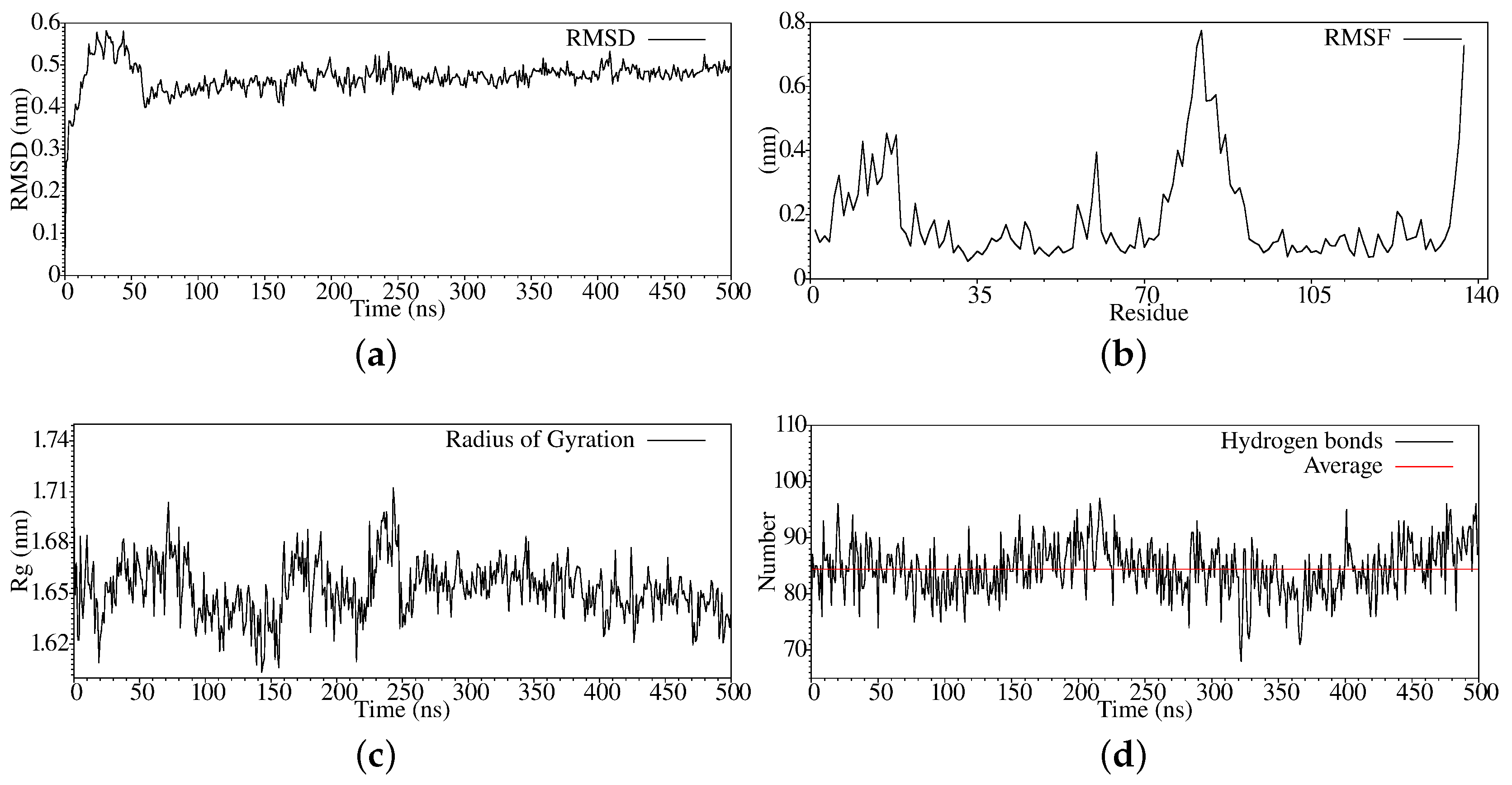
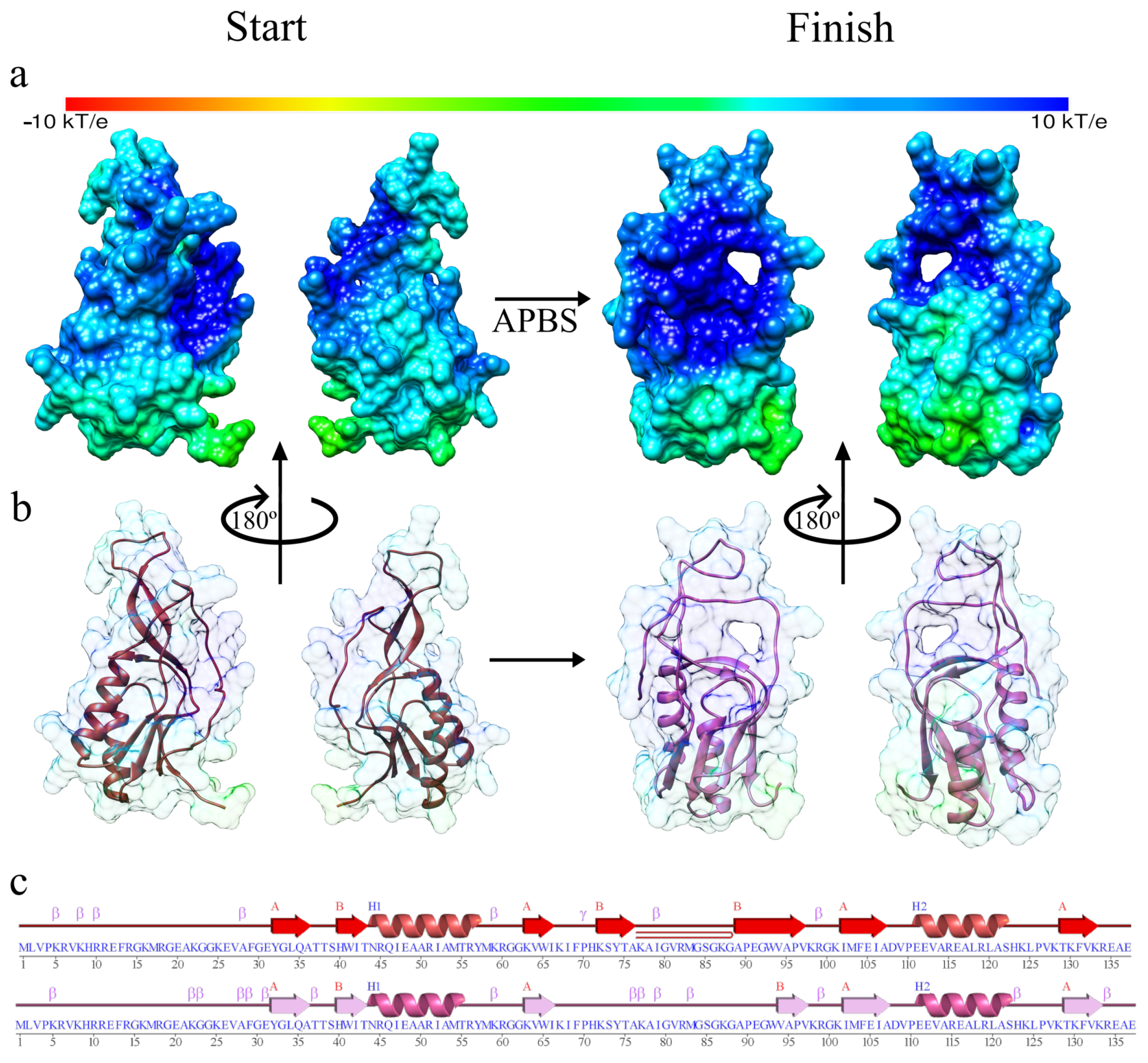
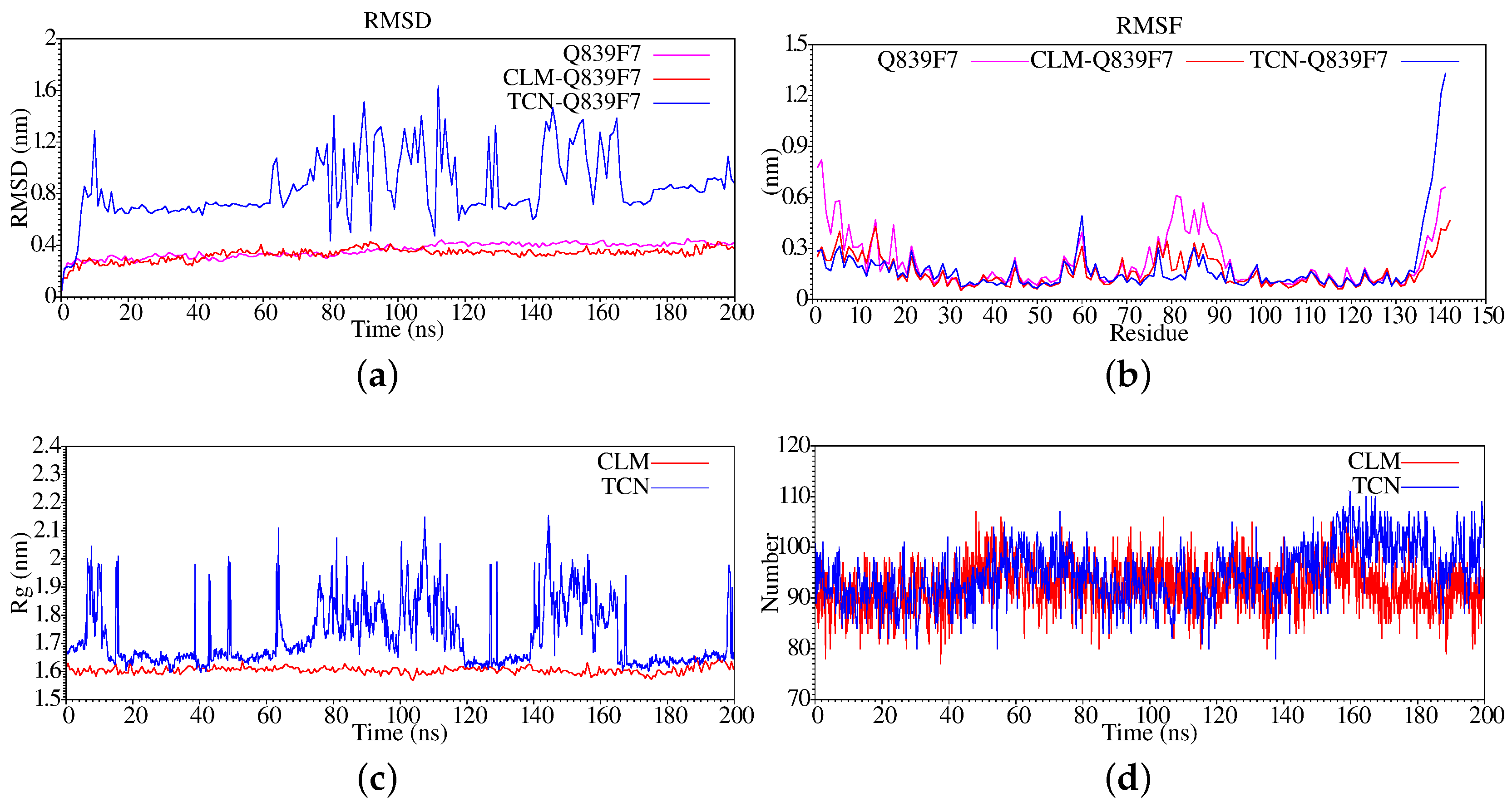
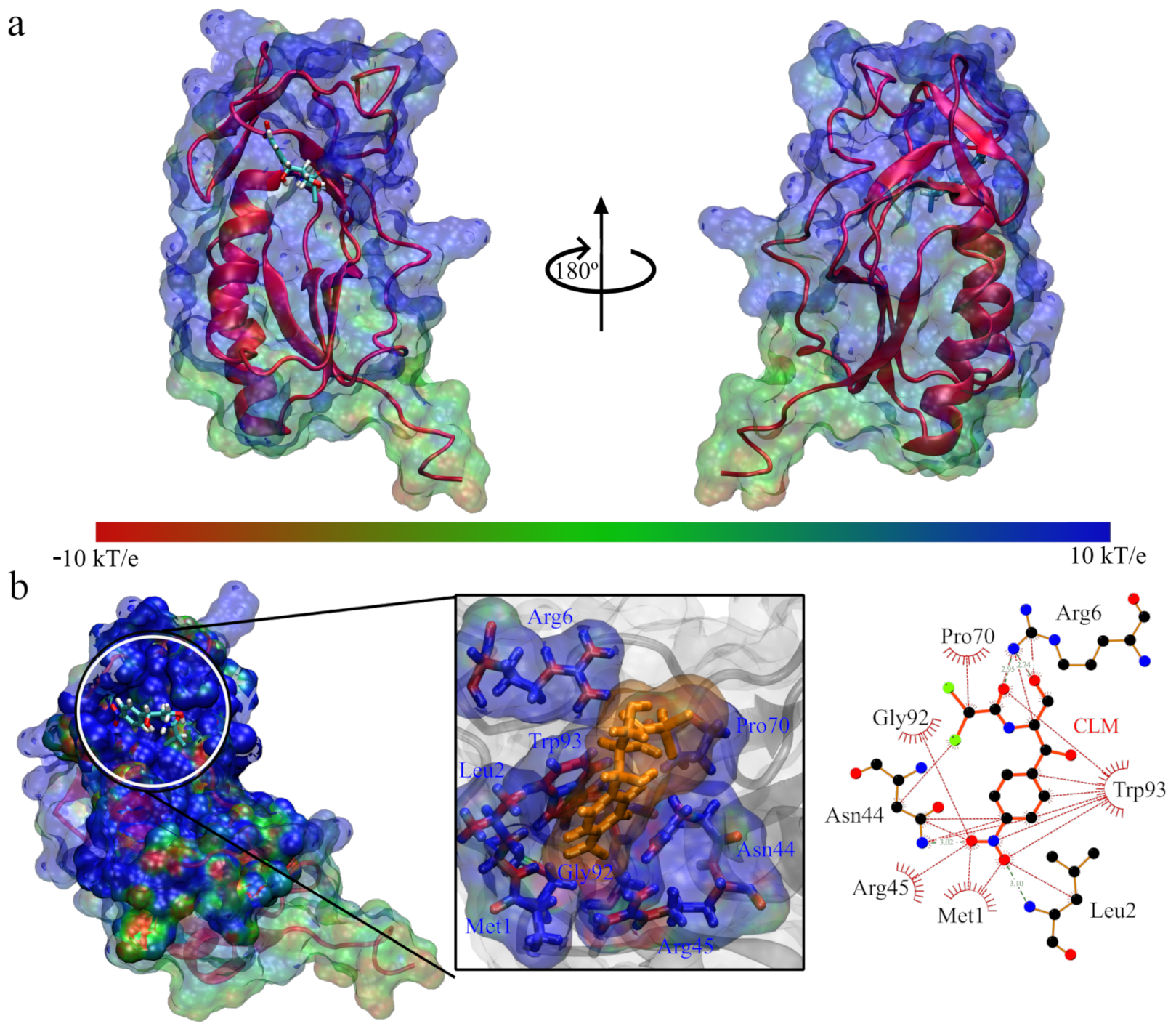
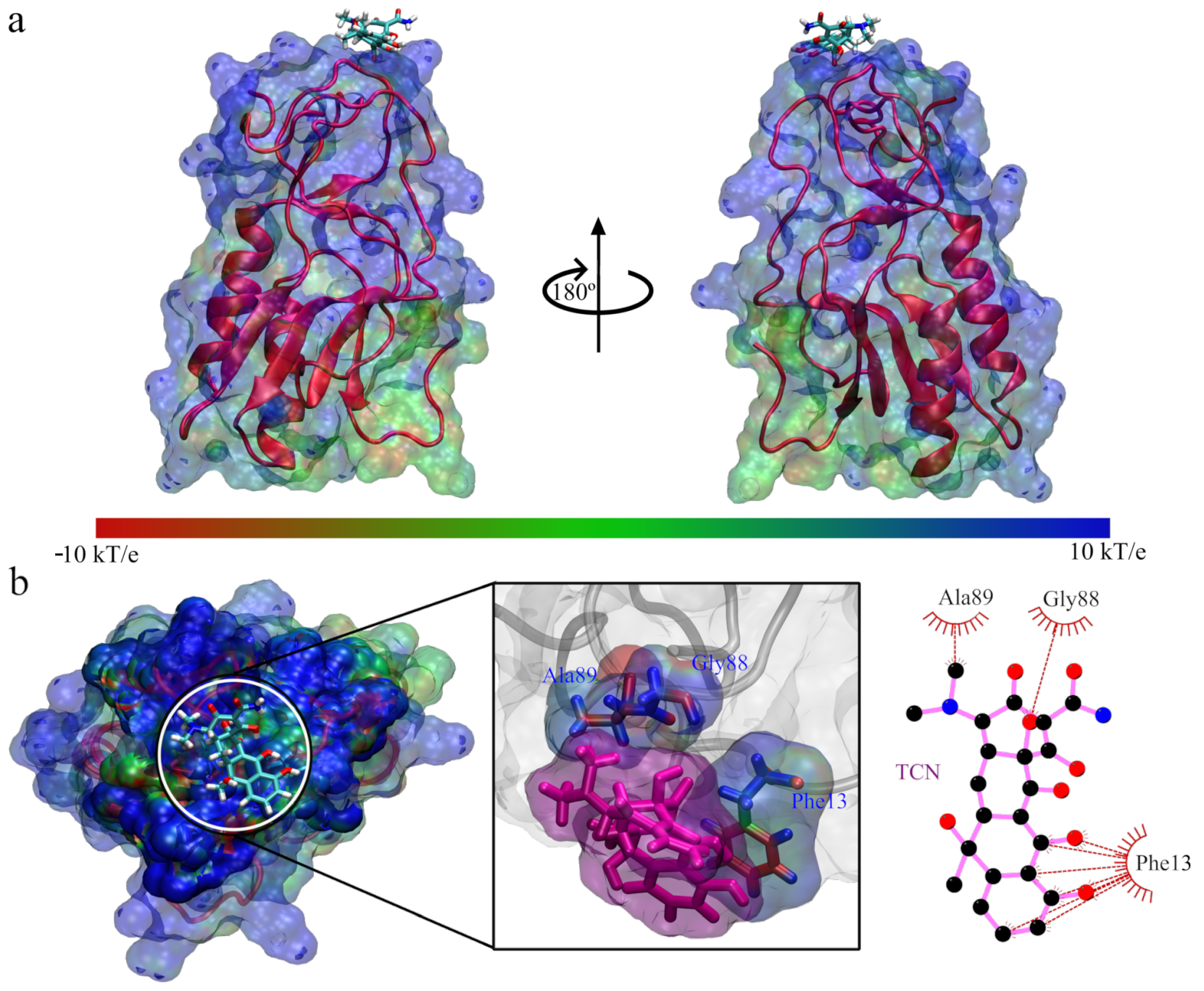
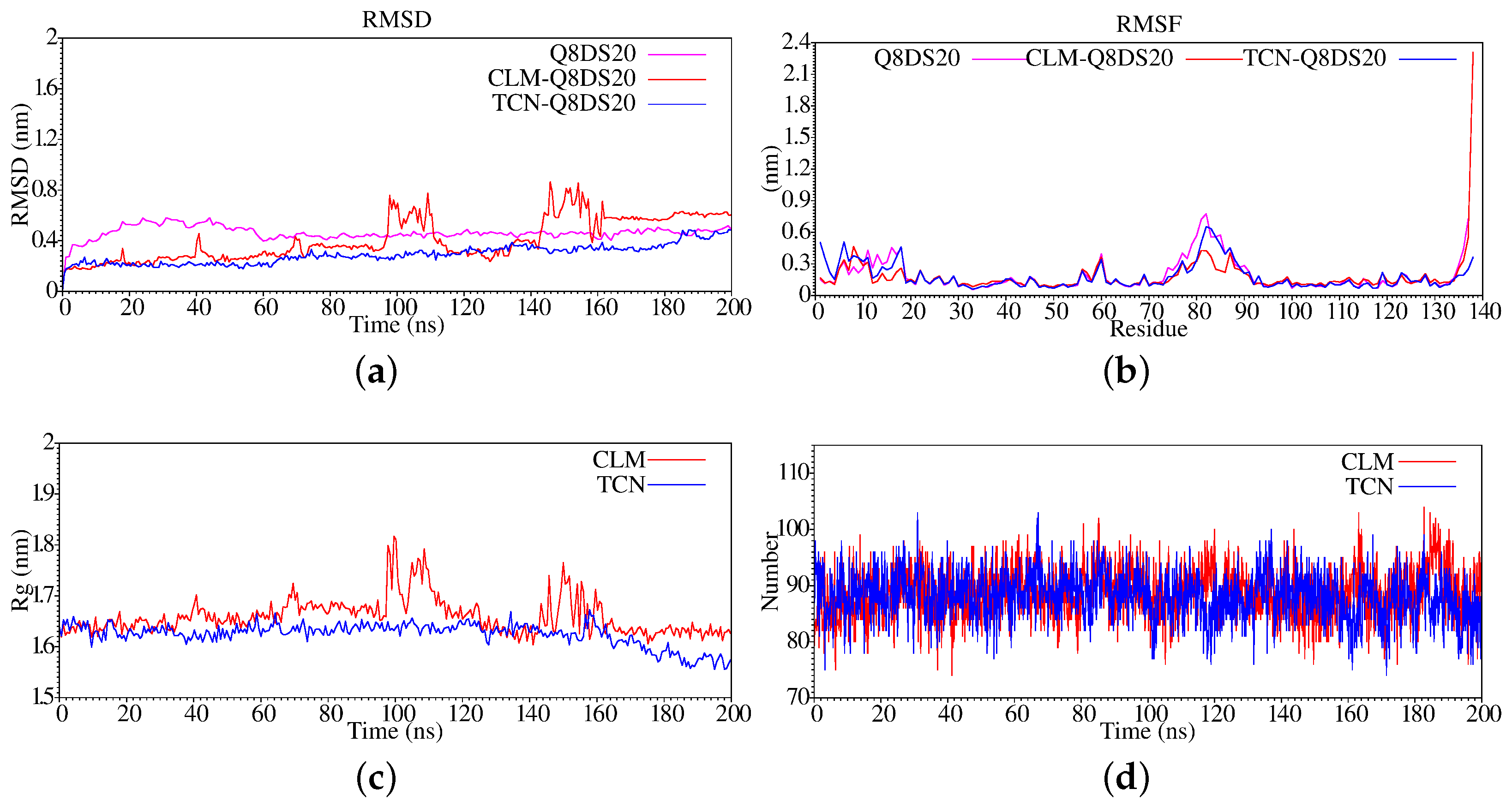
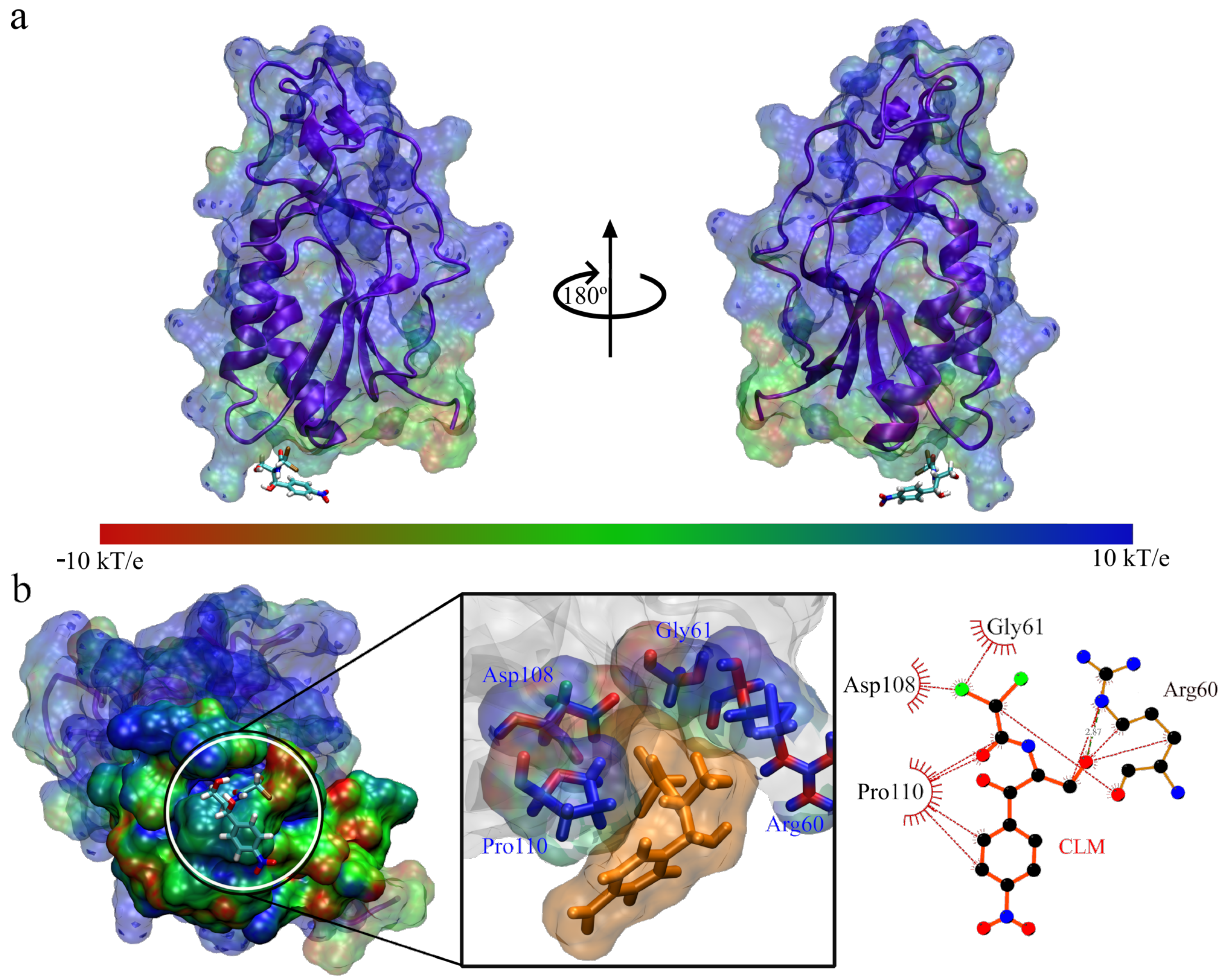
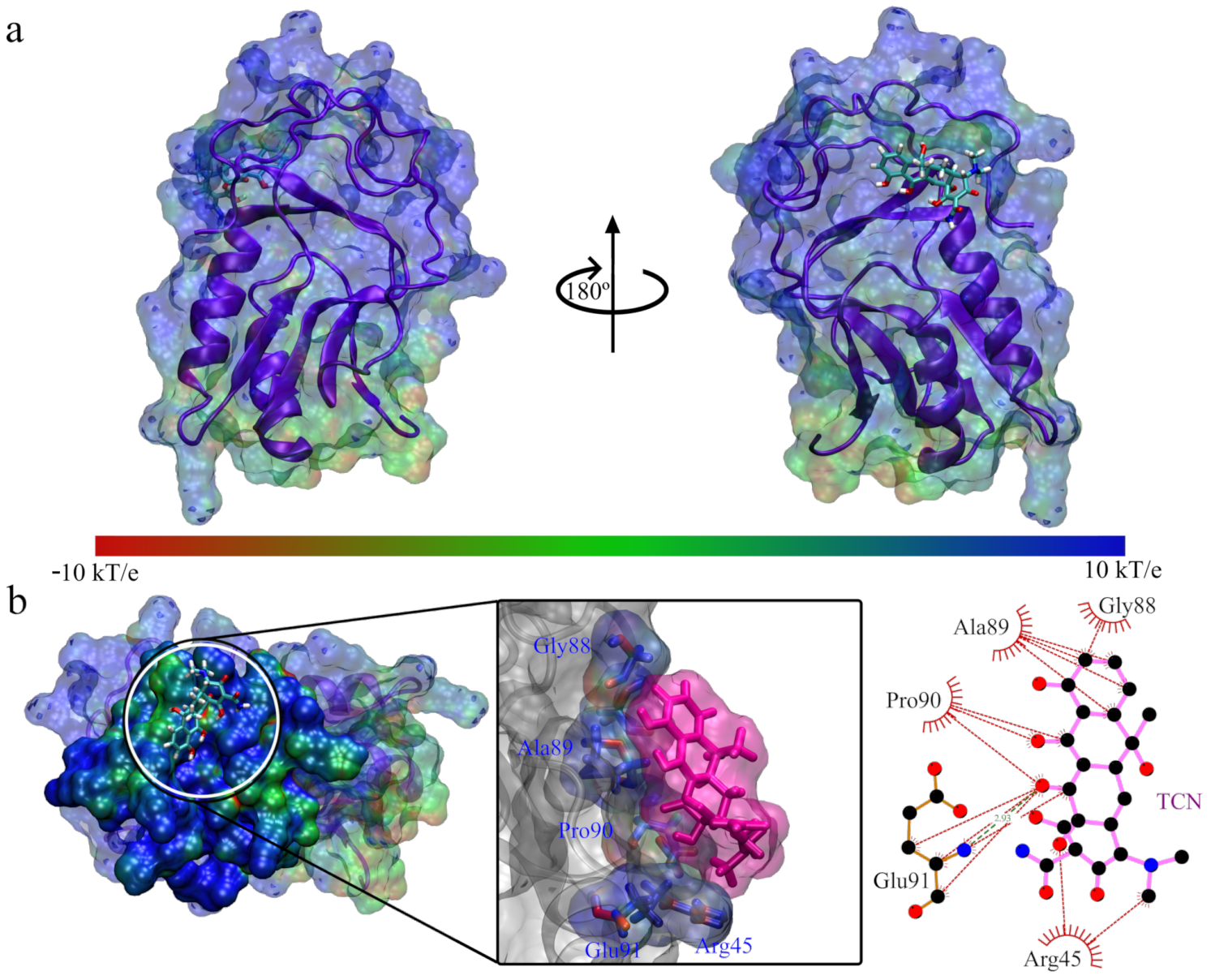
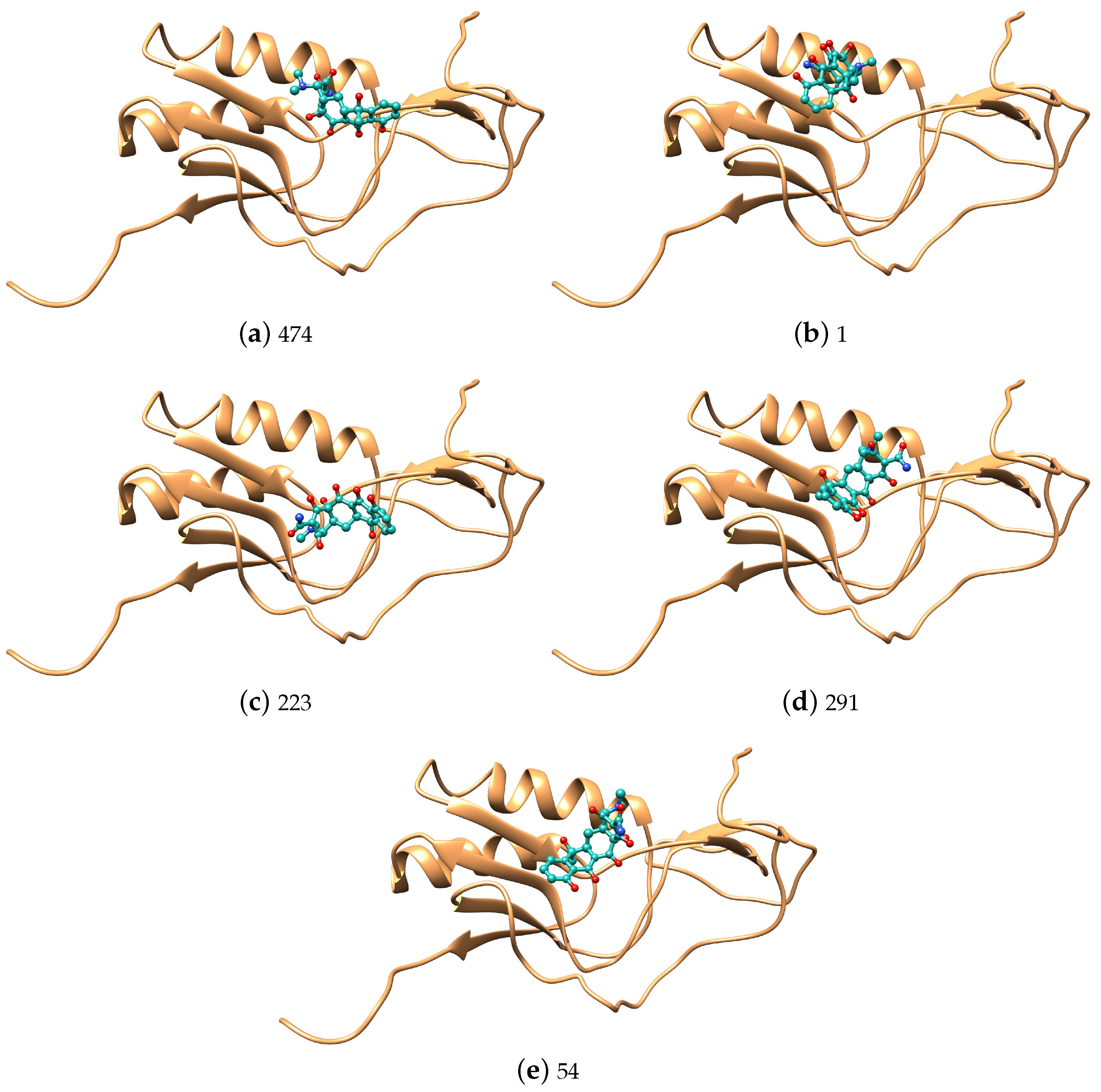
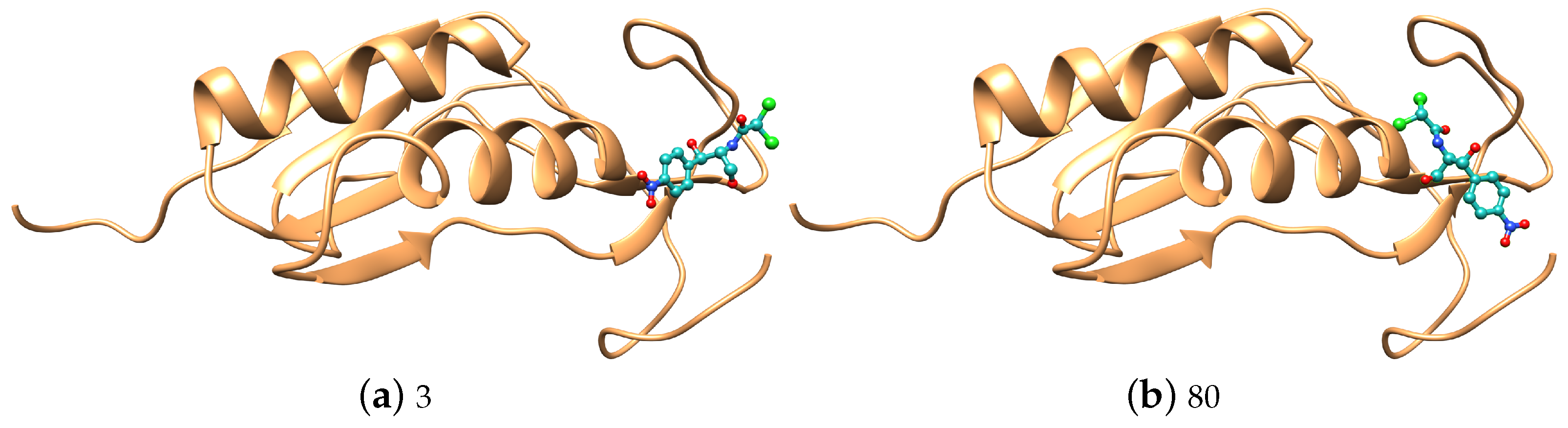
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





References
- Shlaes, D. 3.23—Antibiotics: The Miracle Menaced. In Comprehensive Biotechnology, 2nd ed.; Moo-Young, M., Ed.; Academic Press: Burlington, VT, USA, 2011; pp. 243–254. [Google Scholar] [CrossRef]
- Vallero, D.A. Chapter 1—Environmental Biotechnology: An Overview. In Environmental Biotechnology, 2nd ed.; Vallero, D.A., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 1–40. [Google Scholar] [CrossRef]
- Toledano, M.; Osorio, M.T.; Vallecillo-Rivas, M.; Toledano-Osorio, M.; Rodríguez-Archilla, A.; Toledano, R.; Osorio, R. Efficacy of local antibiotic therapy in the treatment of peri-implantitis: A systematic review and meta-analysis. J. Dent. 2021, 113, 103790. [Google Scholar] [CrossRef]
- Mittal, N.; Jain, J. Antibiotics as an intracanal medicament in endodontics: A review. Indian J. Dent. 2013, 4, 29–34. [Google Scholar] [CrossRef]
- Oberoi, S.S.; Dhingra, C.; Sharma, G.; Sardana, D. Antibiotics in dental practice: How justified are we. Int. Dent. J. 2015, 65, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Satterthwaite, J.; Absi, E.; Lewis, M.; Shepherd, J. Antibiotic prescription for acute dental conditions in the primary care setting. Br. Dent. J. 1996, 181, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Whitten, B.H.; Gardiner, D.L.; Jeansonne, B.G.; Lemon, R.R. Current trends in endodontic treatment: Report of a national survey. J. Am. Dent. Assoc. 1996, 127, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Nunes, L.P.; Nunes, G.P.; Ferrisse, T.M.; Strazzi-Sahyon, H.B.; Cintra, L.T.Â.; Dos Santos, P.H.; Sivieri-Araujo, G. Antimicrobial photodynamic therapy in endodontic reintervention: A systematic review and meta-analysis. Photodiagn. Photodyn. Ther. 2022, 39, 103014. [Google Scholar] [CrossRef]
- Costerton, J.W.; Geesey, G.G.; Cheng, K.J. How bacteria stick. Sci. Am. 1978, 238, 86–95. [Google Scholar] [CrossRef]
- Provoost, C.; Rocca, G.T.; Thibault, A.; Machtou, P.; Bouilllaguet, S. Influence of Needle Design and Irrigant Flow Rate on the Removal of Enterococcus faecalis Biofilms In Vitro. Dent. J. 2022, 10, 59. [Google Scholar] [CrossRef]
- Katalinić, I.; Budimir, A.; Bošnjak, Z.; Jakovljević, S.; Anić, I. The photo-activated and photo-thermal effect of the 445/970 nm diode laser on the mixed biofilm inside root canals of human teeth in vitro: A pilot study. Photodiagn. Photodyn. Ther. 2019, 26, 277–283. [Google Scholar] [CrossRef]
- Madiba, M.; Oluremi, B.B.; Gulube, Z.; Oderinlo, O.O.; Marimani, M.; Osamudiamen, P.M.; Patel, M. Anti-Streptococcus mutans, anti-adherence and anti-acidogenic activity of Uvaria chamae P. Beauv. J. Ethnopharmacol. 2023, 300, 115673. [Google Scholar] [CrossRef]
- Yang, Y.; Qian, Y.; Zhang, M.; Hao, S.; Wang, H.; Fan, Y.; Liu, R.; Xu, D.; Wang, F. Host defense peptide-mimicking beta-peptide polymer displaying strong antibacterial activity against cariogenic Streptococcus mutans. J. Mater. Sci. Technol. 2023, 133, 77–88. [Google Scholar] [CrossRef]
- Chen, F.; Liu, X.; Ge, X.; Wang, Y.; Zhao, Z.; Zhang, X.; Chen, G.Q.; Sun, Y. Porous polydroxyalkanoates (PHA) scaffolds with antibacterial property for oral soft tissue regeneration. Chem. Eng. J. 2023, 451, 138899. [Google Scholar] [CrossRef]
- Kong, C.; Zhang, H.; Li, L.; Liu, Z. Effects of green tea extract epigallocatechin-3-gallate (EGCG) on oral disease-associated microbes: A review. J. Oral Microbiol. 2022, 14, 2131117. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Xu, X.; Peng, X. Transposon mutagenesis in oral streptococcus. J. Oral Microbiol. 2022, 14, 2104951. [Google Scholar] [CrossRef]
- Cruz, M.R.; Cristy, S.; Guha, S.; De Cesare, G.B.; Evdokimova, E.; Sanchez, H.; Borek, D.; Miramon, P.; Yano, J.; Fidel, P.L., Jr.; et al. Structural and functional analysis of EntV reveals a 12 amino acid fragment protective against fungal infections. Nat. Commun. 2022, 13, 6047. [Google Scholar] [CrossRef]
- Jiang, Q.; Jing, Q.; Ren, B.; Cheng, L.; Zhou, X.; Lai, W.; He, J.; Li, M. Culture Supernatant of Enterococcus faecalis Promotes the Hyphal Morphogenesis and Biofilm Formation of Candida albicans. Pathogens 2022, 11, 1177. [Google Scholar] [CrossRef]
- Doub, J.B.; Nandi, S.; Putnam, N. Retention of Minocycline Susceptibility When Gram-Positive Periprosthetic Joint Infection Isolates Are Non-Susceptible to Doxycycline. Infect. Dis. Rep. 2022, 14, 641–645. [Google Scholar] [CrossRef]
- Xia, M.; Zhuo, N.; Ren, S.; Zhang, H.; Yang, Y.; Lei, L.; Hu, T. Enterococcus faecalis rnc gene modulates its susceptibility to disinfection agents: A novel approach against biofilm. BMC Oral Health 2022, 22, 416. [Google Scholar] [CrossRef]
- Verma, N.; Sangwan, P.; Tewari, S.; Duhan, J. Effect of different concentrations of sodium hypochlorite on outcome of primary root canal treatment: A randomized controlled trial. J. Endod. 2019, 45, 357–363. [Google Scholar] [CrossRef]
- Koo, H.; Xiao, J.; Klein, M.; Jeon, J. Exopolysaccharides produced by Streptococcus mutans glucosyltransferases modulate the establishment of microcolonies within multispecies biofilms. J. Bacteriol. 2010, 192, 12. [Google Scholar] [CrossRef]
- Folliero, V.; Dell’Annunziata, F.; Roscetto, E.; Amato, A.; Gasparro, R.; Zannella, C.; Casolaro, V.; De Filippis, A.; Catania, M.R.; Franci, G.; et al. Rhein: A novel antibacterial compound against Streptococcus mutans infection. Microbiol. Res. 2022, 261, 127062. [Google Scholar] [CrossRef] [PubMed]
- Sachidananda, M.P.; Mallya, S. Microbiology and Clinical Implications of Dental Caries—A Review. J. Evol. Med. Dent. Sci. 2020, 9, 3670–3676. [Google Scholar]
- AlEraky, D.M.; Madi, M.; El Tantawi, M.; AlHumaid, J.; Fita, S.; AbdulAzeez, S.; Borgio, J.F.; Al-Harbi, F.A.; Alagl, A.S. Predominance of non-Streptococcus mutans bacteria in dental biofilm and its relation to caries progression. Saudi J. Biol. Sci. 2021, 28, 7390–7395. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Garrido, N.; Lozano, C.P.; Kreth, J.; Giacaman, R.A. Competition and caries on enamel of a dual-species biofilm model with Streptococcus mutans and Streptococcus sanguinis. Appl. Environ. Microbiol. 2020, 86, e01262-20. [Google Scholar] [CrossRef] [PubMed]
- Van Houte, J. Role of micro-organisms in caries etiology. J. Dent. Res. 1994, 73, 672–681. [Google Scholar] [CrossRef]
- Banas, J.A. Virulence properties of Streptococcus mutans. Front. Biosci. Landmark 2004, 9, 1267–1277. [Google Scholar] [CrossRef]
- Bowen, W.; Koo, H. Biology of Streptococcus mutans-derived glucosyltransferases: Role in extracellular matrix formation of cariogenic biofilms. Caries Res. 2011, 45, 69–86. [Google Scholar] [CrossRef]
- Blancas, B.; Lanzagorta, M.d.L.; Jiménez-Garcia, L.F.; Lara, R.; Molinari, J.L.; Fernandez, A.M. Study of the ultrastructure of Enterococcus faecalis and Streptococcus mutans incubated with salivary antimicrobial peptides. Clin. Exp. Dent. Res. 2021, 7, 365–375. [Google Scholar] [CrossRef]
- Dinis, M.; Agnello, M.; Cen, L.; Shokeen, B.; He, X.; Shi, W.; Wong, D.T.; Lux, R.; Tran, N.C. Oral microbiome: Streptococcus mutans/caries concordant-discordant children. Front. Microbiol. 2022, 13, 782825. [Google Scholar] [CrossRef]
- Naka, S.; Wato, K.; Misaki, T.; Ito, S.; Matsuoka, D.; Nagasawa, Y.; Nomura, R.; Matsumoto-Nakano, M.; Nakano, K. Streptococcus mutans induces IgA nephropathy-like glomerulonephritis in rats with severe dental caries. Sci. Rep. 2021, 11, 5784. [Google Scholar] [CrossRef]
- Abranches, J.; Miller, J.H.; Martinez, A.R.; Simpson-Haidaris, P.J.; Burne, R.A.; Lemos, J.A. The collagen-binding protein Cnm is required for Streptococcus mutans adherence to and intracellular invasion of human coronary artery endothelial cells. Infect. Immun. 2011, 79, 2277–2284. [Google Scholar] [CrossRef]
- Doddawad, V.G.; Shivananda, S.; Paul, N.J.; Chandrakala, J. Dental caries: Impact of tobacco product among tobacco chewers and tobacco smokers. J. Oral Biol. Craniofacial Res. 2022, 12, 401–404. [Google Scholar] [CrossRef]
- Zanini, M.; Tenenbaum, A.; Azogui-Lévy, S. La caries dental, un problema de salud pública. EMC Tratado Med. 2022, 26, 1–8. [Google Scholar] [CrossRef]
- Kumari, A.R.; Rao, S.N.; Reddy, P.R. Design of hybrid dental caries segmentation and caries detection with meta-heuristic-based ResneXt-RNN. Biomed. Signal Process. Control 2022, 78, 103961. [Google Scholar] [CrossRef]
- Gönczi, N.N.; Strang, O.; Bagi, Z.; Rákhely, G.; Kovács, K.L. Interactions between probiotic and oral pathogenic strains. Biol. Futur. 2021, 72, 461–471. [Google Scholar] [CrossRef]
- Zoletti, G.O.; Pereira, E.M.; Schuenck, R.P.; Teixeira, L.M.; Siqueira, J.F., Jr.; dos Santos, K.R.N. Characterization of virulence factors and clonal diversity of Enterococcus faecalis isolates from treated dental root canals. Res. Microbiol. 2011, 162, 151–158. [Google Scholar] [CrossRef]
- Siqueira, J.F., Jr.; Rôças, I.N. Polymerase chain reaction–based analysis of microorganisms associated with failed endodontic treatment. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodont. 2004, 97, 85–94. [Google Scholar] [CrossRef]
- Bouillaguet, S.; Manoil, D.; Girard, M.; Louis, J.; Gaïa, N.; Leo, S.; Schrenzel, J.; Lazarevic, V. Root microbiota in primary and secondary apical periodontitis. Front. Microbiol. 2018, 9, 2374. [Google Scholar] [CrossRef]
- Méndez, D.A.C.; Cuéllar, M.R.C.; Pedrinha, V.F.; Espedilla, E.G.V.; de Andrade, F.B.; de Almeida Rodrigues, P.; Cruvinel, T. Effects of curcumin-mediated antimicrobial photodynamic therapy associated to different chelators against Enterococcus faecalis biofilms. Photodiagn. Photodyn. Ther. 2021, 35, 102464. [Google Scholar] [CrossRef]
- Nair, P. On the causes of persistent apical periodontitis: A review. Int. Endod. J. 2006, 39, 249–281. [Google Scholar] [CrossRef]
- Fabretti, F.; Theilacker, C.; Baldassarri, L.; Kaczynski, Z.; Kropec, A.; Holst, O.; Huebner, J. Alanine esters of enterococcal lipoteichoic acid play a role in biofilm formation and resistance to antimicrobial peptides. Infect. Immun. 2006, 74, 4164–4171. [Google Scholar] [CrossRef] [PubMed]
- Byrgazov, K.; Vesper, O.; Moll, I. Ribosome heterogeneity: Another level of complexity in bacterial translation regulation. Curr. Opin. Microbiol. 2013, 16, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Knowles, D.J.; Foloppe, N.; Matassova, N.B.; Murchie, A.I. The bacterial ribosome, a promising focus for structure-based drug design. Curr. Opin. Pharmacol. 2002, 2, 501–506. [Google Scholar] [CrossRef]
- Williamson, J.R. Biophysical studies of bacterial ribosome assembly. Curr. Opin. Struct. Biol. 2008, 18, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Harms, J.; Schluenzen, F.; Zarivach, R.; Bashan, A.; Gat, S.; Agmon, I.; Bartels, H.; Franceschi, F.; Yonath, A. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell 2001, 107, 679–688. [Google Scholar] [CrossRef]
- Yusupov, M.M.; Yusupova, G.Z.; Baucom, A.; Lieberman, K.; Earnest, T.N.; Cate, J.; Noller, H.F. Crystal structure of the ribosome at 5.5 Å resolution. Science 2001, 292, 883–896. [Google Scholar] [CrossRef]
- Teraoka, H.; Nierhaus, K.H. Protein L16 induces a conformational change when incorporated into an L16-deficient core derived from Escherichia coli ribosomes. FEBS Lett. 1978, 88, 223–226. [Google Scholar] [CrossRef]
- Bashan, A.; Agmon, I.; Zarivach, R.; Schluenzen, F.; Harms, J.; Berisio, R.; Bartels, H.; Franceschi, F.; Auerbach, T.; Hansen, H.A.; et al. Structural basis of the ribosomal machinery for peptide bond formation, translocation, and nascent chain progression. Mol. Cell 2003, 11, 91–102. [Google Scholar] [CrossRef]
- Nishimura, M.; Yoshida, T.; Shirouzu, M.; Terada, T.; Kuramitsu, S.; Yokoyama, S.; Ohkubo, T.; Kobayashi, Y. Solution structure of ribosomal protein L16 from Thermus thermophilus HB8. J. Mol. Biol. 2004, 344, 1369–1383. [Google Scholar] [CrossRef]
- Anikaev, A.; Isaev, A.; Korobeinikova, A.; Garber, M.; Gongadze, G. Role of protein L25 and its contact with protein L16 in maintaining the active state of Escherichia coli ribosomes in vivo. Biochemistry 2016, 81, 19–27. [Google Scholar] [CrossRef]
- Golub, L.M.; Lee, H.M. Periodontal therapeutics: Current host-modulation agents and future directions. Periodontology 2000 2020, 82, 186–204. [Google Scholar] [CrossRef]
- Jones, D.S.; Woolfson, A.D.; Brown, A.F.; Coulter, W.A.; McClelland, C.; Irwin, C.R. Design, characterisation and preliminary clinical evaluation of a novel mucoadhesive topical formulation containing tetracycline for the treatment of periodontal disease. J. Control Release 2000, 67, 357–368. [Google Scholar] [CrossRef]
- Qiu, W.; Zhou, Y.; Li, Z.; Huang, T.; Xiao, Y.; Cheng, L.; Peng, X.; Zhang, L.; Ren, B. Application of antibiotics/antimicrobial agents on dental caries. BioMed Res. Int. 2020, 2020, 5658212. [Google Scholar] [CrossRef]
- Markley, J.L.; Wencewicz, T.A. Tetracycline-inactivating enzymes. Front. Microbiol. 2018, 9, 1058. [Google Scholar] [CrossRef]
- Schwarz, S.; Kehrenberg, C.; Doublet, B.; Cloeckaert, A. Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol. Rev. 2004, 28, 519–542. [Google Scholar] [CrossRef]
- Sathya, A.; Prabhu, T.; Ramalingam, S. Structural, biological and pharmaceutical importance of antibiotic agent chloramphenicol. Heliyon 2020, 6, e03433. [Google Scholar] [CrossRef]
- Magoulas, G.E.; Kostopoulou, O.N.; Garnelis, T.; Athanassopoulos, C.M.; Kournoutou, G.G.; Leotsinidis, M.; Dinos, G.P.; Papaioannou, D.; Kalpaxis, D.L. Synthesis and antimicrobial activity of chloramphenicol–polyamine conjugates. Bioorg. Med. Chem. 2015, 23, 3163–3174. [Google Scholar] [CrossRef] [PubMed]
- Crofts, T.S.; Sontha, P.; King, A.O.; Wang, B.; Biddy, B.A.; Zanolli, N.; Gaumnitz, J.; Dantas, G. Discovery and characterization of a nitroreductase capable of conferring bacterial resistance to chloramphenicol. Cell Chem. Biol. 2019, 26, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Mor, A.; Ziv, G.; Levy, Y. Simulations of proteins with inhomogeneous degrees of freedom: The effect of thermostats. J. Comput. Chem. 2008, 29, 1992–1998. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 1995, 240, 283–290. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum new things. Nucleic Acids Res. 2008, 37, D355–D359. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2019. [Google Scholar]
- Williams, T.; Kelley, C.; Broeker, H.B.; Campbell, J.; Cunningham, R.; Denholm, D.; Elber, G.; Fearick, R.; Grammes, C.; Hart, L.; et al. GnuPlot. 2022. Available online: http://gnuplot.info/docs_5.5/gnuplot5.html (accessed on 7 November 2022).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Num | Tetracycline | Num | Chloramphenicol | ||||||
|---|---|---|---|---|---|---|---|---|---|
| GE | A–VdW | R–VdW | ACE e | GE | A–VdW | R–VdW d | ACE e | ||
| 474 | 3.36 | 3 | 2.24 | ||||||
| 1 | 3.46 | 80 | 5.97 | ||||||
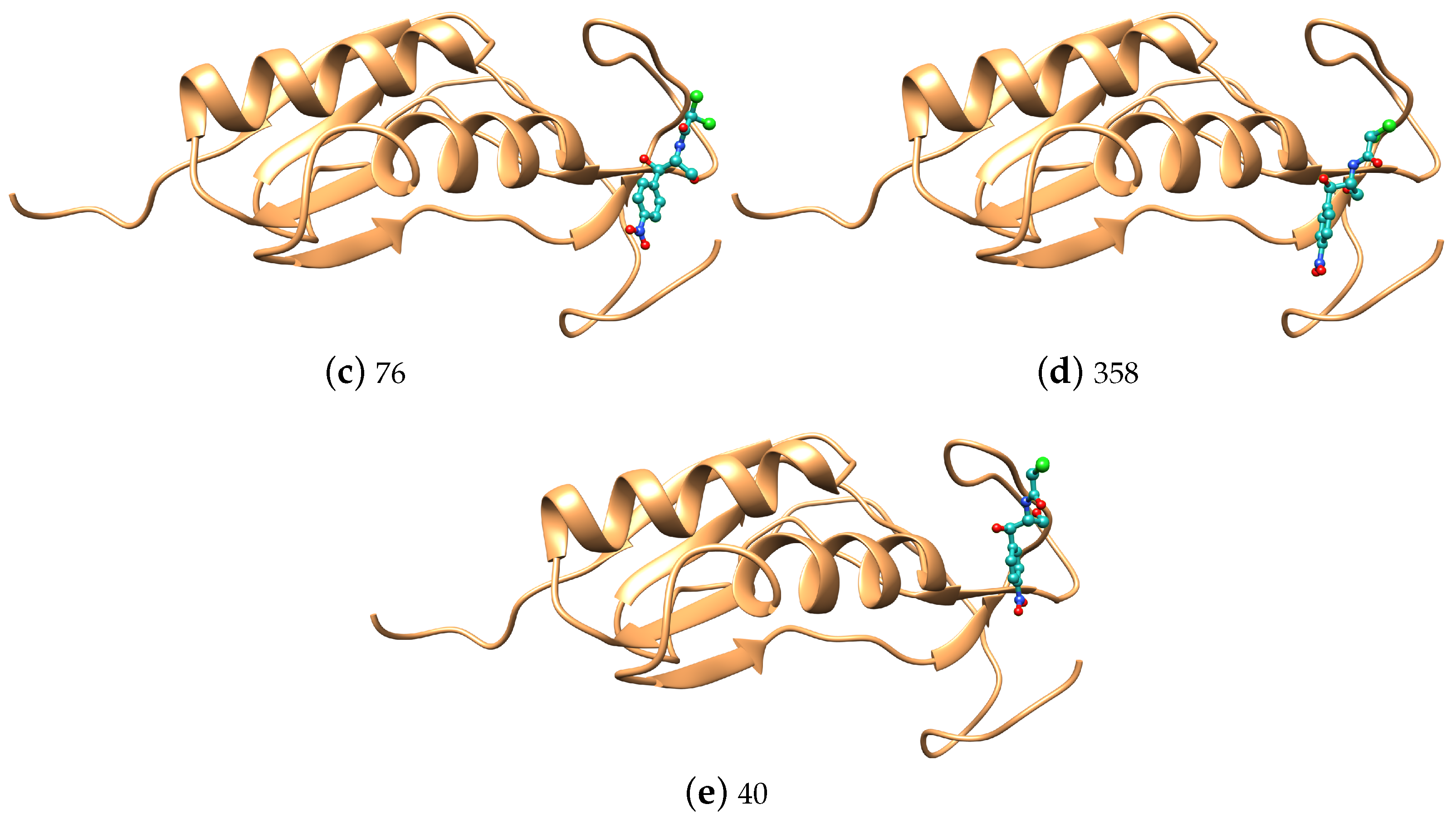
| 223 | 5.59 | 76 | 5.54 | ||||||
| 291 | 2.89 | 358 | 2.34 | ||||||
| 52 | 3.10 | 40 | 6.24 | ||||||
| 122 | 5.31 | 12 | 2.23 | ||||||
| 105 | 7.41 | 70 | 2.34 | ||||||
| 9 | 9.59 | 248 | 4.60 | ||||||
| 207 | 4.91 | 61 | 1.62 | ||||||
| 242 | 5.62 | 114 | 6.36 | ||||||
| 61 | 6.50 | 2 | 5.43 | ||||||
| 47 | −30.07 | −15.12 | 1.59 | −7.21 | 69 | −24.19 | −15.98 | 10.85 | −7.55 |
| 10 | −29.41 | −17.88 | 7.38 | −8.35 | 128 | −24.02 | −12.92 | 2.04 | −8.14 |
| 8 | −29.28 | −17.51 | 3.64 | −7.33 | 35 | −24.01 | −13.96 | 6.98 | −7.73 |
| 49 | −29.27 | −19.22 | 5.16 | −7.79 | 181 | −23.57 | −12.30 | 3.36 | −7.74 |
| 25 | −29.26 | −16.60 | 1.66 | −6.19 | 34 | −23.43 | −12.17 | 3.93 | −7.55 |
| 21 | −29.02 | −16.59 | 5.81 | −7.74 | 18 | −23.37 | −13.30 | 2.61 | −5.42 |
| 140 | −28.85 | −13.43 | 2.50 | −10.29 | 6 | −23.33 | −13.93 | 5.06 | −6.20 |
| 196 | −28.73 | −15.02 | 5.76 | −9.95 | 354 | −23.20 | −14.84 | 10.85 | −7.25 |
| 155 | −28.31 | −16.03 | 4.91 | −8.35 | 78 | −23.03 | −11.30 | 4.89 | −8.78 |
| Energy Component (kJ/mol) | Q839F7— Enterococcus faecalis | |
|---|---|---|
| Tetracycline | Chloramphenicol | |
| van der Waals energy | −68.250 ± 27.109 | −84.947 ± 15.319 |
| Electrostatic energy | 25.815 ± 51.636 | −77.417 ± 29.606 |
| Polar solvation energy | 17.606 ± 23.842 | 66.502 ± 20.757 |
| SASA energy | −9.084 ± 3.356 | −10.697 ± 1.295 |
| SAV energy | −74.343 ± 51.103 | −87.315 ± 16.921 |
| Binding energy | −108.256 ± 80.005 | −193.874 ± 33.456 |
| Num | Tetracycline | Num | Chloramphenicol | ||||||
|---|---|---|---|---|---|---|---|---|---|
| GE | A–VdW | R–VdW | ACE e | GE | A–VdW | R–VdW | ACE e | ||
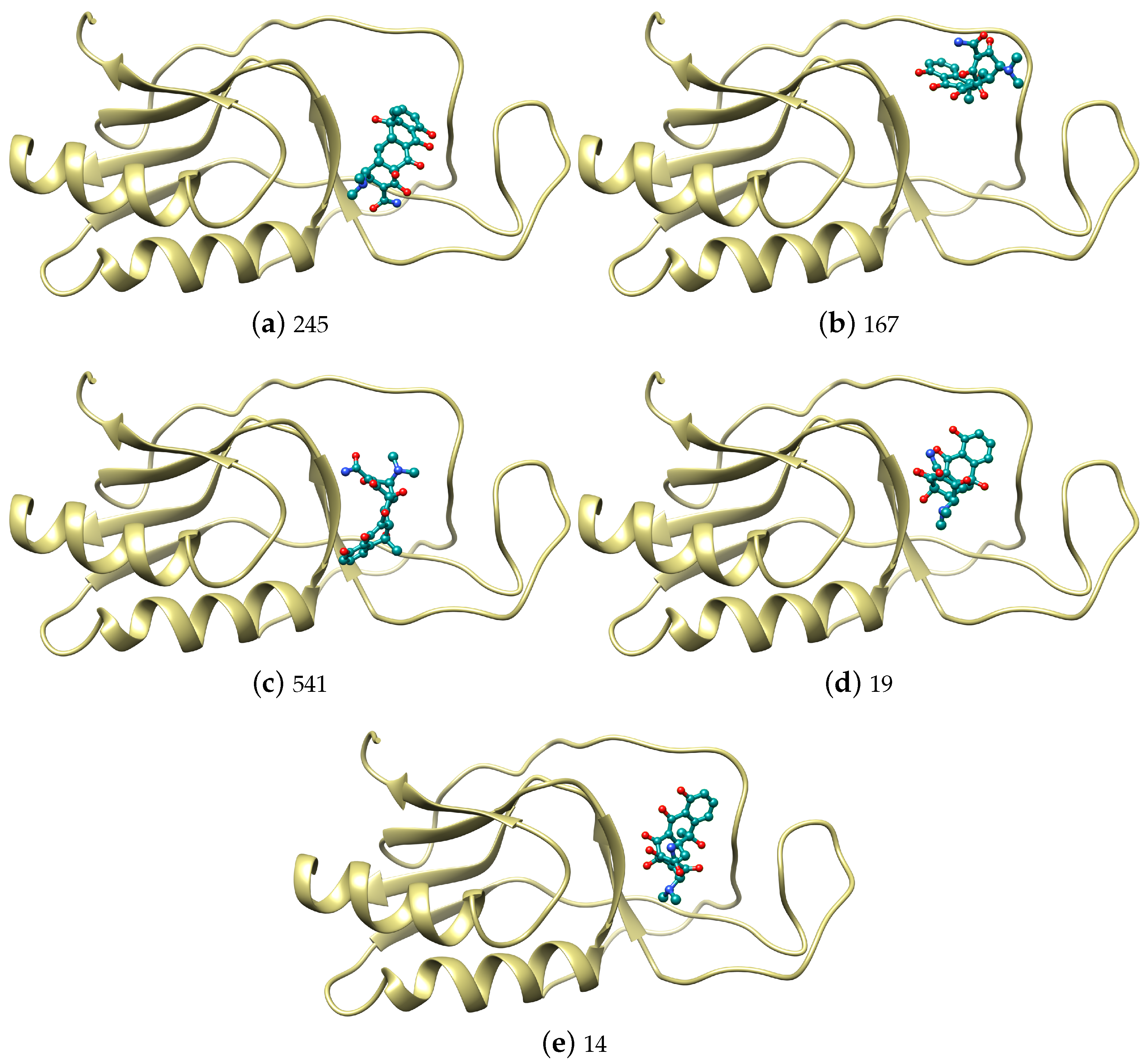
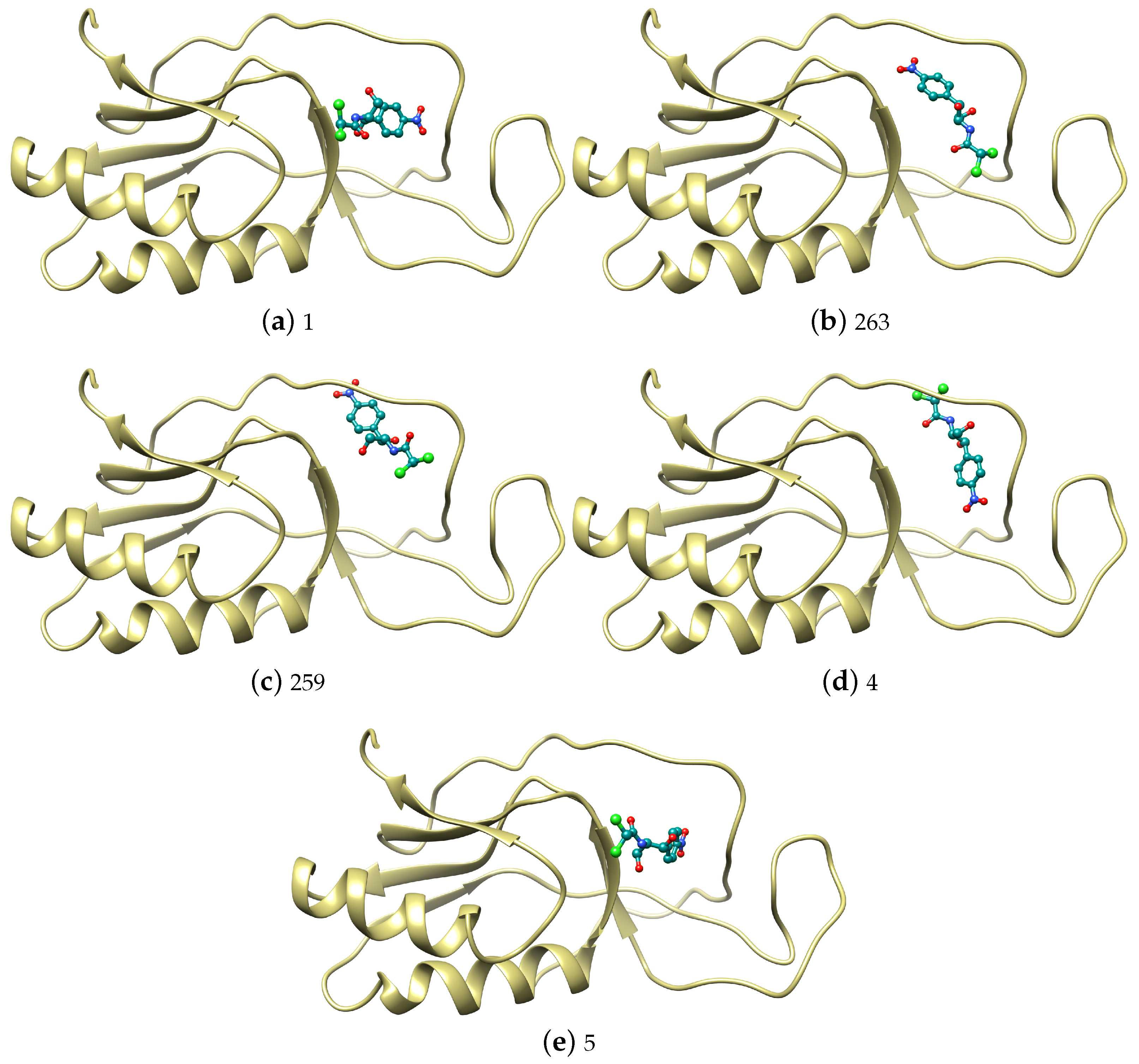
| 245 | −42.02 | −23.66 | 10.83 | −10.43 | 1 | −31.04 | −16.42 | 1.56 | −5.45 |
| 167 | −41.94 | −20.82 | 7.00 | −12.44 | 263 | −30.13 | −15.74 | 1.90 | −5.67 |
| 541 | −39.70 | −19.62 | 6.90 | −11.04 | 259 | −29.31 | −15.00 | 5.39 | −8.54 |
| 19 | −39.32 | −19.30 | 1.98 | −8.27 | 4 | −29.31 | −17.13 | 4.92 | −4.76 |
| 14 | −38.64 | −19.81 | 4.18 | −8.39 | 5 | −28.65 | −13.49 | 1.78 | −6.44 |
| 144 | −37.24 | −19.19 | 2.95 | −9.41 | 190 | −28.55 | −14.66 | 5.51 | −10.88 |
| 5 | −37.06 | −18.26 | 2.28 | −8.65 | 8 | −28.33 | −13.38 | 4.48 | −7.91 |
| 50 | −36.98 | −18.15 | 3.13 | −10.84 | 27 | −27.65 | −13.80 | 3.35 | −9.21 |
| 107 | −36.71 | −19.65 | 4.59 | −10.34 | 6 | −27.42 | −13.48 | 1.11 | −6.55 |
| 208 | −36.71 | −19.70 | 2.66 | −9.27 | 73 | −27.32 | −14.64 | 1.02 | −5.28 |
| 9 | −36.71 | −19.95 | 2.72 | −6.77 | 20 | −27.22 | −14.90 | 5.07 | −7.52 |
| 39 | −36.55 | −17.88 | 2.58 | −8.92 | 101 | −27.14 | −14.94 | 5.67 | −8.59 |
| 12 | −36.38 | −19.83 | 3.48 | −7.48 | 33 | −27.05 | −14.75 | 4.16 | −5.88 |
| 20 | −36.32 | −19.38 | 7.90 | −9.76 | 24 | −26.47 | −13.89 | 6.36 | −7.53 |
| 2 | −36.30 | −23.43 | 7.92 | −5.14 | 51 | −26.10 | −15.79 | 6.83 | −7.52 |
| 181 | −35.58 | −18.06 | 10.45 | −14.43 | 12 | −25.97 | −13.78 | 5.96 | −6.78 |
| 26 | −35.34 | −19.66 | 5.45 | −7.10 | 135 | −25.88 | −13.83 | 6.72 | −9.00 |
| 349 | −34.79 | −20.13 | 11.52 | −11.71 | 47 | −25.73 | −11.88 | 2.89 | −8.63 |
| 8 | −34.75 | −21.05 | 4.38 | −5.11 | 13 | −25.61 | −16.05 | 4.38 | −5.06 |
| 302 | −34.71 | −19.42 | 11.17 | −13.14 | 171 | −25.58 | −15.33 | 4.49 | −6.71 |
| Energy Component (kJ/mol) | Q8DS20—Streptococcus mutans | |
|---|---|---|
| Tetracycline | Chloramphenicol | |
| van der Waals energy | −148.927 ± 22.604 | −61.400 ± 35.589 |
| Electrostatic energy | 2.548 ± 42.704 | −24.125 ± 35.802 |
| Polar solvation energy | 61.456 ± 19.352 | 34.231 ± 31.575 |
| SASA energy | −16.754 ± 1.733 | −8.074 ± 4.459 |
| SAV energy | −146.648 ± 23.330 | −62.666 ± 42.733 |
| Binding energy | −248.326 ± 48.584 | −122.035 ± 71.319 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueroa-Banda, R.A.; Figueroa-Castellanos, K.F.; Chávez-Oblitas, E.A.; Guillen-Nuñez, M.E.; Ayqui-Cueva, F.; Del-Carpio-M, B.A.; Bellido-Vallejo, K.L.; Gómez, B. Theoretical Study at the Molecular Mechanics Level of the Interaction of Tetracycline and Chloramphenicol with the Antibiotic Receptors Present in Enterococcus faecalis (Q839F7) and Streptococcus mutans (Q8DS20). Antibiotics 2022, 11, 1640. https://doi.org/10.3390/antibiotics11111640
Figueroa-Banda RA, Figueroa-Castellanos KF, Chávez-Oblitas EA, Guillen-Nuñez ME, Ayqui-Cueva F, Del-Carpio-M BA, Bellido-Vallejo KL, Gómez B. Theoretical Study at the Molecular Mechanics Level of the Interaction of Tetracycline and Chloramphenicol with the Antibiotic Receptors Present in Enterococcus faecalis (Q839F7) and Streptococcus mutans (Q8DS20). Antibiotics. 2022; 11(11):1640. https://doi.org/10.3390/antibiotics11111640
Chicago/Turabian StyleFigueroa-Banda, Rufo Alberto, Kimberly Francis Figueroa-Castellanos, Edith Angelica Chávez-Oblitas, María Elena Guillen-Nuñez, Flor Ayqui-Cueva, Bruno A. Del-Carpio-M, Karen L. Bellido-Vallejo, and Badhin Gómez. 2022. "Theoretical Study at the Molecular Mechanics Level of the Interaction of Tetracycline and Chloramphenicol with the Antibiotic Receptors Present in Enterococcus faecalis (Q839F7) and Streptococcus mutans (Q8DS20)" Antibiotics 11, no. 11: 1640. https://doi.org/10.3390/antibiotics11111640
APA StyleFigueroa-Banda, R. A., Figueroa-Castellanos, K. F., Chávez-Oblitas, E. A., Guillen-Nuñez, M. E., Ayqui-Cueva, F., Del-Carpio-M, B. A., Bellido-Vallejo, K. L., & Gómez, B. (2022). Theoretical Study at the Molecular Mechanics Level of the Interaction of Tetracycline and Chloramphenicol with the Antibiotic Receptors Present in Enterococcus faecalis (Q839F7) and Streptococcus mutans (Q8DS20). Antibiotics, 11(11), 1640. https://doi.org/10.3390/antibiotics11111640