
Guanidinylated Polymyxins as Outer Membrane Permeabilizers Capable of Potentiating Rifampicin, Erythromycin, Ceftazidime and Aztreonam against Gram-Negative Bacteria

Abstract
1. Introduction
2. Results
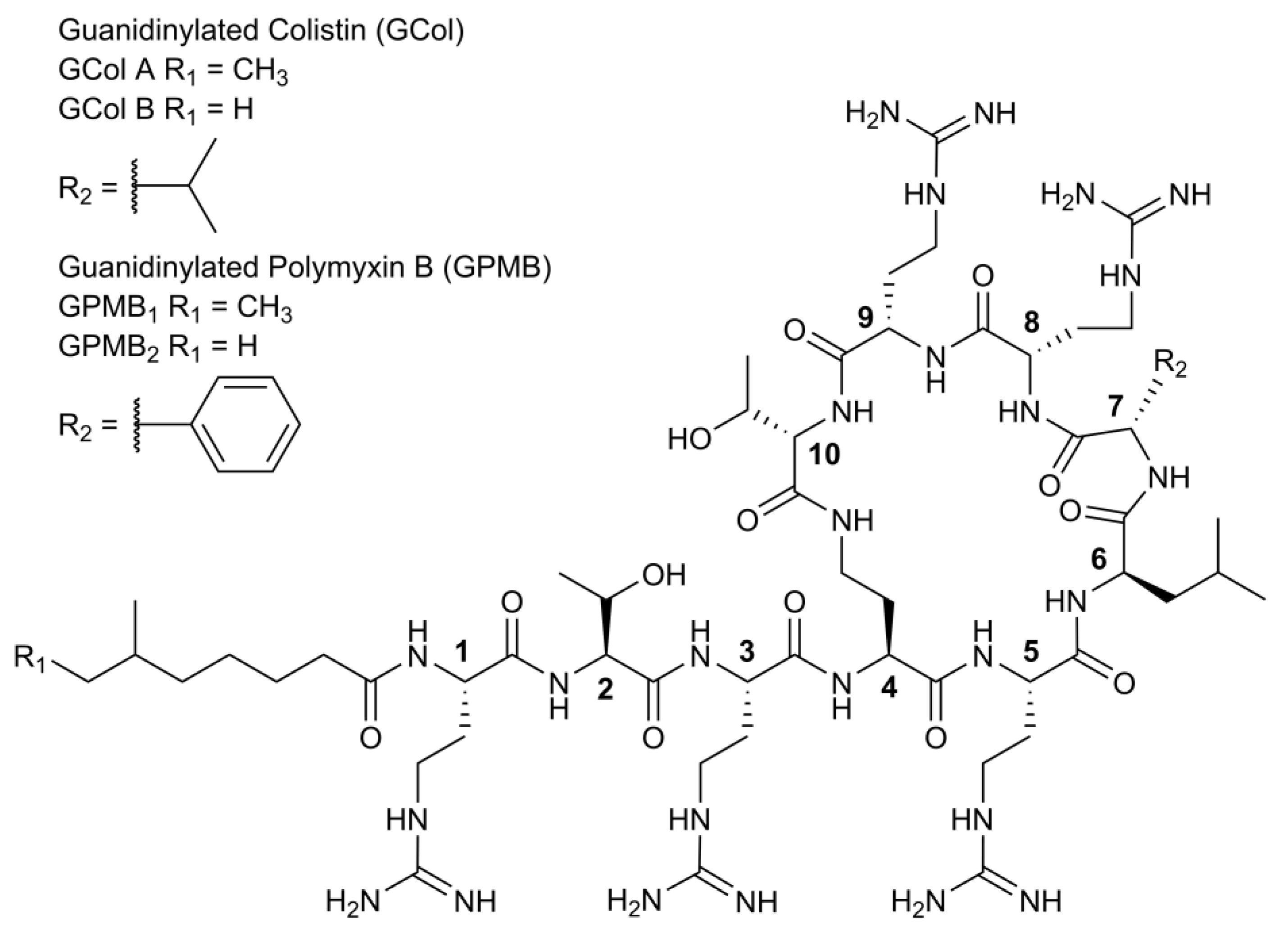
2.1. Synthesis of Guanidinylated Polymyxins
2.2. Antibacterial Activity of GCol and GPMB against Wild-Type Gram-Negative Bacteria
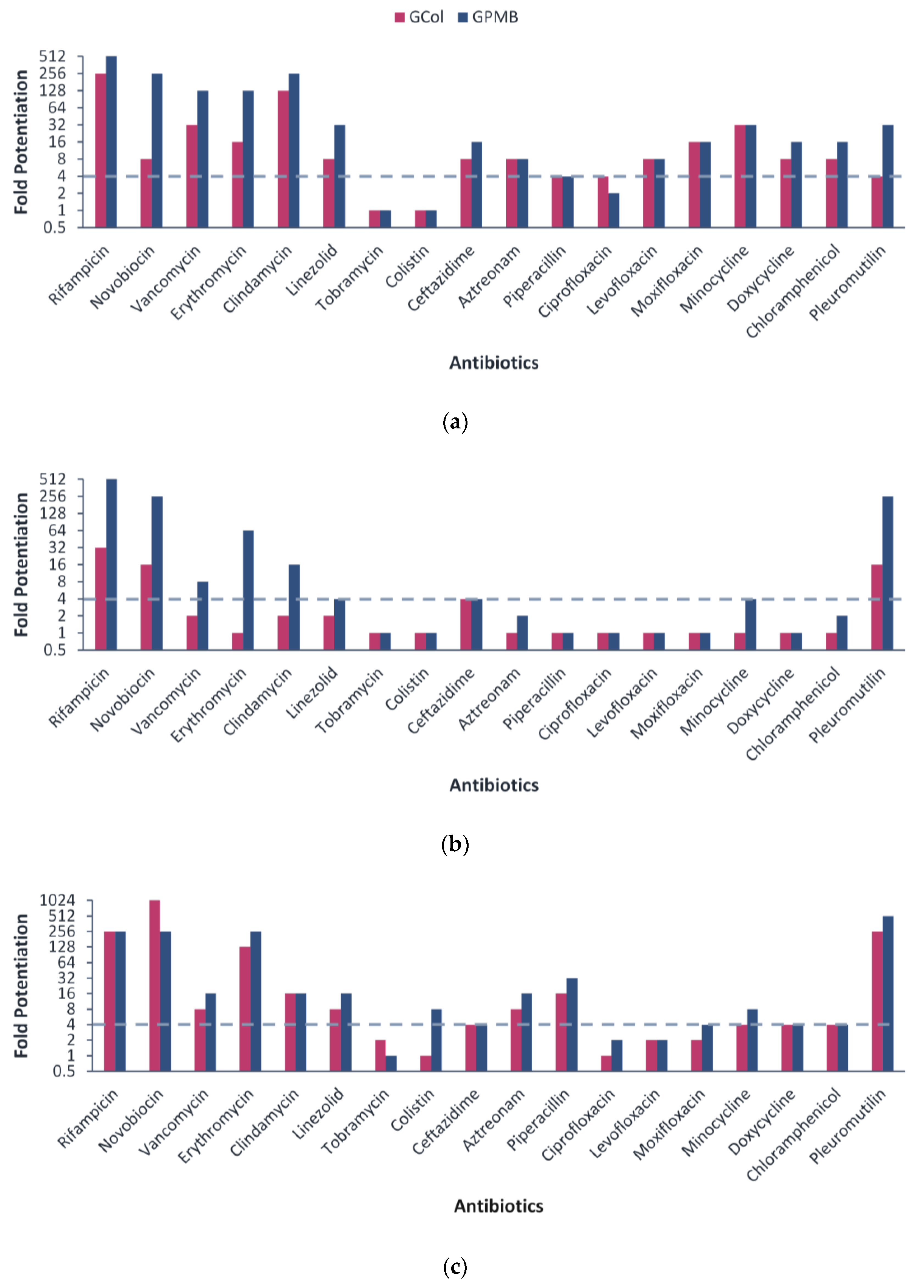
2.3. Synergy with Different Antibiotics against Wild-Type Gram-Negative Bacteria
2.4. Antibacterial Activity of GCol and GPMB against MDR Clinical Isolates of Gram-Negative Bacteria
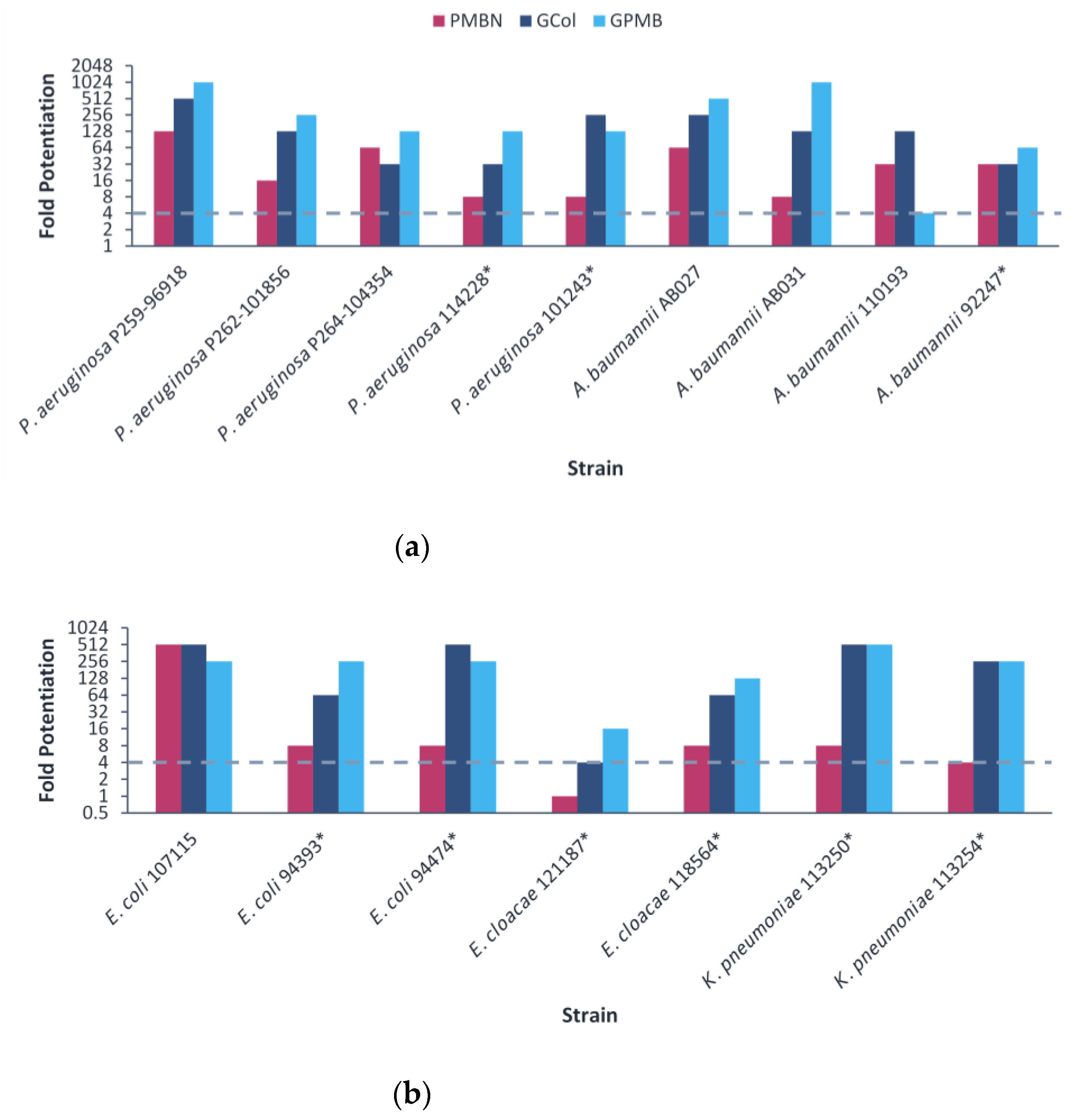
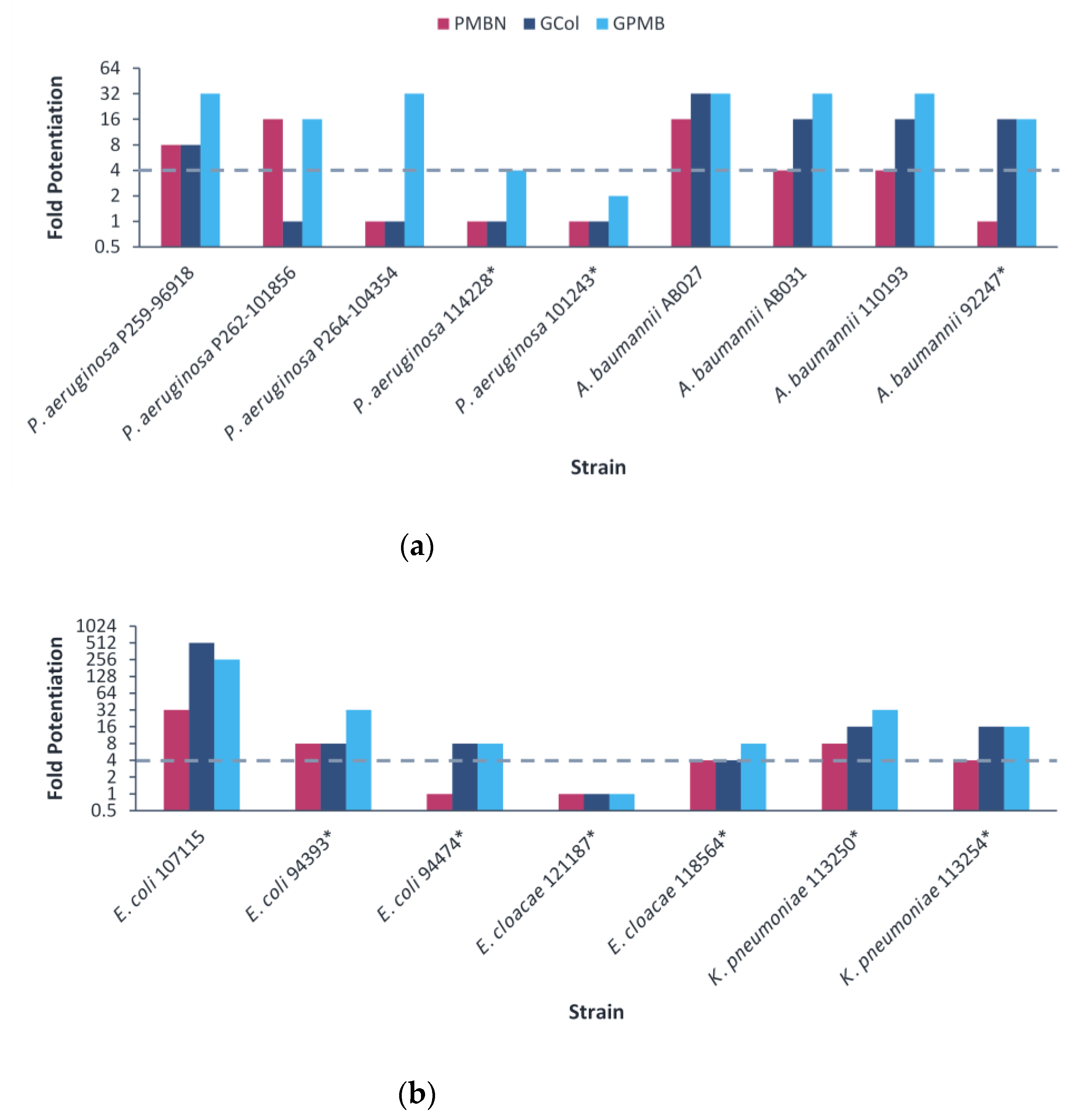
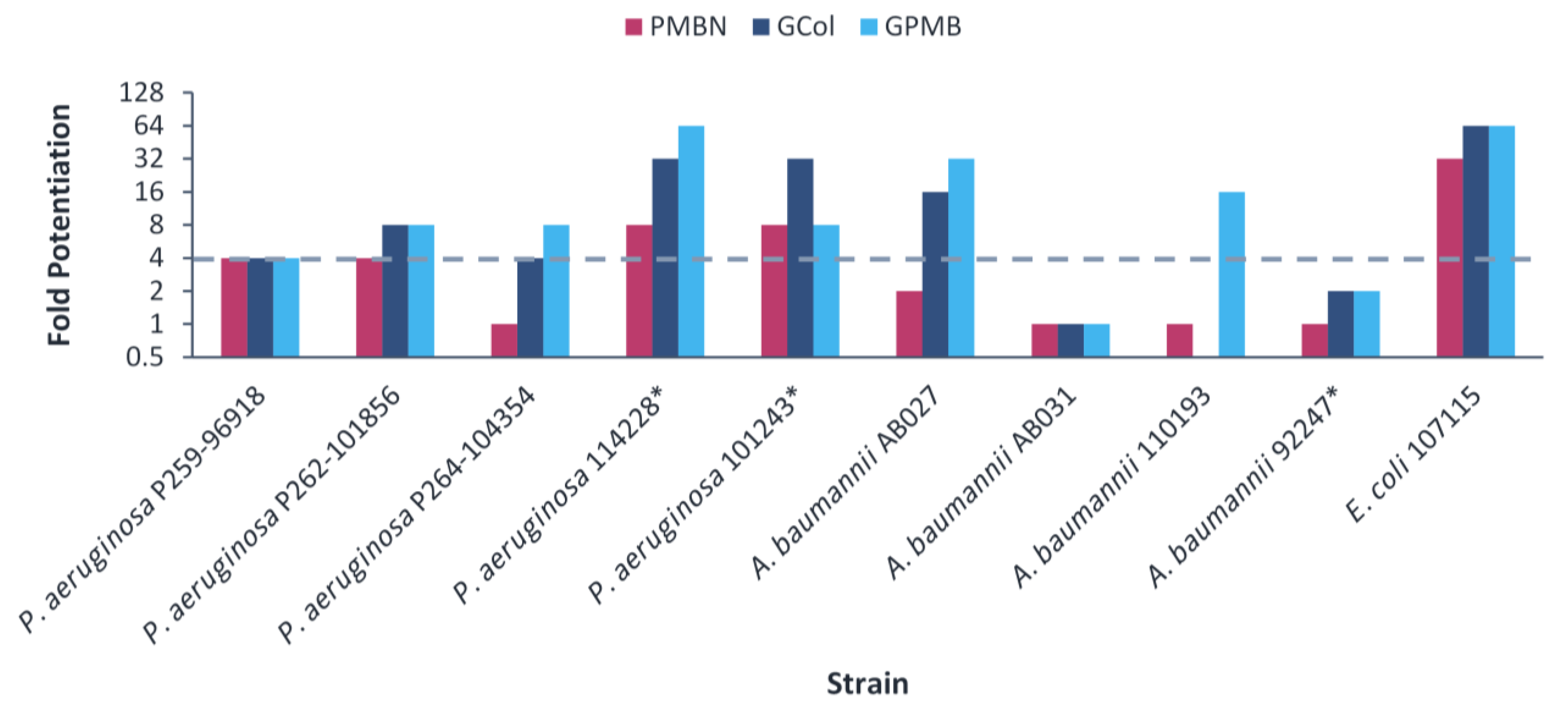
2.5. Synergy of GCol and GPMB with Rifampicin against MDR Clinical Isolates of Gram-Negative Bacteria
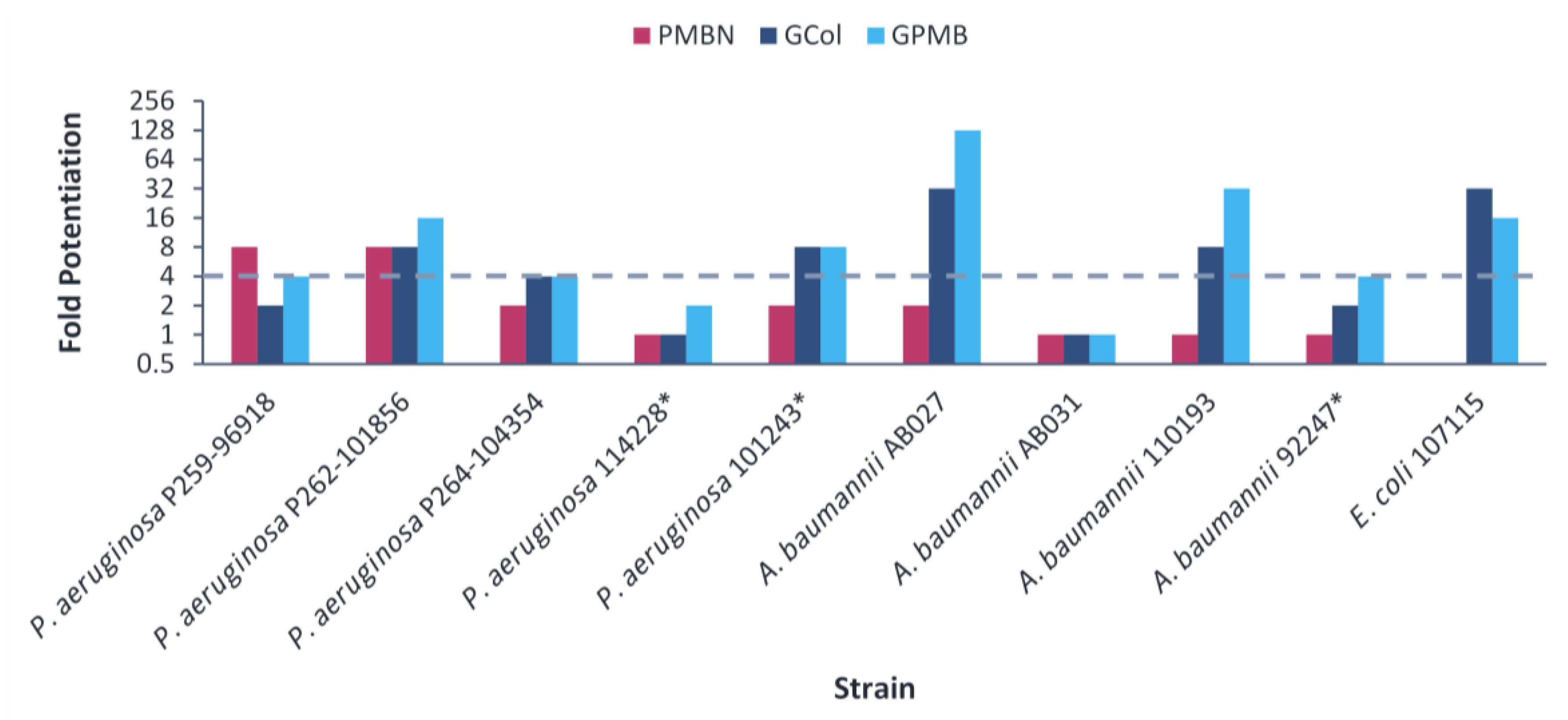
2.6. Synergy of GCol and GPMB with Erythromycin against MDR Clinical Isolates of Gram-Negative Bacteria
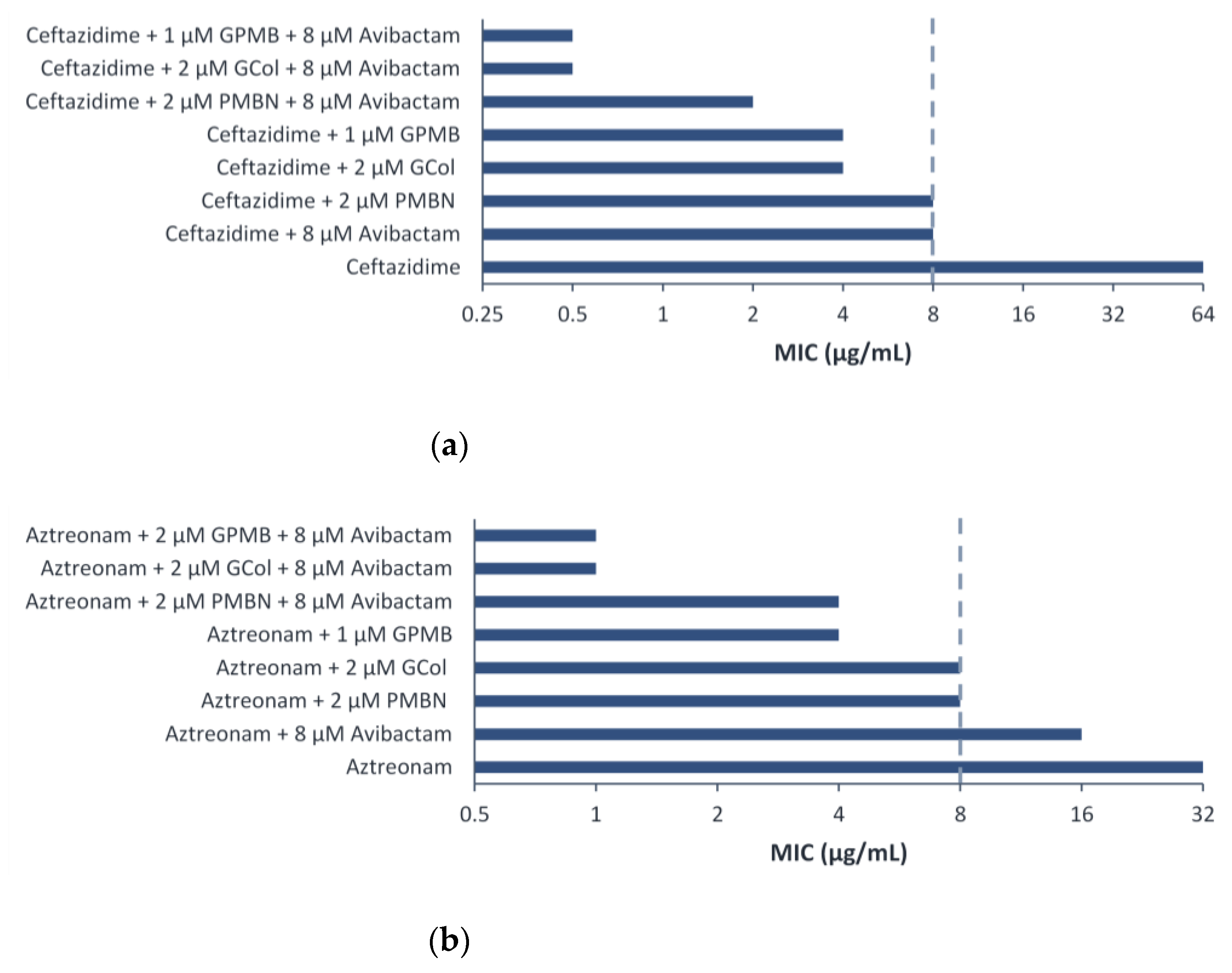
2.7. Synergy of GCol and GPMB with Ceftazidime and Aztreonam against MDR Clinical Isolates of Gram-Negative Bacteria
2.8. Triple Combination Studies against P. aeruginosa Harboring β-Lactamase Clinical Isolates
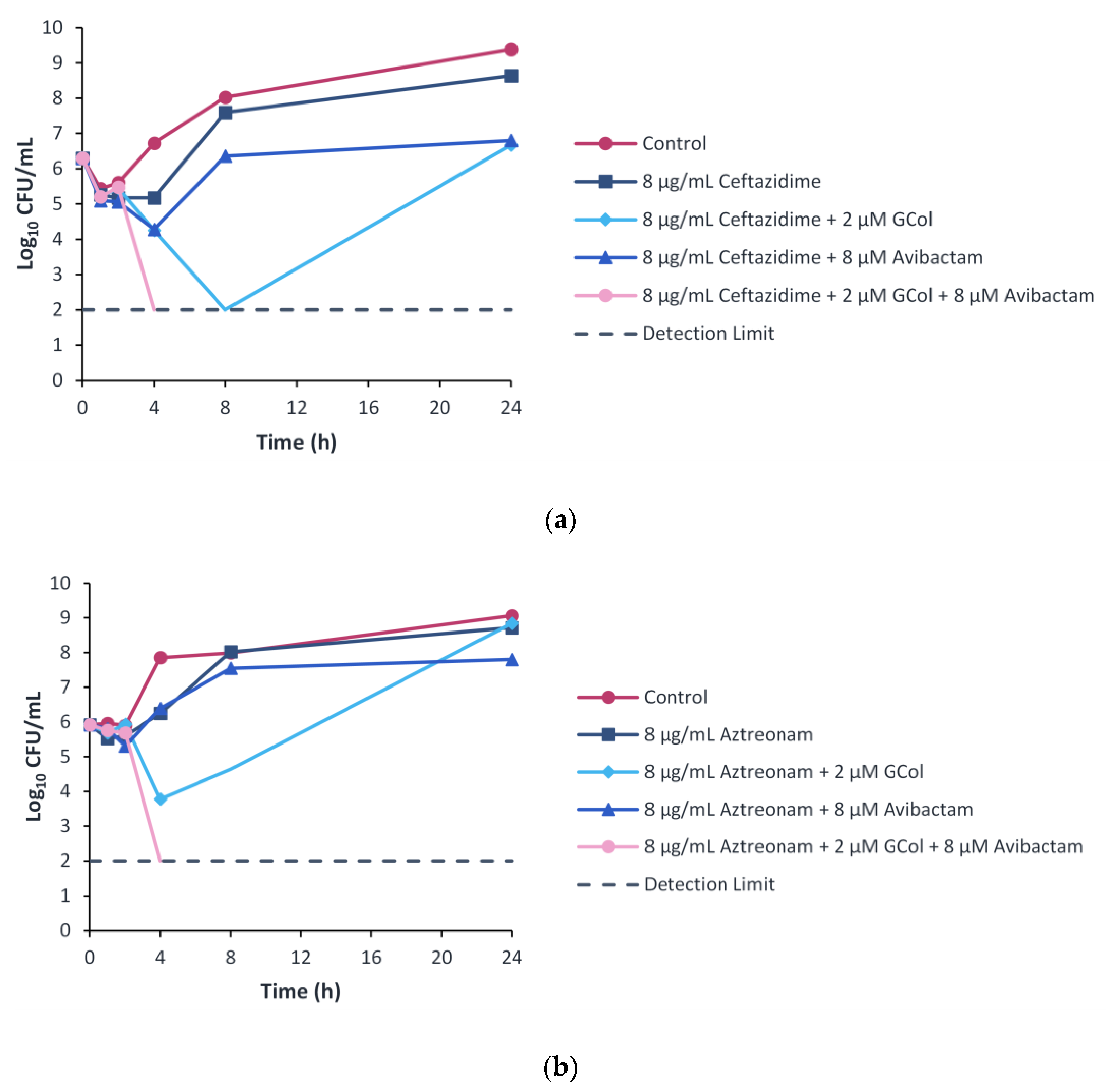
2.9. Time-Kill Kinetics Assay
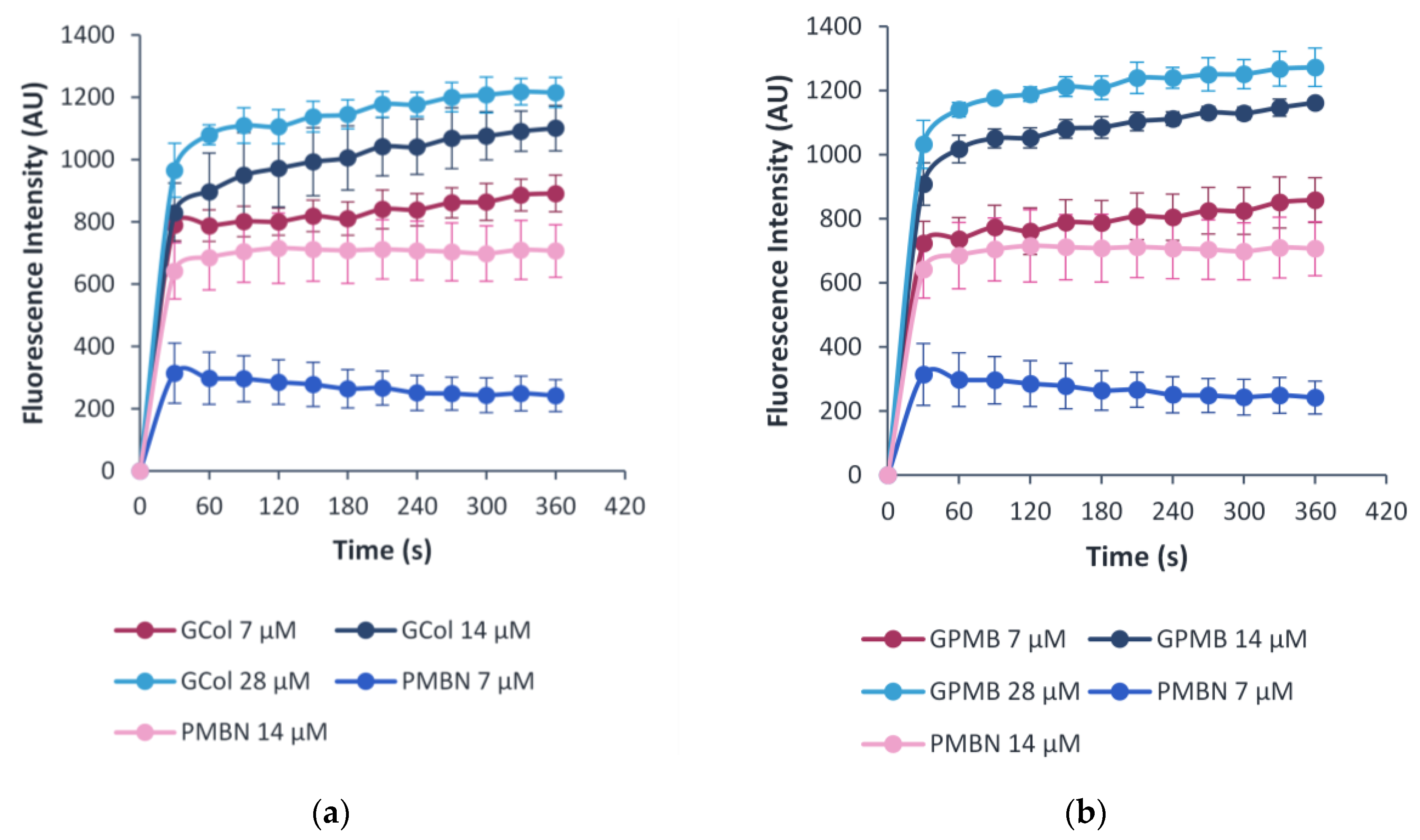
2.10. OM Permeability Assay
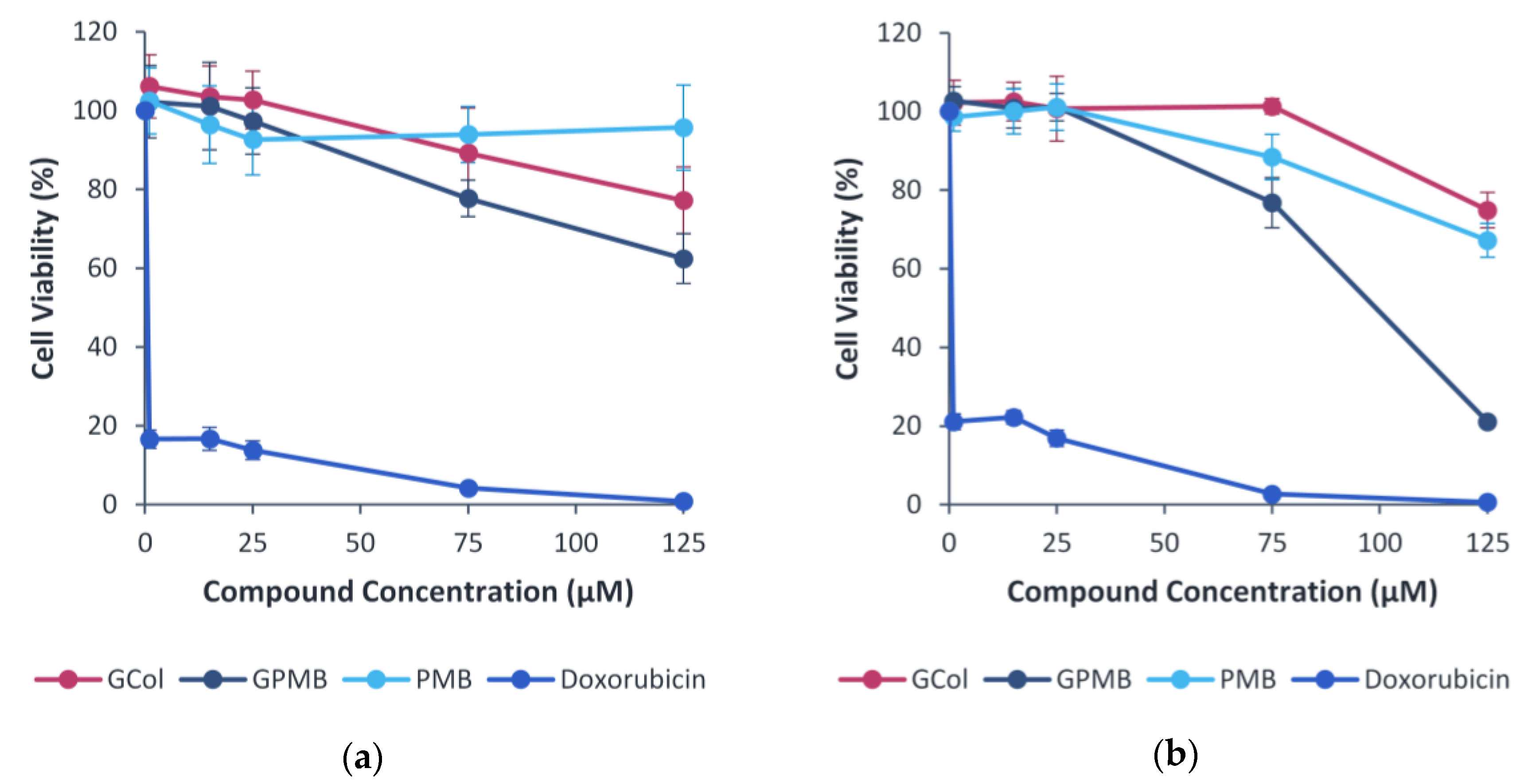
2.11. Cytotoxicity Assay
3. Discussion
4. Materials and Methods
4.1. Preparation of Guanidinylated Polymyxins GPMB and GCol
4.1.1. Chemical Characterization of GCol
4.1.2. Chemical Characterization of GPMB
4.2. Bacterial Isolates and Growth Conditions
4.3. Antimicrobial Susceptibility Assay
4.4. Checkerboard Assay
4.5. Time-Kill Assay
4.6. OM Permeabilization Assay
4.7. Cell Viability Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Fernandes, P. The Global Challenge of New Classes of Antibacterial Agents: An Industry Perspective. Curr. Opin. Pharmacol. 2015, 24, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Melander, R.J.; Melander, C. The Challenge of Overcoming Antibiotic Resistance: An Adjuvant Approach? ACS Infect. Dis. 2017, 3, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Douafer, H.; Andrieu, V.; Phanstiel, O.; Brunel, J.M. Antibiotic Adjuvants: Make Antibiotics Great Again! J. Med. Chem. 2019, 62, 8665–8681. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-H.; Hsieh, Y.-H.; Powers, Z.M.; Kao, C.-Y. Defeating Antibiotic-Resistant Bacteria: Exploring Alternative Therapies for a Post-Antibiotic Era. Int. J. Mol. Sci. 2020, 21, 1061. [Google Scholar] [CrossRef]
- Gill, E.E.; Franco, O.L.; Hancock, R.E.W. Antibiotic Adjuvants: Diverse Strategies for Controlling Drug-Resistant Pathogens. Chem. Biol. Drug Des. 2015, 85, 56–78. [Google Scholar] [CrossRef]
- Ruden, S.; Rieder, A.; Chis Ster, I.; Schwartz, T.; Mikut, R.; Hilpert, K. Synergy Pattern of Short Cationic Antimicrobial Peptides Against Multidrug-Resistant Pseudomonas Aeruginosa. Front. Microbiol. 2019, 10, 2740. [Google Scholar] [CrossRef]
- Berry, L.; Domalaon, R.; Brizuela, M.; Zhanel, G.G.; Schweizer, F. Polybasic Peptide–Levofloxacin Conjugates Potentiate Fluoroquinolones and Other Classes of Antibiotics against Multidrug-Resistant Gram-Negative Bacteria. Medchemcomm 2019, 10, 517–527. [Google Scholar] [CrossRef]
- Ramirez, D.; Berry, L.; Domalaon, R.; Brizuela, M.; Schweizer, F. Dilipid Ultrashort Tetrabasic Peptidomimetics Potentiate Novobiocin and Rifampicin against Multidrug-Resistant Gram-Negative Bacteria. ACS Infect. Dis. 2020, 6, 1413–1426. [Google Scholar] [CrossRef]
- Lyu, Y.; Yang, X.; Goswami, S.; Gorityala, B.K.; Idowu, T.; Domalaon, R.; Zhanel, G.G.; Shan, A.; Schweizer, F. Amphiphilic Tobramycin-Lysine Conjugates Sensitize Multidrug Resistant Gram-Negative Bacteria to Rifampicin and Minocycline. J. Med. Chem. 2017, 60, 3684–3702. [Google Scholar] [CrossRef]
- Idowu, T.; Ammeter, D.; Arthur, G.; Zhanel, G.G.; Schweizer, F. Potentiation of β-Lactam Antibiotics and β-Lactam/β-Lactamase Inhibitor Combinations against MDR and XDR Pseudomonas Aeruginosa Using Non-Ribosomal Tobramycin-Cyclam Conjugates. J. Antimicrob. Chemother. 2019, 74, 2640–2648. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M. Polymyxin Derivatives That Sensitize Gram-Negative Bacteria to Other Antibiotics. Molecules 2019, 24, 249. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Espada, R.; Shahrour, H.; Pitts, B.; Stewart, P.S.; Sánchez-Gómez, S.; Martínez-de-Tejada, G. A Permeability-Increasing Drug Synergizes with Bacterial Efflux Pump Inhibitors and Restores Susceptibility to Antibiotics in Multi-Drug Resistant Pseudomonas Aeruginosa Strains. Sci. Rep. 2019, 9, 3452. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Bell, A. Antibiotic Uptake into Gram-Negative Bacteria. Eur. J. Clin. Microbiol. Infect. Dis. 1988, 7, 713–720. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure-Activity Relationships of Polymyxin Antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef] [PubMed]
- Danner, R.L.; Joiner, K.A.; Rubin, M.; Patterson, W.H.; Johnson, N.; Ayers, K.M.; Parrillo, J.E. Purification, Toxicity, and Antiendotoxin Activity of Polymyxin B Nonapeptide. Antimicrob. Agents Chemother. 1989, 33, 1428–1434. [Google Scholar] [CrossRef]
- Zurawski, D.V.; Reinhart, A.A.; Alamneh, Y.A.; Pucci, M.J.; Si, Y.; Abu-Taleb, R.; Shearer, J.P.; Demons, S.T.; Tyner, S.D.; Lister, T. SPR741, an Antibiotic Adjuvant, Potentiates the In Vitro and In Vivo Activity of Rifampin against Clinically Relevant Extensively Drug-Resistant Acinetobacter Baumannii. Antimicrob. Agents Chemother. 2017, 61, e01239-17. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M.; Siikanen, O.; Apajalahti, J.; Fox, J.; Frimodt-Møller, N.; He, H.; Poudyal, A.; Li, J.; Nation, R.L.; Vaara, T. A Novel Polymyxin Derivative That Lacks the Fatty Acid Tail and Carries Only Three Positive Charges Has Strong Synergism with Agents Excluded by the Intact Outer Membrane. Antimicrob. Agents Chemother. 2010, 54, 3341–3346. [Google Scholar] [CrossRef]
- David, C.; Andrew, W.; Tara, L.; Kirsty, S.; Emily, T.; Stephen, B.; Alain, D.; Stephanie, S.; Jennifer, W.; Peter, W.; et al. Potentiation of Antibiotic Activity by a Novel Cationic Peptide: Potency and Spectrum of Activity of SPR741. Antimicrob. Agents Chemother. 2022, 61, e00200-17. [Google Scholar] [CrossRef]
- Vaara, M. Agents That Increase the Permeability of the Outer Membrane. Microbiol. Rev. 1992, 56, 395–411. [Google Scholar] [CrossRef]
- Nilsson, A.; Goodwin, R.J.A.; Swales, J.G.; Gallagher, R.; Shankaran, H.; Sathe, A.; Pradeepan, S.; Xue, A.; Keirstead, N.; Sasaki, J.C.; et al. Investigating Nephrotoxicity of Polymyxin Derivatives by Mapping Renal Distribution Using Mass Spectrometry Imaging. Chem. Res. Toxicol. 2015, 28, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Blondeau, P.; Segura, M.; Pérez-Fernández, R.; de Mendoza, J. Molecular Recognition of Oxoanions Based on Guanidinium Receptors. Chem. Soc. Rev. 2007, 36, 198–210. [Google Scholar] [CrossRef] [PubMed]
- DeLucia, A.M.; Six, D.A.; Caughlan, R.E.; Gee, P.; Hunt, I.; Lam, J.S.; Dean, C.R. Lipopolysaccharide (LPS) Inner-Core Phosphates Are Required for Complete LPS Synthesis and Transport to the Outer Membrane in Pseudomonas Aeruginosa PAO1. MBio 2011, 2, e00142-11. [Google Scholar] [CrossRef]
- Baker, T.J.; Luedtke, N.W.; Tor, Y.; Goodman, M. Synthesis and Anti-HIV Activity of Guanidinoglycosides. J. Org. Chem. 2000, 65, 9054–9058. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, CLSI Supplement M100, 31st ed.; Clinical and Laboratory Standards Institute: Wayne, NJ, USA, 2021. [Google Scholar]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-Resistant, Extensively Drug-Resistant and Pandrug-Resistant Bacteria: An International Expert Proposal for Interim Standard Definitions for Acquired Resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef]
- Akhoundsadegh, N.; Belanger, C.R.; Hancock, R.E.W. Outer Membrane Interaction Kinetics of New Polymyxin B Analogs in Gram-Negative Bacilli. Antimicrob. Agents Chemother. 2019, 63, e00935-19. [Google Scholar] [CrossRef]
- Gallardo-Godoy, A.; Muldoon, C.; Becker, B.; Elliott, A.G.; Lash, L.H.; Huang, J.X.; Butler, M.S.; Pelingon, R.; Kavanagh, A.M.; Ramu, S.; et al. Activity and Predicted Nephrotoxicity of Synthetic Antibiotics Based on Polymyxin B. J. Med. Chem. 2016, 59, 1068–1077. [Google Scholar] [CrossRef]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef]
- Bulitta, J.B.; Ly, N.S.; Landersdorfer, C.B.; Wanigaratne, N.A.; Velkov, T.; Yadav, R.; Oliver, A.; Martin, L.; Shin, B.S.; Forrest, A.; et al. Two Mechanisms of Killing of Pseudomonas Aeruginosa by Tobramycin Assessed at Multiple Inocula via Mechanism-Based Modeling. Antimicrob. Agents Chemother. 2015, 59, 2315–2327. [Google Scholar] [CrossRef]
- Langendonk, R.F.; Neill, D.R.; Fothergill, J.L. The Building Blocks of Antimicrobial Resistance in Pseudomonas Aeruginosa: Implications for Current Resistance-Breaking Therapies. Front. Cell. Infect. Microbiol. 2021, 11, 665759. [Google Scholar] [CrossRef]
- Zgurskaya, H.I.; López, C.A.; Gnanakaran, S. Permeability Barrier of Gram-Negative Cell Envelopes and Approaches to Bypass It. ACS Infect. Dis. 2015, 1, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Sébastien, C.; Patrice, C.; Bruno, P. Efflux-Mediated Antibiotic Resistance in Acinetobacter spp. Antimicrob. Agents Chemother. 2011, 55, 947–953. [Google Scholar] [CrossRef]
- Rothstein, D.M. Rifamycins, Alone and in Combination. Cold Spring Harb. Perspect. Med. 2016, 6, a027011. [Google Scholar] [CrossRef] [PubMed]
- Drapeau, C.M.J.; Grilli, E.; Petrosillo, N. Rifampicin Combined Regimens for Gram-Negative Infections: Data from the Literature. Int. J. Antimicrob. Agents 2010, 35, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Dueck, M.; Hoban, D.J.; Vercaigne, L.M.; Embil, J.M.; Gin, A.S.; Karlowsky, J.A. Review of Macrolides and Ketolides. Drugs 2001, 61, 443–498. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.G.; Clark, R.B. Discovery of Macrolide Antibiotics Effective against Multi-Drug Resistant Gram-Negative Pathogens. Acc. Chem. Res. 2021, 54, 1635–1645. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Ceftazidime-Avibactam: A Review in the Treatment of Serious Gram-Negative Bacterial Infections. Drugs 2018, 78, 675–692. [Google Scholar] [CrossRef]
- Novelli, A.; Conti, S.; Cassetta, M.I.; Fallani, S. Cephalosporins: A Pharmacological Update. Clin. Microbiol. Infect. 2000, 6, 50–52. [Google Scholar] [CrossRef]
- Ramsey, C.; MacGowan, A.P. A Review of the Pharmacokinetics and Pharmacodynamics of Aztreonam. J. Antimicrob. Chemother. 2016, 71, 2704–2712. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Epidemiology of β-Lactamase-Producing Pathogens. Clin. Microbiol. Rev. 2022, 33, e00047-19. [Google Scholar] [CrossRef]
- Boyd, S.; Livermore, D.; Hooper, D.; Hope, W. Metallo-β-Lactamases: Structure, Function, Epidemiology, Treatment Options, and the Development Pipeline. Antimicrob. Agents Chemother. 2022, 64, e00397-20. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.; Hujer, A.M.; Rojas, L.J.; Papp-Wallace, K.M.; Humphries, R.M.; Spellberg, B.; Hujer, K.M.; Marshall, E.K.; Rudin, S.D.; Perez, F.; et al. Can Ceftazidime-Avibactam and Aztreonam Overcome β-Lactam Resistance Conferred by Metallo-β-Lactamases in Enterobacteriaceae? Antimicrob. Agents Chemother. 2022, 61, e02243-16. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Kern, G.; Walkup, G.K.; Fisher, S.L. Avibactam Is a Covalent, Reversible, Non–β-Lactam β-Lactamase Inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 11663–11668. [Google Scholar] [CrossRef]
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Durand-Réville, T.F.; Lahiri, S.; Thresher, J.; Livchak, S.; Gao, N.; et al. Kinetics of Avibactam Inhibition against Class A, C, and D β-Lactamases. J. Biol. Chem. 2013, 288, 27960–27971. [Google Scholar] [CrossRef]
- Cornely, O.A.; Cisneros, J.M.; Torre-Cisneros, J.; Rodríguez-Hernández, M.J.; Tallón-Aguilar, L.; Calbo, E.; Horcajada, J.P.; Queckenberg, C.; Zettelmeyer, U.; Arenz, D.; et al. Pharmacokinetics and Safety of Aztreonam/Avibactam for the Treatment of Complicated Intra-Abdominal Infections in Hospitalized Adults: Results from the REJUVENATE Study. J. Antimicrob. Chemother. 2020, 75, 618–627. [Google Scholar] [CrossRef]
- Mauri, C.; Maraolo, A.E.; Di Bella, S.; Luzzaro, F.; Principe, L. The Revival of Aztreonam in Combination with Avibactam against Metallo-β-Lactamase-Producing Gram-Negatives: A Systematic Review of In Vitro Studies and Clinical Cases. Antibiotics 2021, 10, 1012. [Google Scholar] [CrossRef]
- Poirel, L.; Jayol, A.; Nordmann, P. Polymyxins: Antibacterial Activity, Susceptibility Testing, and Resistance Mechanisms Encoded by Plasmids or Chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef] [PubMed]
- Ah, Y.-M.; Kim, A.-J.; Lee, J.-Y. Colistin Resistance in Klebsiella Pneumoniae. Int. J. Antimicrob. Agents 2014, 44, 8–15. [Google Scholar] [CrossRef]
- Vester, B.; Douthwaite, S. Macrolide Resistance Conferred by Base Substitutions in 23S RRNA. Antimicrob. Agents Chemother. 2001, 45, 1–12. [Google Scholar] [CrossRef]
- Roberts, M.C. Update on Macrolide–Lincosamide–Streptogramin, Ketolide, and Oxazolidinone Resistance Genes. FEMS Microbiol. Lett. 2008, 282, 147–159. [Google Scholar] [CrossRef]
- Nobuhisa, M.; Eiko, S.; Satoshi, O.; Naomasa, G.; Hideto, T.; Takeshi, N. Substrate Specificities of MexAB-OprM, MexCD-OprJ, and MexXY-OprM Efflux Pumps in Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3322–3327. [Google Scholar] [CrossRef]
- Gai, Z.; Samodelov, S.L.; Kullak-Ublick, G.A.; Visentin, M. Molecular Mechanisms of Colistin-Induced Nephrotoxicity. Molecules 2019, 24, 653. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamaguchi, H.; Ogura, J.; Kobayashi, M.; Yamada, T.; Iseki, K. Megalin Contributes to Kidney Accumulation and Nephrotoxicity of Colistin. Antimicrob. Agents Chemother. 2013, 57, 6319–6324. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M. Novel Derivatives of Polymyxins. J. Antimicrob. Chemother. 2013, 68, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, K.; Robertson, J.H. Improved High-Performance Liquid Chromatographic Method for Polypeptide Antibiotics and Its Application to Study the Effects of Treatments to Reduce Microbial Levels in Bacitracin Powder. J. Chromatogr. A 1975, 112, 663–672. [Google Scholar] [CrossRef]
- Zhanel, G.G.; DeCorby, M.; Laing, N.; Weshnoweski, B.; Vashisht, R.; Tailor, F.; Nichol, K.A.; Wierzbowski, A.; Baudry, P.J.; Karlowsky, J.A.; et al. Antimicrobial-Resistant Pathogens in Intensive Care Units in Canada: Results of the Canadian National Intensive Care Unit (CAN-ICU) Study, 2005-2006. Antimicrob. Agents Chemother. 2008, 52, 1430–1437. [Google Scholar] [CrossRef]
- Hoban, D.J.; Zhanel, G.G. Introduction to the CANWARD Study (2007–11). J. Antimicrob. Chemother. 2013, 68, i3–i5. [Google Scholar] [CrossRef]
- Idowu, T.; Arthur, G.; Zhanel, G.G.; Schweizer, F. Heterodimeric Rifampicin–Tobramycin Conjugates Break Intrinsic Resistance of Pseudomonas Aeruginosa to Doxycycline and Chloramphenicol in Vitro and in a Galleria Mellonella in Vivo Model. Eur. J. Med. Chem. 2019, 174, 16–32. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | MIC (μg/mL) | ||||
|---|---|---|---|---|---|
| PMBN | GCol | GPMB | Colistin | PMB | |
| P. aeruginosa PAO1 | 128 | >128 | 32 | 1 | 0.5 |
| A. baumannii ATCC 17978 | >256 | >128 | 32 | 0.5 | 2 |
| E. coli ATCC 25922 | >256 | 16 | 8 | 0.125 | 1 |
| Strain | MIC (μg/mL) | ||||
|---|---|---|---|---|---|
| PMBN | GCol | GPMB | Colistin | PMB | |
| P. aeruginosa P259-96918 | >256 | 32 | 32 | 0.5 | ≤0.0625 |
| P. aeruginosa P262-101856 | >256 | 128 | 16 | 2 | 0.5 |
| P. aeruginosa P264-104354 | >256 | 32 | 16 | 1 | 2 |
| A. baumannii AB027 | >256 | >128 | >128 | 0.25 | 0.25 |
| A. baumannii AB031 | >256 | 8 | 2 | 0.25 | ≤0.125 |
| A. baumannii LAC-4 | >256 | 2 | 0.5 | 0.125 | 0.5 |
| A. baumannii 110193 | >256 | 64 | 4 | 0.5 | 1 |
| E. coli 107115 | >256 | 8 | 8 | 0.125 | ≤0.125 |
| Strain | MIC (μg/mL) | ||||
|---|---|---|---|---|---|
| PMBN | GCol | GPMB | Colistin | PMB | |
| P. aeruginosa 114228 | >256 | >128 | 32 | 4 | 64 |
| P. aeruginosa 101243 | >256 | >128 | >128 | 1024 | >128 |
| A. baumannii 92247 | >256 | >128 | 128 | 4 | 8 |
| E. coli 94474 | >256 | 32 | 16 | 16 | 16 |
| E. coli 94393 | >256 | 16 | 8 | 4 | 2 |
| E. cloacae 121187 | >256 | >128 | >128 | >128 | >64 |
| E. cloacae 118564 | >256 | >128 | >128 | >128 | >64 |
| K. pneumoniae 113250 | >256 | >128 | >128 | 256 | >64 |
| K. pneumoniae 113254 | >256 | >128 | >128 | 256 | >64 |
| Strain | MIC (μg/mL) | ||||
|---|---|---|---|---|---|
| PMBN | GCol | GPMB | Colistin | PMB | |
| PA 107092 | 64 | 16 | 8 | 0.5 | 0.25 |
| PA 109084 | 32 | 4 | 4 | 0.5 | 0.25 |
| PA 86052 | 16 | 2 | 1 | 1 | 0.25 |
| PA 88949 | 16 | 16 | 4 | 1 | 0.5 |
| PA 108590 | 1 | 2 | 1 | 1 | ≤0.125 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramirez, D.M.; Ramirez, D.; Arthur, G.; Zhanel, G.; Schweizer, F. Guanidinylated Polymyxins as Outer Membrane Permeabilizers Capable of Potentiating Rifampicin, Erythromycin, Ceftazidime and Aztreonam against Gram-Negative Bacteria. Antibiotics 2022, 11, 1277. https://doi.org/10.3390/antibiotics11101277
Ramirez DM, Ramirez D, Arthur G, Zhanel G, Schweizer F. Guanidinylated Polymyxins as Outer Membrane Permeabilizers Capable of Potentiating Rifampicin, Erythromycin, Ceftazidime and Aztreonam against Gram-Negative Bacteria. Antibiotics. 2022; 11(10):1277. https://doi.org/10.3390/antibiotics11101277
Chicago/Turabian StyleRamirez, Danzel Marie, Danyel Ramirez, Gilbert Arthur, George Zhanel, and Frank Schweizer. 2022. "Guanidinylated Polymyxins as Outer Membrane Permeabilizers Capable of Potentiating Rifampicin, Erythromycin, Ceftazidime and Aztreonam against Gram-Negative Bacteria" Antibiotics 11, no. 10: 1277. https://doi.org/10.3390/antibiotics11101277
APA StyleRamirez, D. M., Ramirez, D., Arthur, G., Zhanel, G., & Schweizer, F. (2022). Guanidinylated Polymyxins as Outer Membrane Permeabilizers Capable of Potentiating Rifampicin, Erythromycin, Ceftazidime and Aztreonam against Gram-Negative Bacteria. Antibiotics, 11(10), 1277. https://doi.org/10.3390/antibiotics11101277