
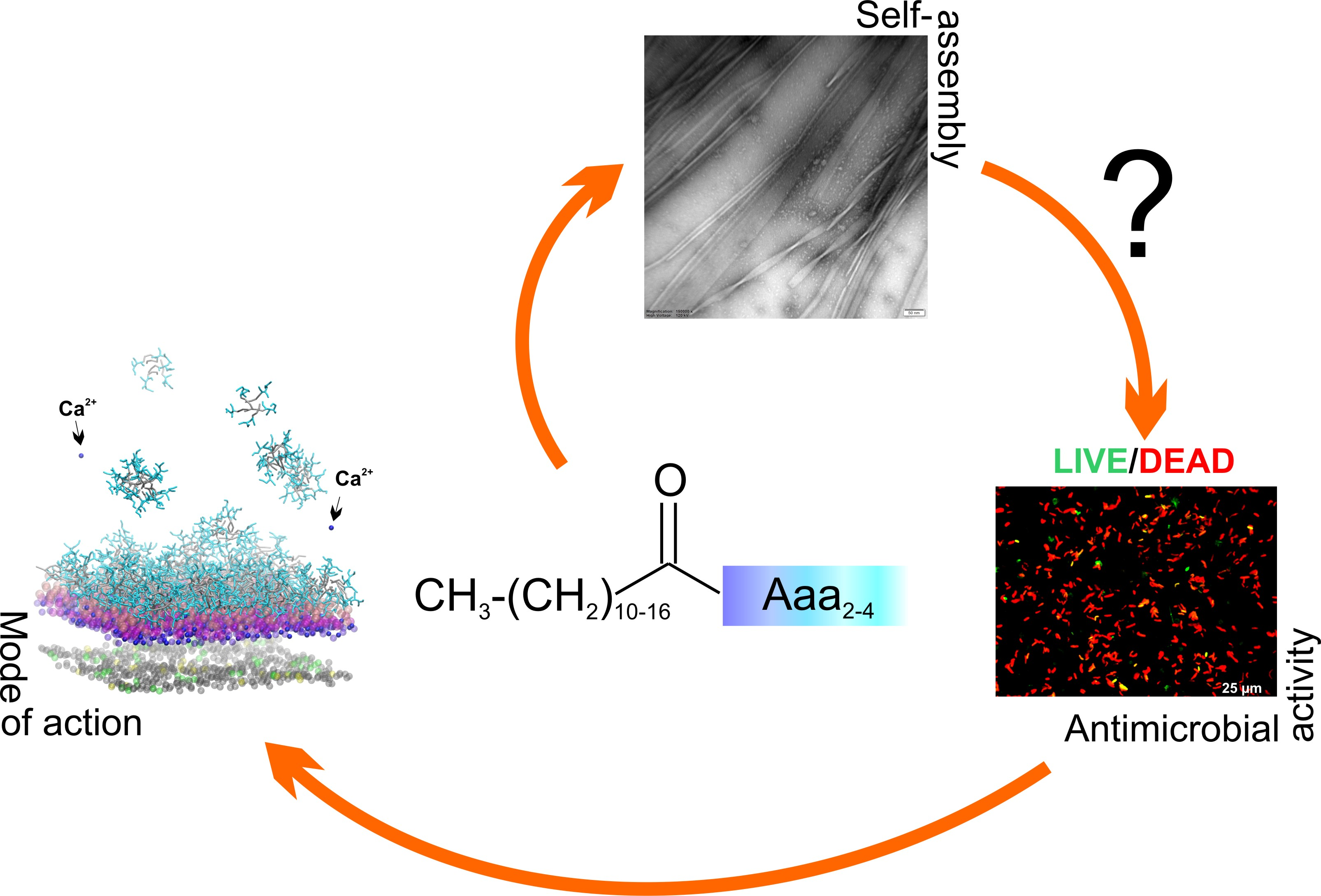
Understanding the Role of Self-Assembly and Interaction with Biological Membranes of Short Cationic Lipopeptides in the Effective Design of New Antibiotics

 , , , , ,
, , , , , 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Antimicrobial Activity and Cytotoxicity
2.2. Evaluation of Bacterial Cell Viability
2.3. Stability in Serum
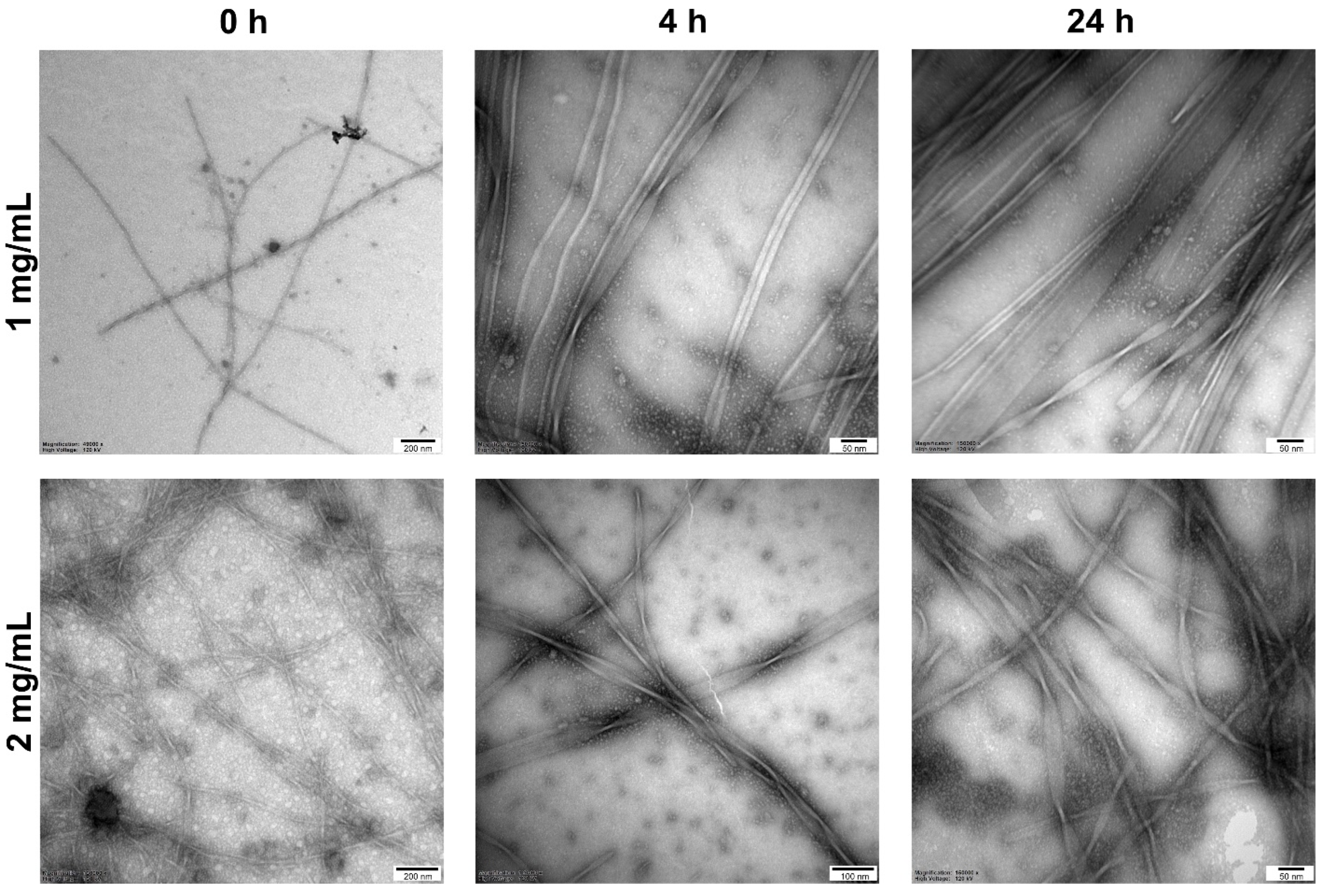
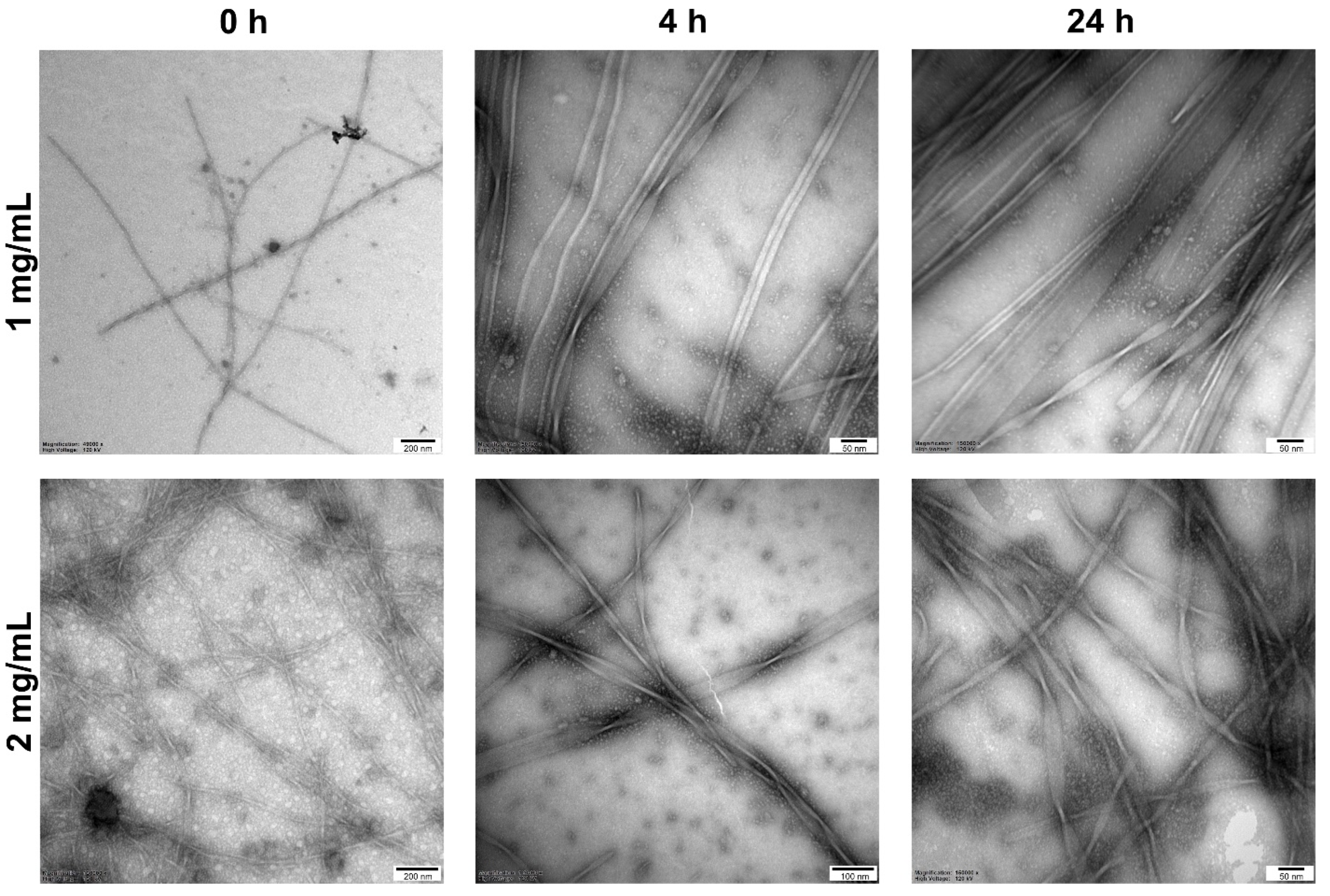
2.4. Self-Assembly
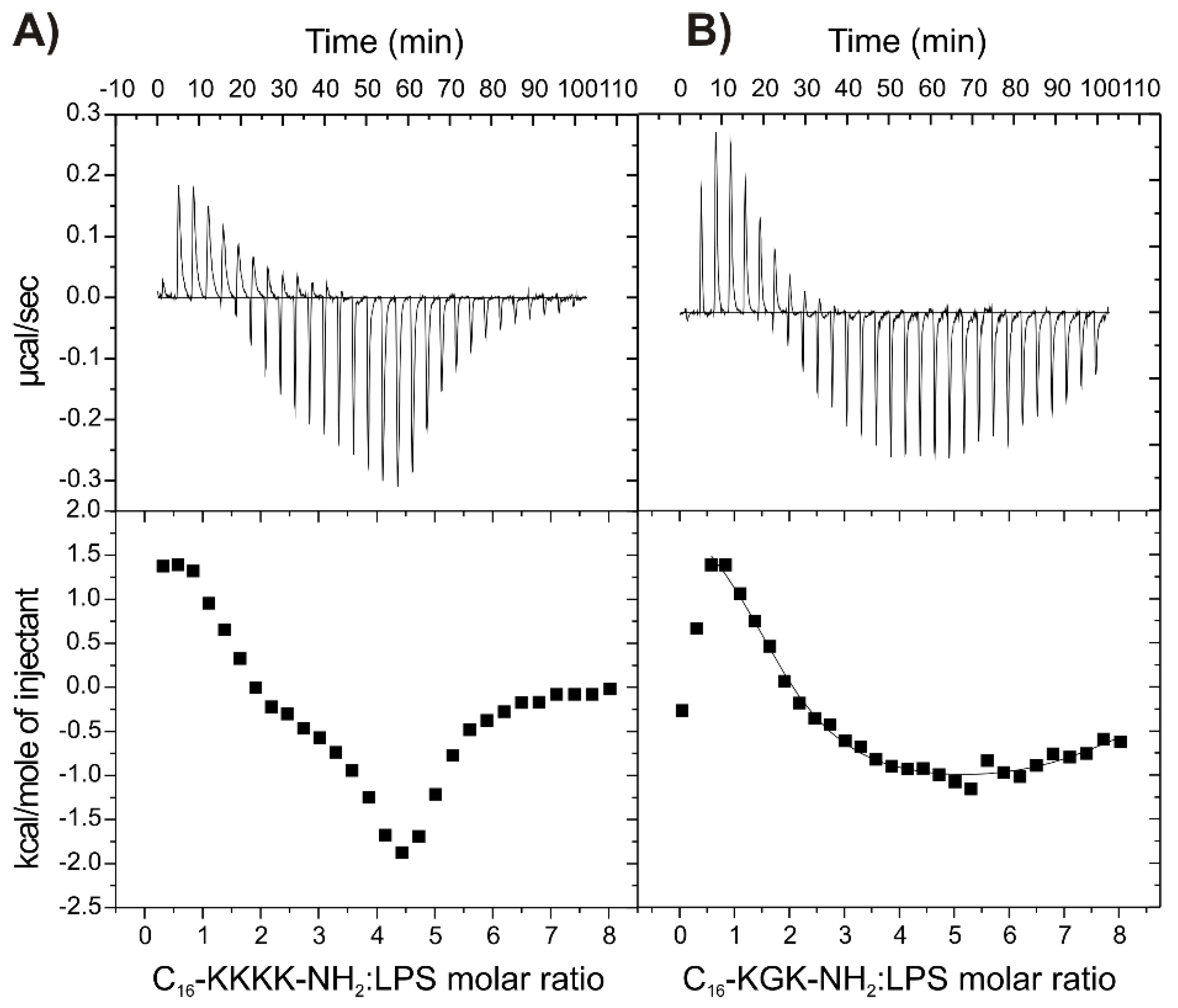
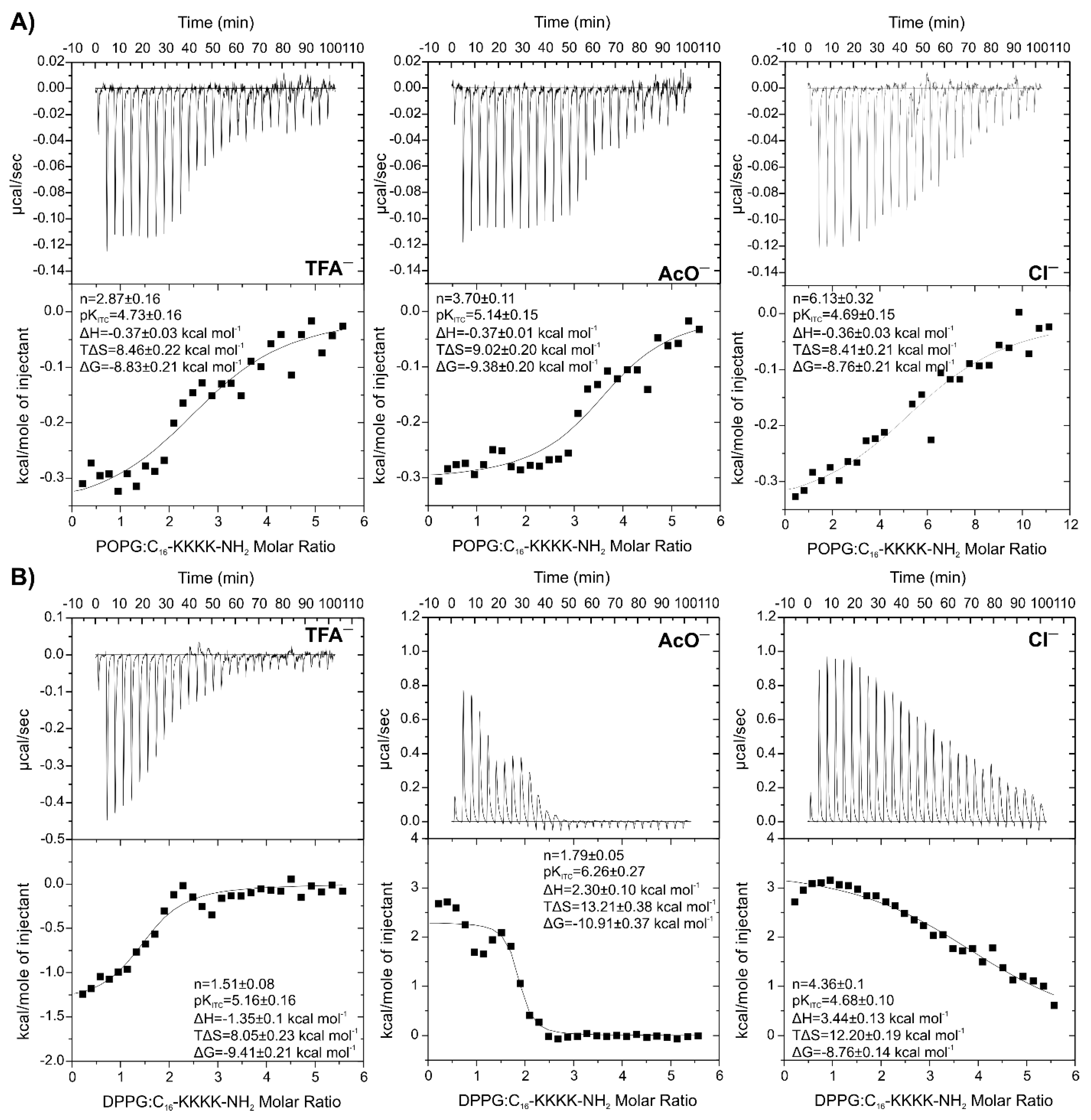
2.5. Isothermal Titration Calorimetry to Study Lipopeptide-Lipid Interaction
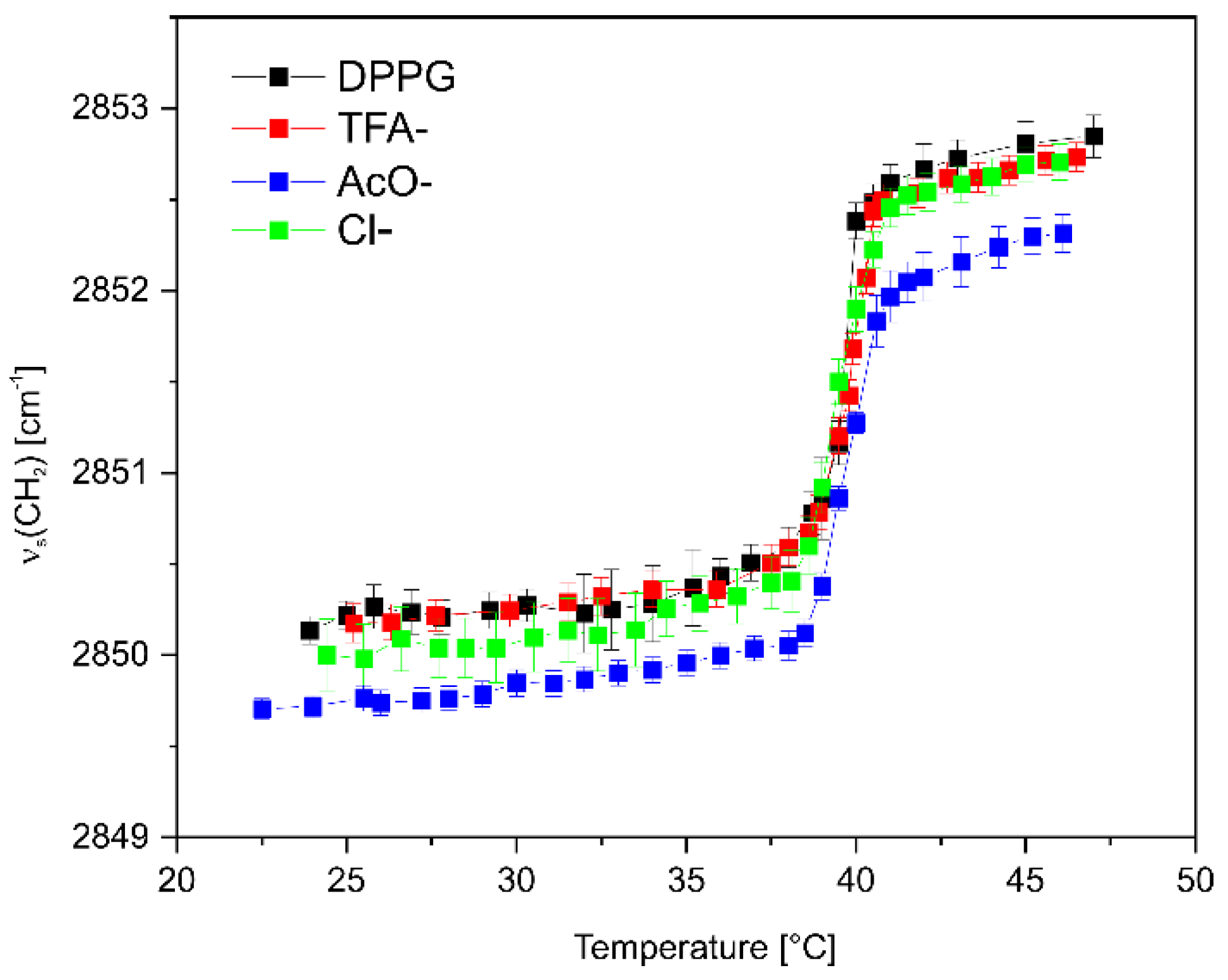
2.6. Effect of Net Peptide and Counterions on Lipopeptide-Lipid Interactions
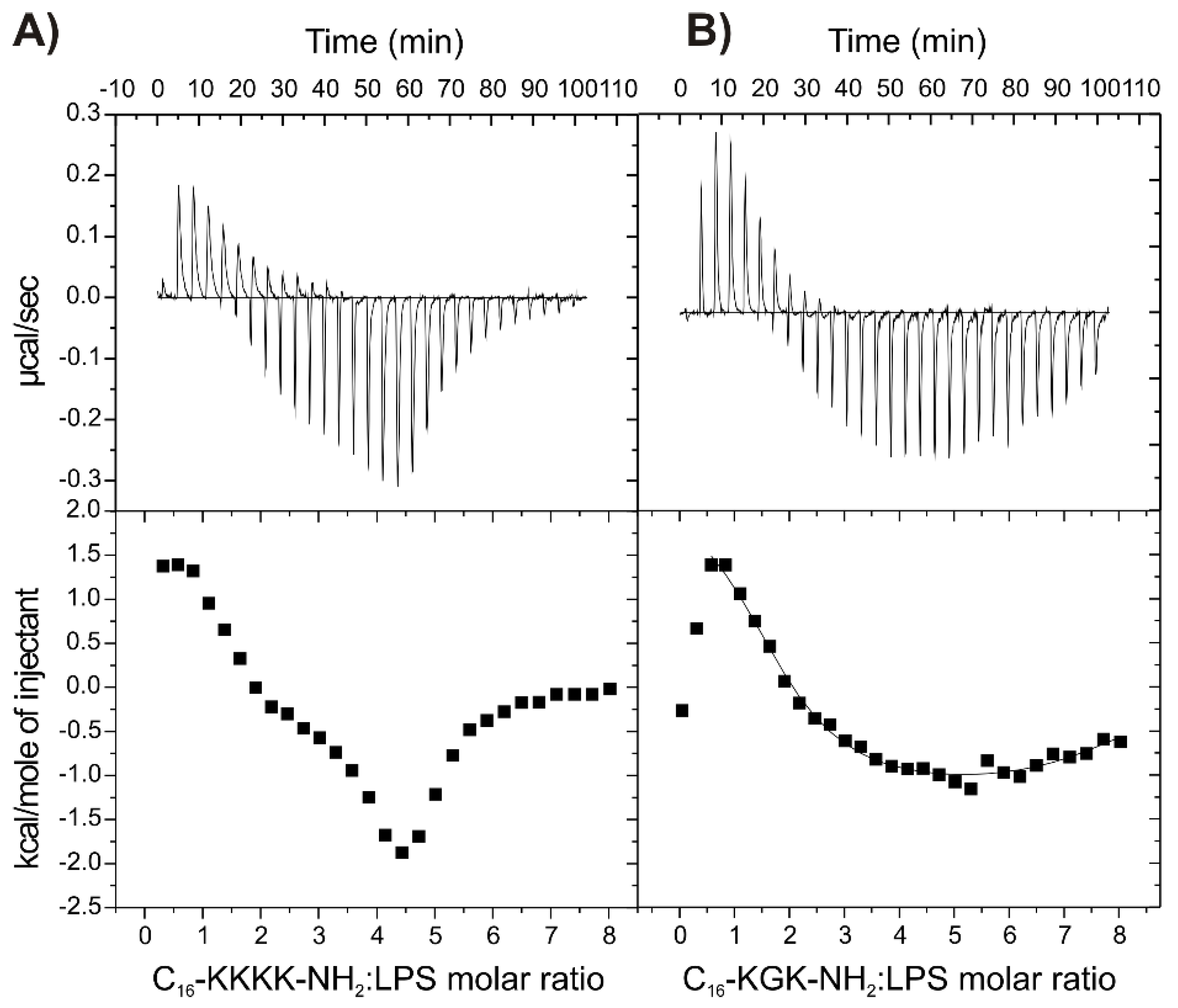
2.7. Binding of the Peptides to LPS
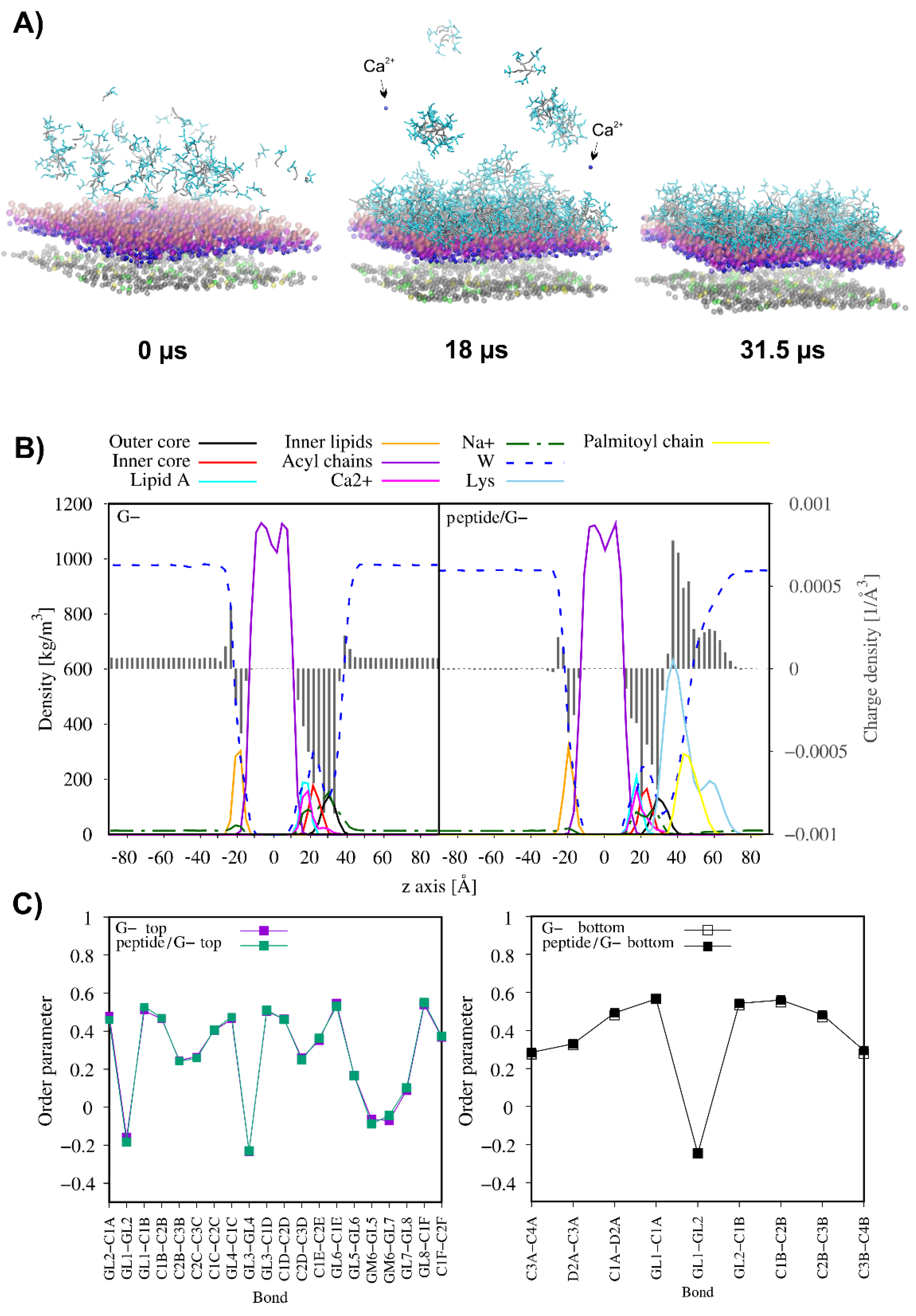
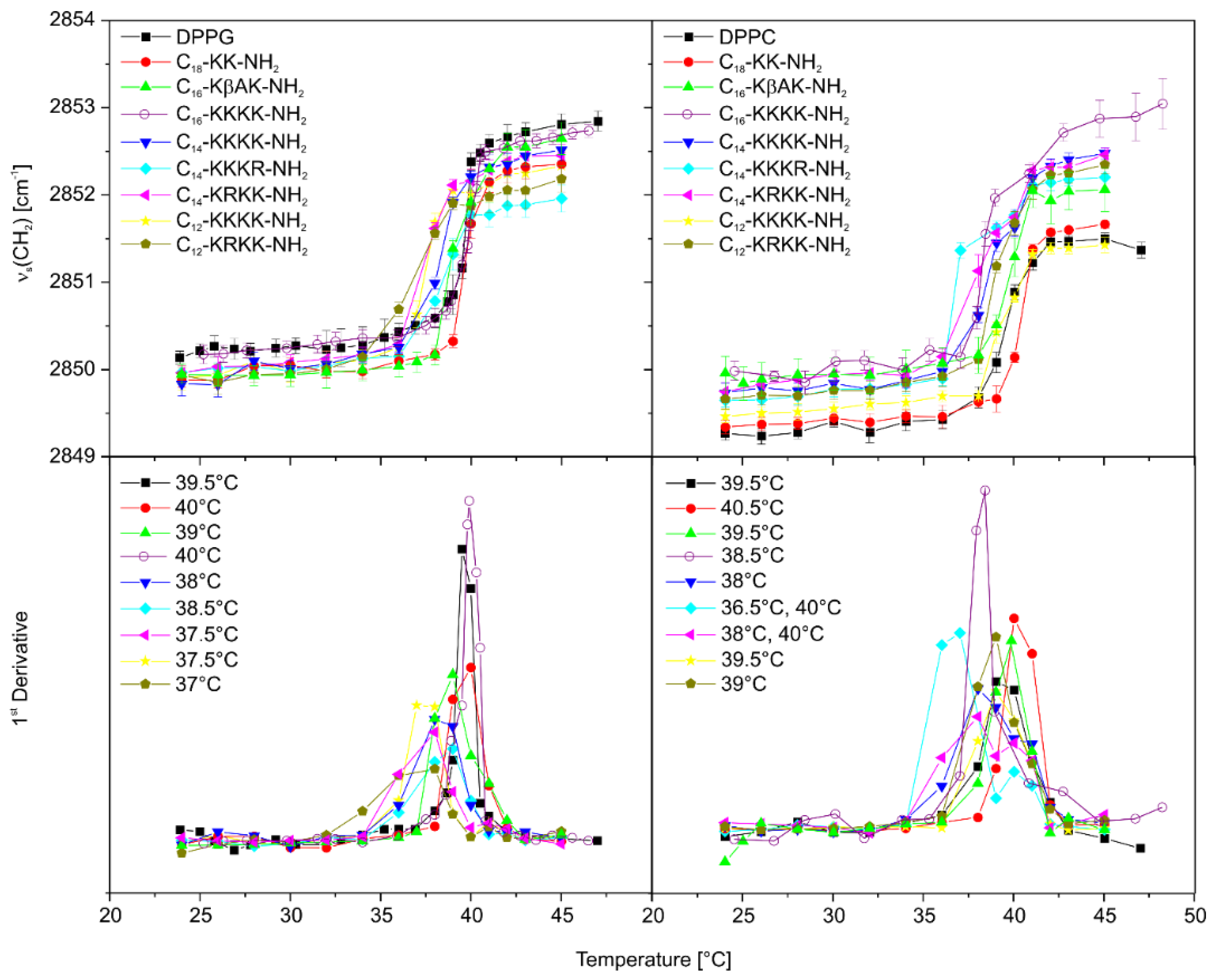
2.8. The Effect of Lipopeptide Binding on the Lipid Acyl Chain Order
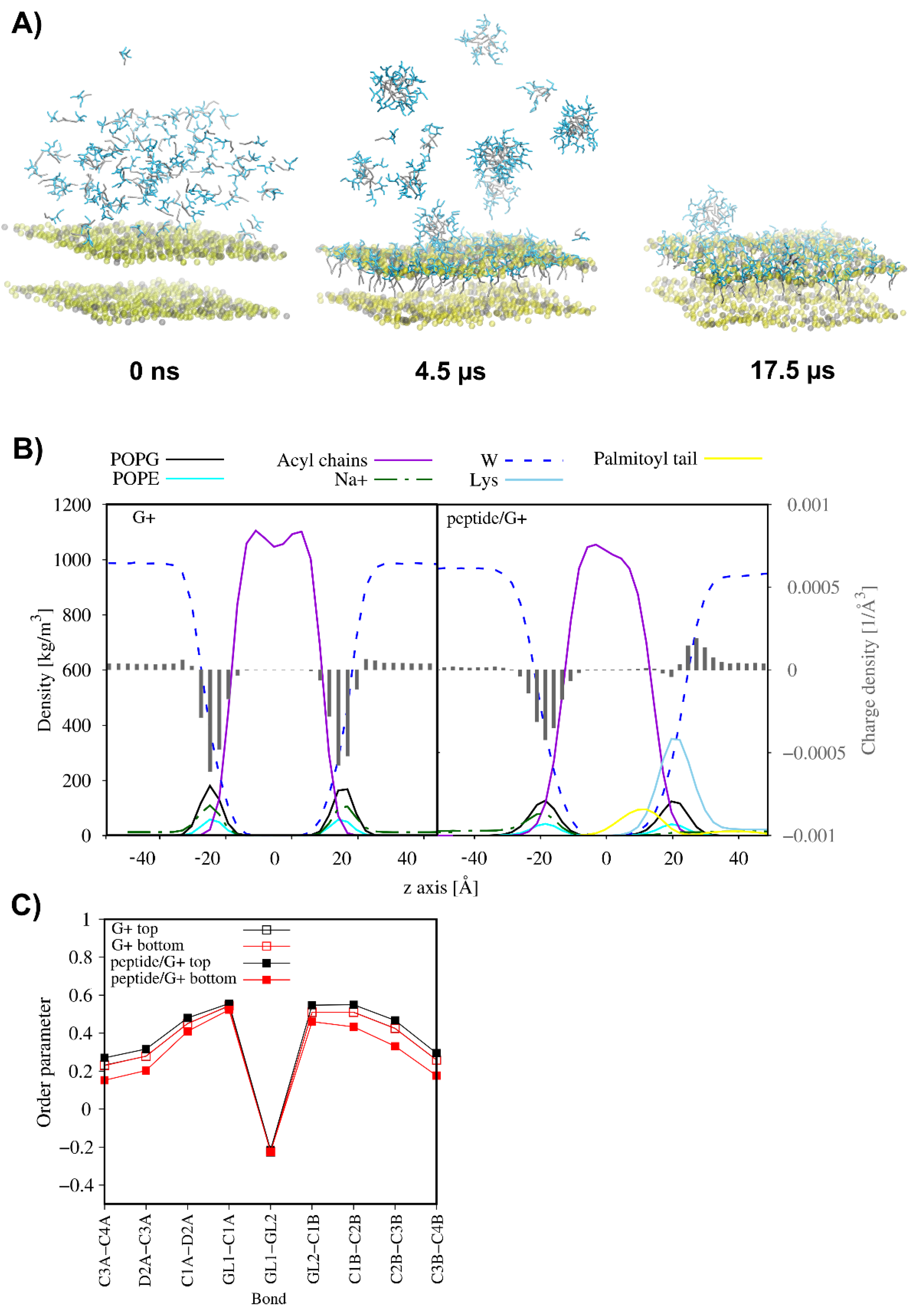
2.9. MD Simulations
3. Materials and Methods
3.1. Reagents
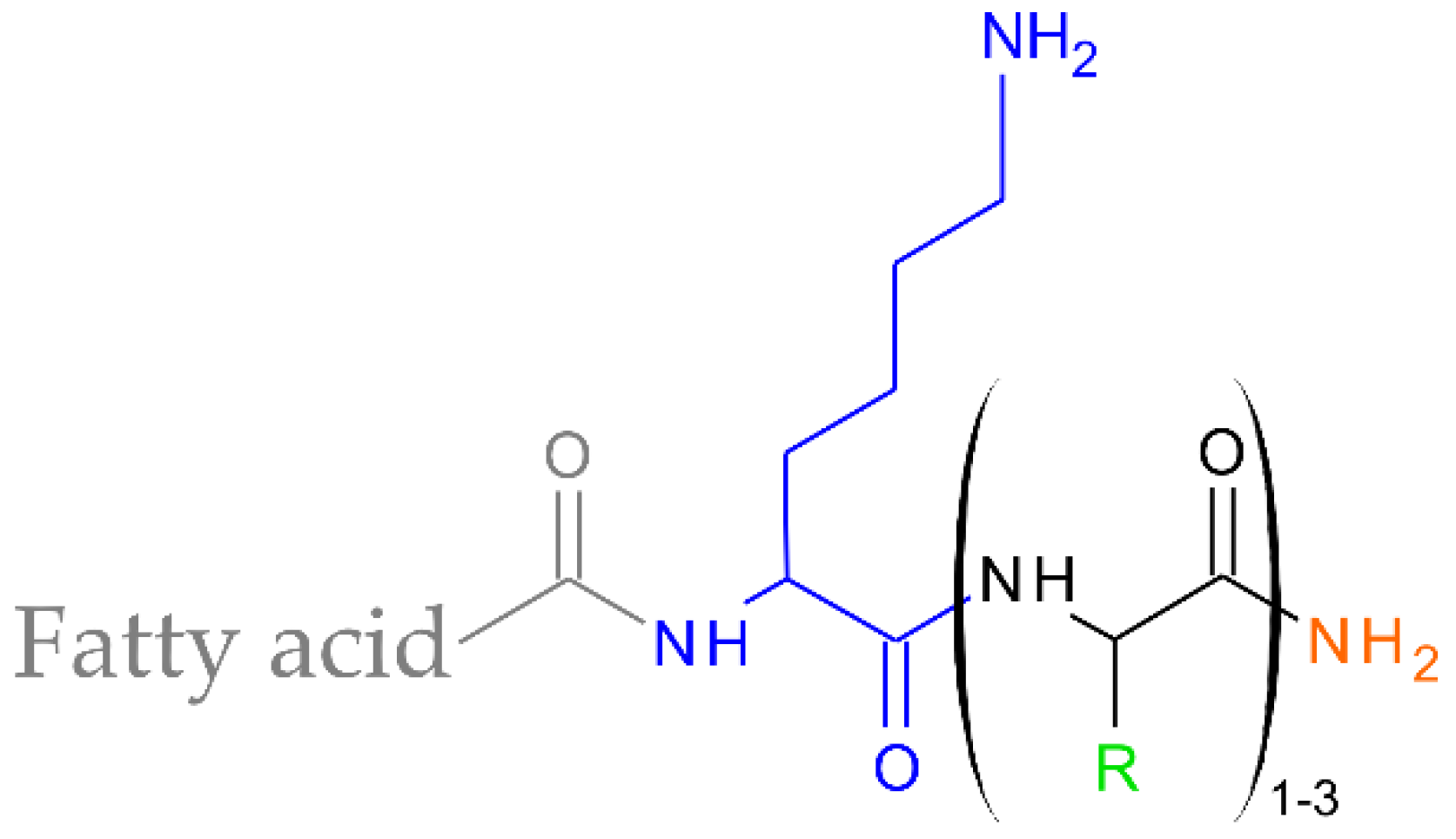
3.2. Lipopeptide Synthesis
3.3. Counterion Exchange
3.4. Microbiological Studies
3.5. Evaluation of Haemolytic and Cytotoxic Activities
3.6. Serum Stability Studies
3.7. Critical Aggregation Concentration Measurements
3.8. Circular Dichroism Spectroscopy
3.9. Thioflavin T (ThT) Assay
3.10. Transmission Electron Microscopy (TEM)
3.11. Fluorescence Spectroscopy
3.12. FTIR Measurements
3.13. ITC Measurements
3.14. Coarse-Grained Molecular Dynamic Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAC | Critical aggregation concentration |
| C18 | Stearic acid |
| C16 | Palmitic acid |
| C14 | Myristic acid |
| C12 | Lauric acid |
| CDL2 | Cardiolipin 2 |
| CG MD | Coarse-grained molecular dynamics |
| DPPC | 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine |
| DPPG | 1,2-Dipalmitoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] |
| LUV | Large unilamellar vesicle |
| MLV | Multilayer vesicle |
| POPC | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPE | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine |
| POPG | 1-Palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] |
| RaLPS | Ra mutant rough chemotype lipopolysaccharide |
References
- Rosenblatt-Farrell, N. The landscape of antibiotic resistance. Environ. Health Perspect. 2009, 17, A244–A250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullough, A.; Parekh, S.; Rathbone, J.; Del Mar, C.; Hoffmann, T. A systematic review of the public’s knowledge and beliefs about antibiotic resistance. J. Antimicrob. Chemother. 2016, 71, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadgostar, P. Antimicrobial resistance: Implications and costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straus, S.K.; Hancock, R.E. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: Comparison with cationic antimicrobial peptides and lipopeptides. Biochim. Biophys. Acta 2006, 1758, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Grossfield, A. Thermodynamics of antimicrobial lipopeptide binding to membranes: Origins of affinity and selectivity. Biophys. J. 2014, 107, 1862–1872. [Google Scholar] [CrossRef] [Green Version]
- Andrushchenko, V.V.; Aarabi, M.H.; Nguyen, L.T.; Prenner, E.J.; Vogel, H.J. Thermodynamics of the interactions of tryptophan-rich cathelicidin antimicrobial peptides with model and natural membranes. Biochim. Biophys. Acta 2008, 1778, 1004–1014. [Google Scholar] [CrossRef] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Narayana, J.L.; Chen, J.Y. Antimicrobial peptides: Possible anti-infective agents. Peptides 2015, 72, 88–94. [Google Scholar] [CrossRef]
- Haney, E.F.; Hancock, R.E. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100, 572–583. [Google Scholar] [CrossRef] [Green Version]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef]
- Kowalczyk, R.; Harris, P.W.R.; Williams, G.M.; Yang, S.H.; Brimble, M.A. Peptide lipidation—A synthetic strategy to afford peptide based therapeutics. Adv. Exp. Med. Biol. 2017, 1030, 185–227. [Google Scholar] [PubMed]
- Avrahami, D.; Shai, Y. A new group of antifungal and antibacterial lipopeptides derived from non-membrane active peptides conjugated to palmitic acid. J. Biol. Chem. 2004, 279, 12277–12285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radzishevsky, I.S.; Rotem, S.; Bourdetsky, D.; Navon-Venezia, S.; Carmeli, Y.; Mor, A. Improved antimicrobial peptides based on acyl-lysine oligomers. Nat. Biotechnol. 2007, 25, 657–659. [Google Scholar] [CrossRef]
- Radzishevsky, I.S.; Kovachi, T.; Porat, Y.; Ziserman, L.; Zaknoon, F.; Danino, D.; Mor, A. Structure-activity relationships of antibacterial acyl-lysine oligomers. Chem. Biol. 2008, 15, 354–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makovitzki, A.; Viterbo, A.; Brotman, Y.; Chet, I.; Shai, Y. Inhibition of fungal and bacterial plant pathogens in vitro and in planta with ultrashort cationic lipopeptides. Appl. Environ. Microb. 2007, 73, 6629–6636. [Google Scholar] [CrossRef] [Green Version]
- Makovitzki, A.; Baram, J.; Shai, Y. Antimicrobial lipopolypeptides composed of palmitoyl di- and tricationic peptides: In vitro and in vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry 2008, 47, 10630–10636. [Google Scholar] [CrossRef]
- Kamysz, W.; Silvestri, C.; Cirioni, O.; Giacometti, A.; Licci, A.; Della Vittoria, A.; Okroj, M.; Scalise, G. In vitro activities of the lipopeptides palmitoyl (Pal)-Lys-Lys-NH2 and Pal-Lys-Lys alone and in combination with antimicrobial agents against multiresistant Gram-positive cocci. Antimicrob. Agents Chemother. 2007, 51, 354–358. [Google Scholar] [CrossRef] [Green Version]
- Cirioni, O.; Giacometti, A.; Ghiselli, R.; Kamysz, W.; Silvestri, C.; Orlando, F.; Mocchegiani, F.; Della Vittoria, A.; Kamysz, E.; Saba, V.; et al. The lipopeptides Pal–Lys–Lys–NH2 and Pal–Lys–Lys soaking alone and in combination with intraperitoneal vancomycin prevent vascular graft biofilm in a subcutaneous rat pouch model of staphylococcal infection. Peptides 2007, 28, 1299–1303. [Google Scholar] [CrossRef]
- Barchiesi, F.; Giacometti, A.; Cirioni, O.; Arzeni, D.; Silvestri, C.; Kamysz, W.; Abbruzzetti, A.; Riva, A.; Kamysz, E.; Scalise, G. In vitro activity of the synthetic lipopeptide PAL-Lys-Lys-NH2 alone and in combination with antifungal agents against clinical isolates of Cryptococcus neoformans. Peptides 2007, 28, 1509–1513. [Google Scholar] [CrossRef]
- Jerala, R. Synthetic lipopeptides: A novel class of anti-infectives. Expert Opin. Investig. Drugs 2007, 16, 1159–1169. [Google Scholar] [CrossRef]
- Radzishevsky, I.S.; Rotem, S.; Zaknoon, F.; Gaidukov, L.; Dagan, A.; Mor, A. Effects of acyl versus aminoacyl conjugation on the properties of antimicrobial peptides. Antimicrob. Agents Chemother. 2005, 49, 2412–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avrahami, D.; Shai, Y. Conjugation of a magainin analogue with lipophilic acids controls hydrophobicity, solution assembly, and cell selectivity. Biochemistry 2002, 41, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, D.; Shai, Y. Bestowing antifungal and antibacterial activities by lipophilic acid conjugation to D, L-amino acid-containing antimicrobial peptides: A plausible mode of action. Biochemistry 2003, 42, 14946–14956. [Google Scholar] [CrossRef]
- Majerle, A.; Kidrič, J.; Jerala, R. Enhancement of antibacterial and lipopolysaccharide binding activities of a human lactoferrin peptide fragment by the addition of acyl chain. J. Antimicrob. Chemoth. 2003, 51, 1159–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simerska, P.; Moyle, P.M.; Toth, I. Modern lipid-, carbohydrate-, and peptide-based delivery systems for peptide, vaccine, and gene products. Med. Res. Rev. 2011, 31, 520–547. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Riis, B.J.; Mehta, N.; Stern, W.; Arbit, E.; Christiansen, C.; Henriksen, K. Lessons learned from the clinical development of oral peptides. Br. J. Clin. Pharmacol. 2015, 79, 720–732. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Bulaj, G. Converting peptides into drug leads by lipidation. Curr. Med. Chem. 2012, 19, 1602–1618. [Google Scholar] [CrossRef]
- Schneider, T.; Müller, A.; Miess, H.; Gross, H. Cyclic lipopeptides as antibacterial agents—Potent antibiotic activity mediated by intriguing mode of actions. Int. J. Med. Microbiol. 2014, 304, 37–43. [Google Scholar] [CrossRef]
- Letscher-Bru, V.; Herbrecht, R. Caspofungin: The first representative of a new antifungal class. J. Antimicrob. Chemother. 2003, 51, 513–521. [Google Scholar] [CrossRef] [Green Version]
- Meir, O.; Zaknoon, F.; Cogan, U.; Mor, A. A broad-spectrum bactericidal lipopeptide with anti-biofilm properties. Sci. Rep. 2017, 7, 2198. [Google Scholar] [CrossRef]
- Mishra, B.; Lushnikova, T.; Wang, G. Small lipopeptides possess anti-biofilm capability comparable to daptomycin and vancomycin. RSC Adv. 2015, 5, 59758–59769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paduszynska, M.A.; Maciejewska, M.; Neubauer, D.; Golacki, K.; Szymukowicz, M.; Bauer, M.; Kamysz, W. Influence of short cationic lipopeptides with fatty acids of different chain lengths on bacterial biofilms formed on polystyrene and hydrogel surfaces. Pharmaceutics 2019, 11, 506. [Google Scholar] [CrossRef] [Green Version]
- Paduszynska, M.A.; Greber, K.E.; Paduszynski, W.; Sawicki, W.; Kamysz, W. Activity of Temporin A and short lipopeptides combined with gentamicin against biofilm formed by Staphylococcus aureus and Pseudomonas aeruginosa. Antibiotics 2020, 9, 566. [Google Scholar] [CrossRef] [PubMed]
- Vaezi, Z.; Bortolotti, A.; Luca, V.; Perilli, G.; Mangoni, M.L.; Khosravi-Far, R.; Bobone, S.; Stella, L. Aggregation determines the selectivity of membrane-active anticancer and antimicrobial peptides: The case of killerFLIP. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikorska, E.; Stachurski, O.; Neubauer, D.; Małuch, I.; Wyrzykowski, D.; Bauer, M.; Brzozowski, B.; Kamysz, W. Short arginine-rich lipopeptides: From self-assembly to antimicrobial activity. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2242–2251. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, E.; Dawgul, M.; Greber, K.; Iłowska, E.; Pogorzelska, A.; Kamysz, W. Self-assembly and interactions of short antimicrobial cationic lipopeptides with membrane lipids: ITC, FTIR and molecular dynamics studies. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2625–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greber, K.E. Synthesis and surface activity of cationic amino acid-based surfactants in aqueous solution. J. Surfact. Deterg. 2017, 20, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Stachurski, O.; Neubauer, D.; Małuch, I.; Wyrzykowski, D.; Bauer, M.; Bartoszewska, S.; Kamysz, W.; Sikorska, E. Effect of self-assembly on antimicrobial activity of double-chain short cationic lipopeptides. Bioorg. Med. Chem. 2019, 27, 115129. [Google Scholar] [CrossRef]
- Miyake, M.; Yamada, K.; Oyama, N. Self-assembling of guanidine-type surfactant. Langmuir 2008, 24, 8527–8532. [Google Scholar] [CrossRef]
- Harms, M.J.; Schlessman, J.L.; Sue, G.R.; García-Moreno, B. Arginine residues at internal positions in a protein are always charged. Proc. Natl. Acad. Sci. USA 2011, 108, 18954–18959. [Google Scholar] [CrossRef]
- Lee, D.; Lee, J.; Seok, C. What stabilizes close arginine pairing in proteins? Phys. Chem. Chem. Phys. 2013, 15, 5844–5853. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, M.F.; Herrera, M.G.; Dodero, V.I. Evaluation of peptide/protein self-assembly and aggregation by spectroscopic methods. Molecules 2020, 25, 4854. [Google Scholar] [CrossRef]
- Castelletto, V.; Edwards-Gayle, C.J.C.; Greco, F.; Hamley, I.W.; Seitsonen, J.; Ruokolainen, J. Self-assembly, tunable hydrogel properties, and selective anti-cancer activity of a carnosine-derived lipidated peptide. ACS Appl. Mater. Interfaces 2019, 11, 33573–33580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, D.J.; Steinman, L.; Kim, D.; Fathman, C.; Rothbard, J. Polyarginine enters cells more efficiently than other polycationic homopolymers. Chem. Biol. Drug Des. 2000, 56, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Rothbard, J.B.; Jessop, T.C.; Lewis, R.S.; Murray, B.A.; Wender, P.A. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J. Am. Chem Soc. 2004, 126, 9506–9507. [Google Scholar] [CrossRef]
- Schwieger, C.; Blume, A. Interaction of poly (L-arginine) with negatively charged DPPG membranes: Calorimetric and monolayer studies. Biomacromolecules 2009, 10, 2152–2161. [Google Scholar] [CrossRef]
- Arouri, A.; Dathe, M.; Blume, A. The helical propensity of KLA amphipathic peptides enhances their binding to gel-state lipid membranes. Biophys. Chem. 2013, 180, 10–21. [Google Scholar] [CrossRef]
- Hoernke, M.; Schwieger, C.; Kerth, A.; Blume, A. Binding of cationic pentapeptides with modified side chain lengths to negatively charged lipid membranes: Complex interplay of electrostatic and hydrophobic interactions. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Giribaldi, J.; Haufe, Y.; Evans, E.R.J.; Wilson, D.T.; Daly, N.L.; Enjalbal, C.; Nicke, A.; Dutertre, S. Synthesis, structural and pharmacological characterizations of CIC, a novel α-conotoxin with an extended N-Terminal tail. Mar. Drugs 2021, 19, 141. [Google Scholar] [CrossRef]
- Vemuri, S. Comparison of assays for determination of peptide content for lyophilized thymalfasin. J. Pept. Res. 2005, 65, 433–439. [Google Scholar] [CrossRef]
- Schmidt, M.; Toplak, A.; Rozeboom, H.J.; Wijma, H.J.; Quaedflieg, P.J.L.M.; van Maarseveen, J.H.; Janssen, D.B.; Nuijens, T. Design of a substrate-tailored peptiligase variant for the efficient synthesis of thymosin-α 1. Org. Biomol. Chem. 2018, 16, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Giribaldi, J.; Haufe, Y.; Evans, E.R.; Amar, M.; Durner, A.; Schmidt, C.; Faucherre, A.; Moha Ou Maati, H.; Enjalbal, C.; Molgó, J.; et al. Backbone cyclization turns a venom peptide into a stable and equipotent ligand at both muscle and neuronal nicotinic receptors. J. Med. Chem. 2020, 63, 12682–12692. [Google Scholar] [CrossRef] [PubMed]
- Sikora, K.; Jaśkiewicz, M.; Neubauer, D.; Bauer, M.; Bartoszewska, S.; Barańska-Rybak, W.; Kamysz, W. Counter-ion effect on antistaphylococcal activity and cytotoxicity of selected antimicrobial peptides. Amino Acids 2018, 50, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Abraham, T.; Lewis, R.N.; Hodges, R.S.; McElhaney, R.N. Isothermal titration calorimetry studies of the binding of the antimicrobial peptide gramicidin S to phospholipid bilayer membranes. Biochemistry 2005, 44, 11279–11285. [Google Scholar] [CrossRef] [PubMed]
- Sikora, K.; Jaśkiewicz, M.; Neubauer, D.; Migoń, D.; Kamysz, W. The role of counter-ions in peptides-an overview. Pharmaceuticals 2020, 13, 442. [Google Scholar] [CrossRef]
- Paterová, J.; Rembert, K.B.; Heyda, J.; Kurra, Y.; Okur, H.I.; Liu, W.R.; Hilty, C.; Cremer, P.S.; Jungwirth, P. Reversal of the hofmeister series: Specific ion effects on peptides. J. Phys. Chem. B 2013, 117, 8150–8158. [Google Scholar] [CrossRef] [PubMed]
- Dér, A. Salts, interfacial water and protein conformation. Biotechnol. Biotechnol. Equip. 2008, 22, 629–633. [Google Scholar] [CrossRef] [Green Version]
- Shang, D.; Zhang, Q.; Dong, W.; Liang, H.; Bi, X. The effects of LPS on the activity of Trp-containing antimicrobial peptides against Gram-negative bacteria and endotoxin neutralization. Acta Biomater. 2016, 33, 153–165. [Google Scholar] [CrossRef]
- Domingues, M.M.; Bianconi, M.L.; Barbosa, L.R.; Santiago, P.S.; Tabak, M.; Castanho, M.A.R.B.; Itri, R.; Santos, N.C. rBPI21 interacts with negative membranes endothermically promoting the formation of rigid multilamellar structures. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2419–2427. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, K.; David, A.; Howe, J.; Koch, M.H.; Andrä, J.; Garidel, P. Temperature dependence of the binding of endotoxins to the polycationic peptides polymyxin B and its nonapeptide. Biophys. J. 2005, 88, 1845–1858. [Google Scholar] [CrossRef]
- Moorman, M.A.; Thelemann, C.A.; Zhou, S.; Pestka, J.J.; Linz, J.E.; Ryser, E.T. Altered hydrophobicity and membrane composition in stress-adapted Listeria innocua. J. Food Prot. 2008, 71, 182–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royce, L.A.; Liu, P.; Stebbins, M.J.; Hanson, B.C.; Jarboe, L.R. The damaging effects of short chain fatty acids on Escherichia coli membranes. Appl. Microbiol. Biotechnol. 2013, 97, 8317–8327. [Google Scholar] [CrossRef] [Green Version]
- Melo, M.N.; Ferre, R.; Castanho, M.A. Antimicrobial peptides: Linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 2009, 7, 245–250. [Google Scholar] [CrossRef]
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Antimicrobial peptides: Interaction with model and biological membranes and synergism with chemical antibiotics. Front. Chem. 2018, 6, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifton, L.A.; Skoda, M.W.; Le Brun, A.P.; Ciesielski, F.; Kuzmenko, I.; Holt, S.A.; Lakey, J.H. Effect of divalent cation removal on the structure of gram-negative bacterial outer membrane models. Langmuir 2015, 31, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikora, K.; Neubauer, D.; Jaśkiewicz, M.; Kamysz, W. Citropin 1.1 trifluoroacetate to chloride counter-ion exchange in hcl-saturated organic solutions: An alternative approach. Int. J. Pept. Res. Ther. 2018, 24, 265–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Validation of Analytical Procedures: Text and Methodology Q2(R1); ICH Expert Working Group: Geneva, Switzerland, 2005; pp. 1–13. Available online: https://www.gmp-compliance.org/files/guidemgr/Q2(R1).pdf (accessed on 11 October 2022).
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, 3rd ed.; Approved standard; CLSI document M27-A3; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008. [Google Scholar]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 10th ed.; Approved standard; CLSI document M07-A10; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2009. [Google Scholar]
- Małuch, I.; Stachurski, O.; Kosikowska-Adamus, P.; Makowska, M.; Bauer, M.; Wyrzykowski, D.; Hać, A.; Kamysz, W.; Deptuła, M.; Pikuła, M.; et al. Double-headed cationic lipopeptides: An emerging class of antimicrobials. Int. J. Mol. Sci. 2020, 21, 8944. [Google Scholar] [CrossRef]
- Periole, X.; Marrink, S.J. The Martini coarse-grained force field. Methods Mol. Biol. 2013, 924, 533–565. [Google Scholar]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, K.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Chugunov, A.; Pyrkova, D.; Nolde, D.; Polyansky, A.; Pentkovsky, V.; Efremov, R. Lipid-II forms potential “landing terrain” for lantibiotics in simulated bacterial membrane. Sci. Rep. 2013, 3, 1678. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.C.; Bruininks, B.M.H.; Jefferies, D.; de Souza, P.C.T.; Lee, J.; Patel, D.S.; Marrink, S.J.; Qi, Y.; Khalid, S.; Im, W. CHARMM-GUI Martini Maker for modeling and simulation of complex bacterial membranes with lipopolysaccharides. J. Comput. Chem. 2017, 38, 2354–2363. [Google Scholar] [CrossRef] [Green Version]
- Winger, M.; Trzesniak, D.; Baron, R.; van Gunsteren, W.F. On using a too large integration time step in molecular dynamics simulations of coarse-grained molecular models. Phys. Chem. Chem. Phys. 2009, 11, 1934–1941. [Google Scholar] [CrossRef] [Green Version]
- Allen, W.J.; Lemkul, J.A.; Bevan, D.R. GridMAT-MD: A grid-based membrane analysis tool for use with molecular dynamics. J. Comput. Chem. 2009, 30, 1952–1958. [Google Scholar] [CrossRef]
- Greber, K.E.; Dawgul, M.; Kamysz, W.; Sawicki, W. Cationic net charge and counter ion type as antimicrobial activity determinant factors of short lipopeptides. Front. Microbiol. 2017, 8, 123. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
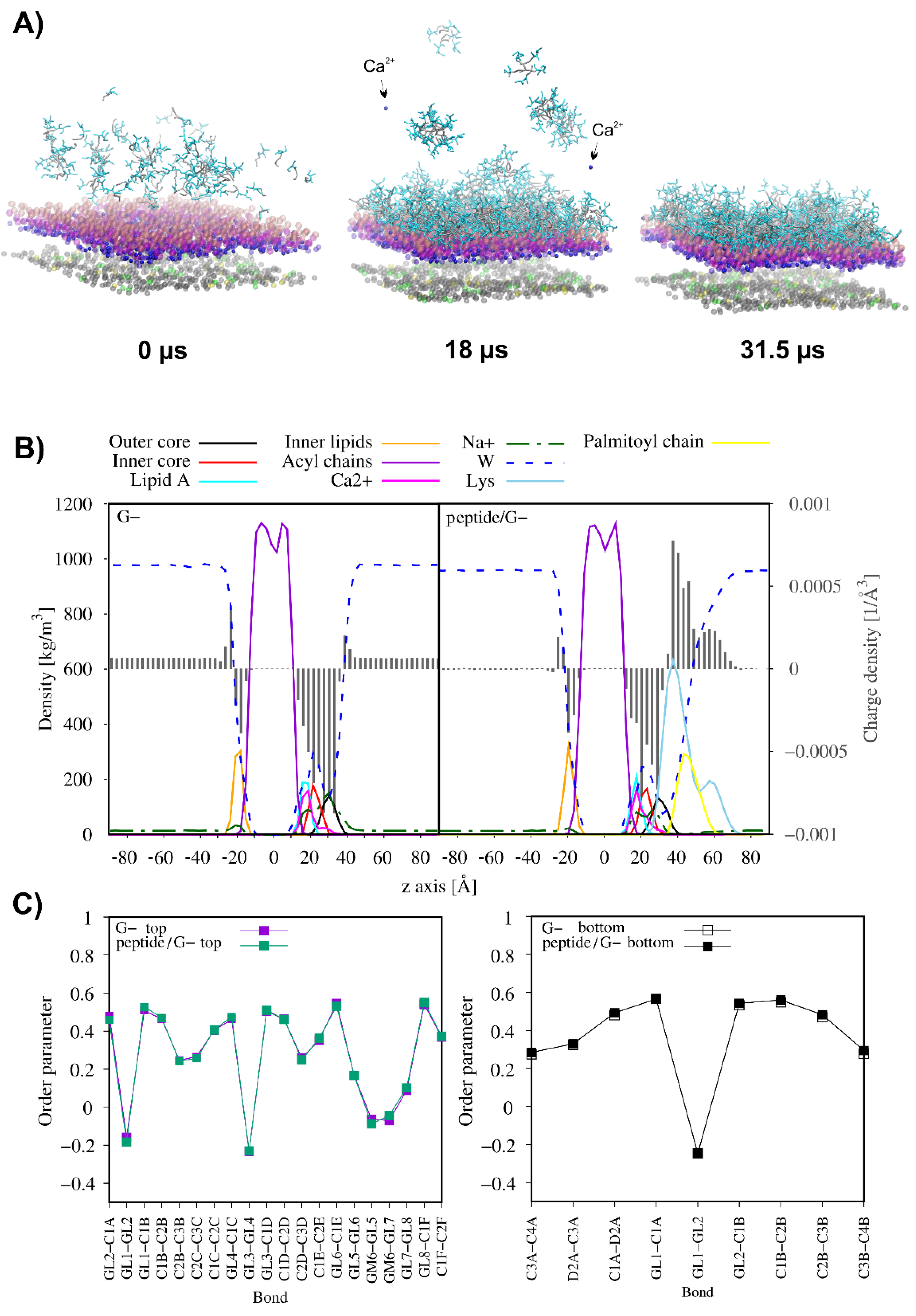{kind=link}
| Peptide | MIC μg/mL | IC50 (μg/mL) | ||||
|---|---|---|---|---|---|---|
| S. aureus | S. epidermidis | E. coli | P. aeruginosa | C. albicans | ||
| C18-KK-NH2 | 64 | 4 | 128 | 64 | 4 | 0.69 ± 0.001 |
| C16-KK-NH2 | 8 | 4 | 8 | 128 | 4 | 2.5 ± 0.8 |
| C16-KKKK-NH2 | 8 | 8 | 16 | 16 | 16 | 23.52 ± 1.30 |
| C16-KGK-NH2 | 16 | 4 | 8 | 64 | 8 | 7.41 ± 3.05 |
| C16-KβAK-NH2 | 128 | 2 | 64 | 64 | 4 | 25.11 ± 6.17 |
| C14-KKKK-NH2 | 32 | 2 | 128 | 64 | 128 | 15.63 ± 0.74 |
| C14-KKKR-NH2 | 32 | ≤1 | 128 | 128 | 128 | 32.5 ± 10.0 |
| C14-KRKK-NH2 | 16 | ≤1 | 64 | 64 | 128 | 20.38 ± 4.12 |
| C12-KKKK-NH2 | 64 | 16 | 512 | 256 | >512 | - |
| C12-KRKK-NH2 | 64 | ≤1 | 512 | 512 | 512 | - |
| Peptide | CAC mM | CAC μg/mL | γCAC mN/m |
|---|---|---|---|
| C18-KK-NH2 | 0.06 | 34 | 45 |
| C16-KK-NH2 | 0.09 (1.07) | 45 | 43 |
| C16-KKKK-NH2 | 0.75 (14.6) | 575 | 46 |
| C16-KGK-NH2 | 0.16 (1.89) | 92 | 47 |
| C16-KβAK-NH2 | Not determined | ||
| C14-KKKK-NH2 | 1.22 (18.04) | 905 | 40 |
| C14-KKKR-NH2 | 1.01 | 779 | 42 |
| C14-KRKK-NH2 | 1.25 | 962 | 37 |
| C12-KKKK-NH2 | 3.10 | 2216 | 37 |
| C12-KRKK-NH2 | 3.60 | 2658 | 40 |
| Peptide | n | pKITC | ΔH kcal mol−1 | TΔS kcal mol−1 | ΔG kcal mol−1 | |
|---|---|---|---|---|---|---|
| POPG | ||||||
| C18-KK-NH2 | 0.91 ± 0.03 | 5.90 ± 0.22 | −0.67 ± 0.03 | 9.74 ± 0.23 | −10.41 ± 0.23 | |
| C16-KK-NH2 | 1.08 ± 0.04 | 5.32 ± 0.11 | −0.49 ± 0.03 | 9.14 ± 0.15 | −9.62 ± 0.15 | |
| C16-KGK-NH2 | 0.76 ± 0.05 | 5.61 ± 0.21 | −0.46 ± 0.04 | 9.56 ± 0.29 | −10.02 ± 0.29 | |
| C16-KβAK-NH2 | 1.54 ± 0.05 | 5.34 ± 0.11 | −0.52 ± 0.02 | 9.13 ± 0.16 | −9.65 ± 0.16 | |
| C16-KKKK-NH2 | ||||||
| TFA− | 2.87 ± 0.16 | 4.73 ± 0.16 | −0.37 ± 0.03 | 8.46 ± 0.22 | −8.83 ± 0.21 | |
| AcO− | 3.70 ± 0.11 | 5.14 ± 0.15 | −0.37 ± 0.01 | 9.02 ± 0.20 | −9.38 ± 0.20 | |
| Cl− | 6.13 ± 0.32 | 4.69 ± 0.15 | −0.36 ± 0.03 | 8.41 ± 0.21 | −8.76 ± 0.21 | |
| C14-KKKK-NH2 | 3.94 ± 0.14 | 6.10 ± 0.42 | −0.09 ± 0.01 | 10.60 ± 0.58 | −10.69 ± 0.58 | |
| C14-KKKR-NH2 | 3.44 ± 0.08 | 5.97 ± 0.22 | −0.14 ± 0.01 | 10.37 ± 0.30 | −10.51 ± 0.30 | |
| C14-KRKK-NH2 | 4.01 ± 0.07 | 6.05 ± 0.20 | −0.29 ± 0.01 | 10.34 ± 0.27 | −10.63 ± 0.27 | |
| DPPG | ||||||
| C16-KKKK-NH2 | ||||||
| TFA− | 1.51 ± 0.08 | 5.16 ± 0.16 | −1.35 ± 0.10 | 8.05 ± 0.21 | −9.41 ± 0.21 | |
| AcO− | 1.79 ± 0.05 | 6.26 ± 0.27 | 2.30 ± 0.10 | 13.21 ± 0.38 | −10.91 ± 0.37 | |
| Cl− | 4.36 ± 0.10 | 4.68 ± 0.10 | 3.44 ± 0.13 | 12.20 ± 0.17 | −8.76 ± 0.14 | |
| C14-KKKK-NH2 | 3.39 ± 0.03 | 5.39 ± 0.06 | 1.71 ± 0.02 | 11.44 ± 0.22 | −9.72 ± 0.08 | |
| C12-KKKK-NH2 | 2.61 ± 0.03 | 5.19 ± 0.04 | 1.84 ± 0.03 | 11.29 ± 0.06 | −9.45 ± 0.06 | |
| LPS E. coli 055:B5 | ||||||
| C16-KGK-NH2 | 1.67 ± 0.09 6.31 ± 0.26 | 6.74 ± 0.41 5.48 ± 0.34 | 2.4 ± 0.36 −1.31 ± 0.15 | 13.96 ± 0.56 8.54 ± 0.59 | −11.56 ± 0.56 −9.84 ± 0.47 | |
| Counterion | Mass Fraction mg/mg | RSD % | Relative Content |
|---|---|---|---|
| No counterion | 0.965 | 2.343 | 100% |
| TFA− | 0.596 | 5.633 | 62% |
| AcO− | 0.691 | 5.065 | 72% |
| Cl− | 0.749 | 5.138 | 78% |
| Model | APL [Å2] | Thickness [Å] | |||||
|---|---|---|---|---|---|---|---|
| Top Leaflet | Bottom Leaflet | ||||||
| RaLPS | POPG | POPE | CDL2 | POPG | POPE | ||
| G− | 182.03 ± 0.30 | - | - | 73.54 ± 2.93 | 64.74 ± 3.17 | 62.98 ± 0.27 | 36.60 ± 0.01 |
| G−/peptide | 179.83 ± 0.25 | 74.15 ± 2.32 | 64.04 ± 2.65 | 62.13 ± 0.23 | 36.73 ± 0.09 | ||
| G+ | 63.39 ± 0.53 | 58.58 ± 1.06 | - | 63.40 ± 0.50 | 58.53 ± 1.18 | 39.98 ± 0.02 | |
| G+/peptide | 34.56 ± 0.77 | 39.50 ± 0.98 | 67.63 ± 6.14 | 63.07 ± 1.31 | 39.29 ± 0.17 | ||
| D [10−7 cm2/s] | |||||||
| G− | 0.014 ± 0.008 | 3.94 ± 0.38 | 5.16 ± 0.17 | 5.23 ± 0.25 | |||
| G−/peptide | 0.013 ± 0.006 | 2.66 ± 0.43 | 6.46 ± 0.79 | 4.71 ± 0.03 | |||
| G+ | - | 4.33 ± 0.87 | 4.68 ± 0.89 | - | 4.56 ± 0.64 | 5.01 ± 0.66 | |
| G+/peptide | - | 4.21 ± 0.21 | 4.24 ± 0.09 | - | 6.55 ± 0.27 | 5.65 ± 0.81 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stachurski, O.; Neubauer, D.; Walewska, A.; Iłowska, E.; Bauer, M.; Bartoszewska, S.; Sikora, K.; Hać, A.; Wyrzykowski, D.; Prahl, A.; et al. Understanding the Role of Self-Assembly and Interaction with Biological Membranes of Short Cationic Lipopeptides in the Effective Design of New Antibiotics. Antibiotics 2022, 11, 1491. https://doi.org/10.3390/antibiotics11111491
Stachurski O, Neubauer D, Walewska A, Iłowska E, Bauer M, Bartoszewska S, Sikora K, Hać A, Wyrzykowski D, Prahl A, et al. Understanding the Role of Self-Assembly and Interaction with Biological Membranes of Short Cationic Lipopeptides in the Effective Design of New Antibiotics. Antibiotics. 2022; 11(11):1491. https://doi.org/10.3390/antibiotics11111491
Chicago/Turabian StyleStachurski, Oktawian, Damian Neubauer, Aleksandra Walewska, Emilia Iłowska, Marta Bauer, Sylwia Bartoszewska, Karol Sikora, Aleksandra Hać, Dariusz Wyrzykowski, Adam Prahl, and et al. 2022. "Understanding the Role of Self-Assembly and Interaction with Biological Membranes of Short Cationic Lipopeptides in the Effective Design of New Antibiotics" Antibiotics 11, no. 11: 1491. https://doi.org/10.3390/antibiotics11111491
APA StyleStachurski, O., Neubauer, D., Walewska, A., Iłowska, E., Bauer, M., Bartoszewska, S., Sikora, K., Hać, A., Wyrzykowski, D., Prahl, A., Kamysz, W., & Sikorska, E. (2022). Understanding the Role of Self-Assembly and Interaction with Biological Membranes of Short Cationic Lipopeptides in the Effective Design of New Antibiotics. Antibiotics, 11(11), 1491. https://doi.org/10.3390/antibiotics11111491