
Computational Drug Repurposing for Antituberculosis Therapy: Discovery of Multi-Strain Inhibitors




Abstract
:1. Introduction
2. Results and Discussion
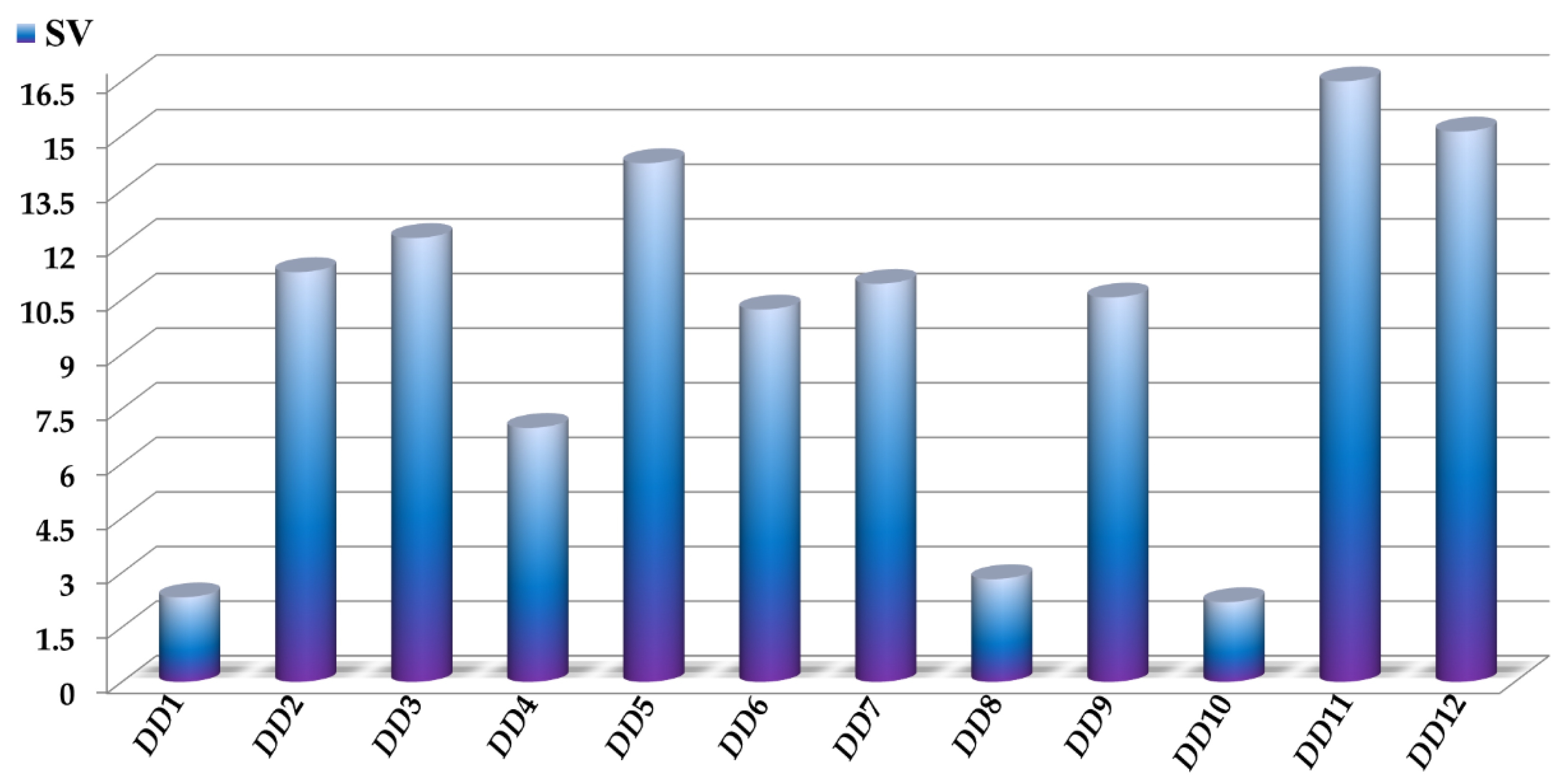
2.1. Performance of the Mtc-QSAR-EL Model and Applicability Domain
2.2. Molecular Descriptors and Their Physicochemical and Structural Meanings
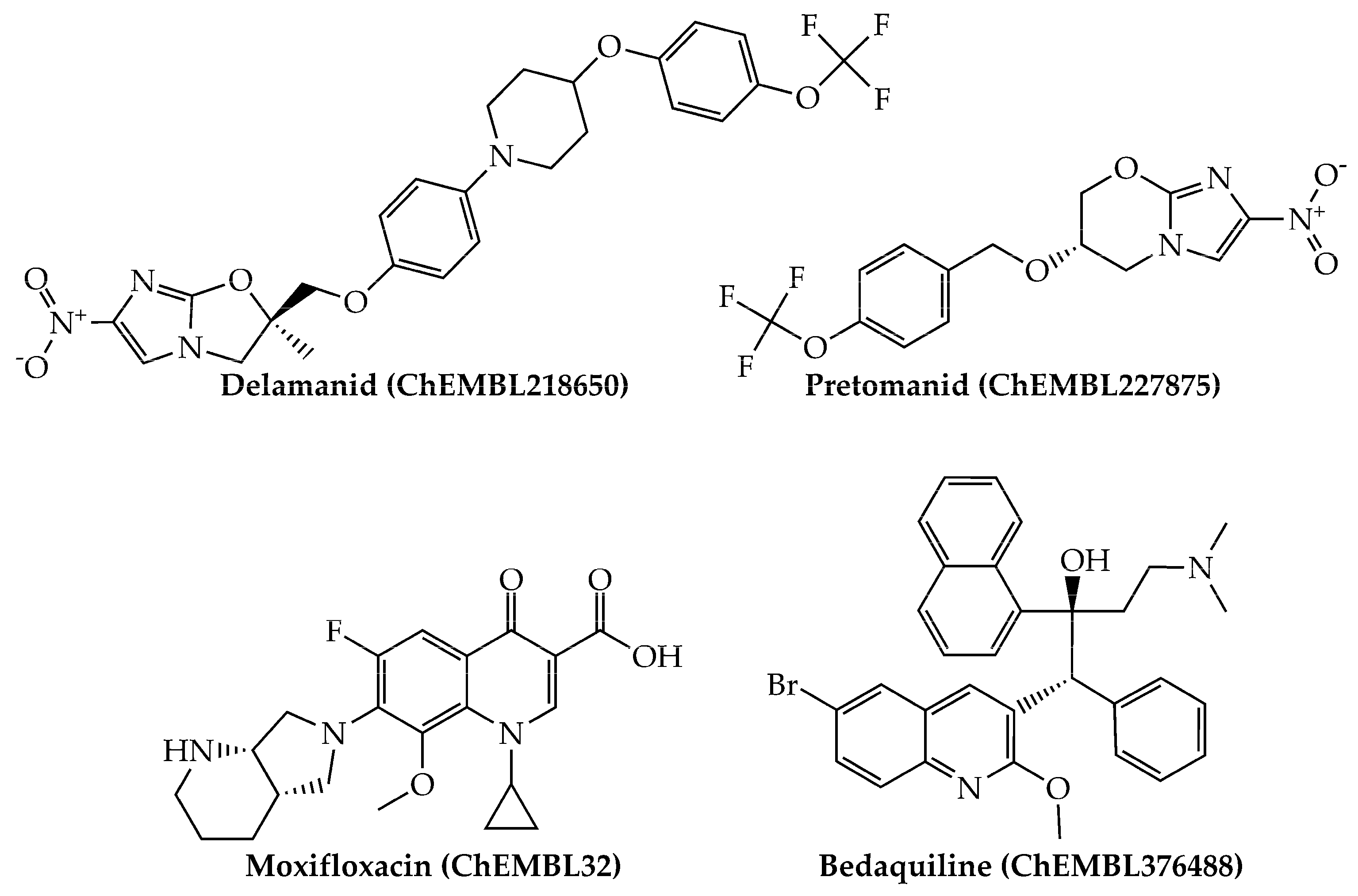
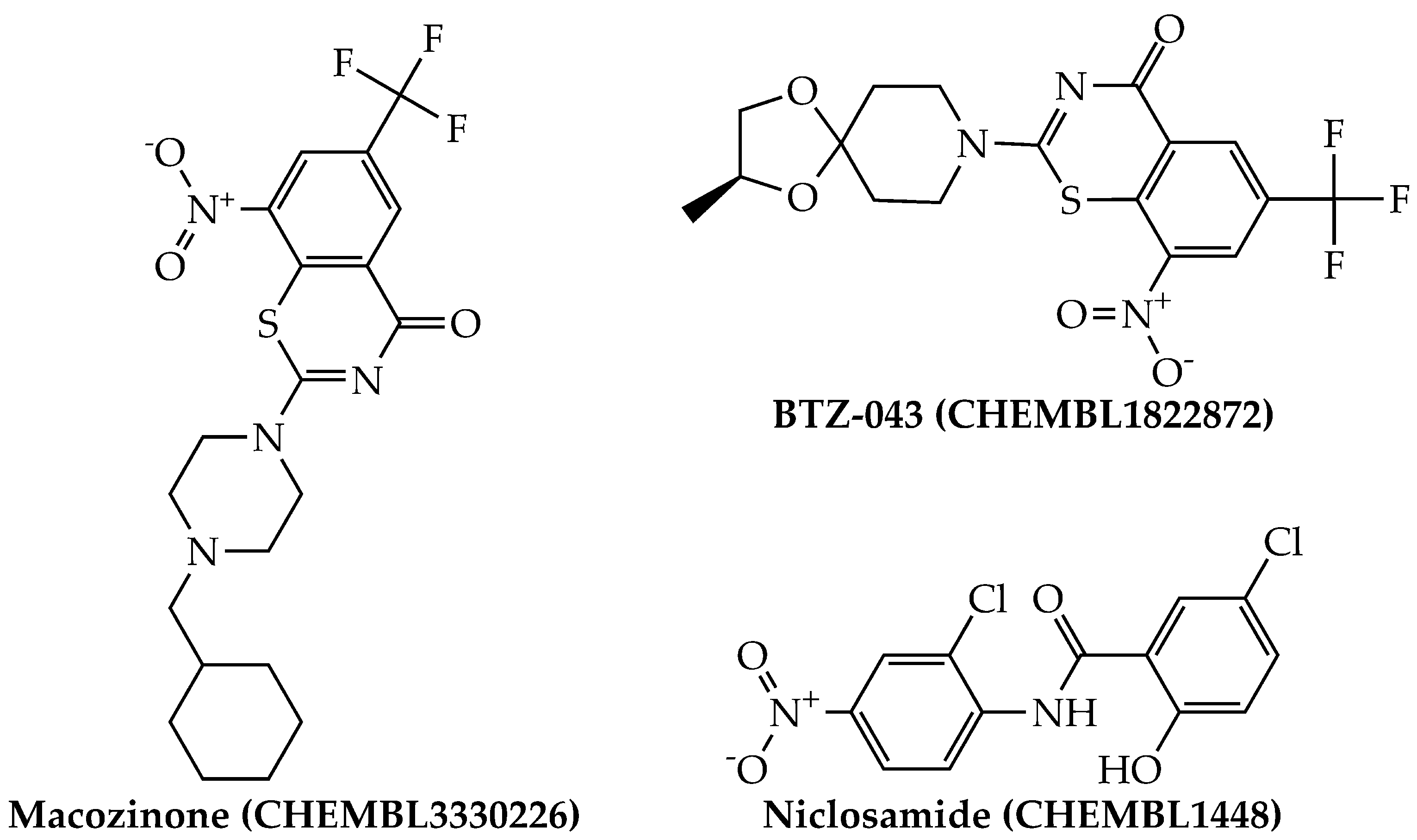
2.3. Computational Drug Repurposing of Agency-Regulated Chemicals as Anti-TB Agents
3. Materials and Methods
3.1. Dataset and Computation of the Molecular Descriptors
3.2. Building the Mtc-QSAR-EL Model
3.3. Applicability Domain
3.4. Interpretation of the Molecular Descriptors in the Mtc-QSAR-EL Model
3.5. Virtual Screening of Agency-Regulated Chemicals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Global Tuberculosis Report. Available online: https://www.who.int/tb/publications/global_report/tb19_Exec_Sum_12Nov2019.pdf?ua=1 (accessed on 20 July 2021).
- Louw, G.E.; Warren, R.M.; Gey van Pittius, N.C.; McEvoy, C.R.; Van Helden, P.D.; Victor, T.C. A balancing act: Efflux/influx in mycobacterial drug resistance. Antimicrob. Agents Chemother. 2009, 53, 3181–3189. [Google Scholar] [CrossRef] [Green Version]
- Grzelak, E.M.; Choules, M.P.; Gao, W.; Cai, G.; Wan, B.; Wang, Y.; McAlpine, J.B.; Cheng, J.; Jin, Y.; Lee, H.; et al. Strategies in anti-Mycobacterium tuberculosis drug discovery based on phenotypic screening. J. Antibiot. 2019, 72, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.T.; Liu, J.; Lee, R.B.; Lee, R.E. New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv. Drug Deliv. Rev. 2016, 102, 55–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Simultaneous modeling of antimycobacterial activities and ADMET profiles: A chemoinformatic approach to medicinal chemistry. Curr. Top. Med. Chem. 2013, 13, 1656–1665. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Cordeiro, M.N.D.S. New insights toward the discovery of antibacterial agents: Multi-tasking QSBER model for the simultaneous prediction of anti-tuberculosis activity and toxicological profiles of drugs. Eur. J. Pharm. Sci. 2013, 48, 812–818. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. In silico discovery and virtual screening of multi-target inhibitors for proteins in Mycobacterium tuberculosis. Comb. Chem. High Throughput Screen. 2012, 15, 666–673. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Scotti, M.T.; García-López, A.; Emerenciano, V.P.; Molina-Pérez, E.; Uriarte, E. Design of novel antituberculosis compounds using graph-theoretical and substructural approaches. Mol. Divers. 2009, 13, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Prathipati, P.; Ma, N.L.; Keller, T.H. Global Bayesian models for the prioritization of antitubercular agents. J. Chem. Inf. Model. 2008, 48, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, P.M.; Geetha Babu, S.K.; Mukesh, D. QSAR studies on chalcones and flavonoids as anti-tuberculosis agents using genetic function approximation (GFA) method. Chem. Pharm. Bull. 2007, 55, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Ragno, R.; Marshall, G.R.; Di Santo, R.; Costi, R.; Massa, S.; Rompei, R.; Artico, M. Antimycobacterial pyrroles: Synthesis, anti-Mycobacterium tuberculosis activity and QSAR studies. Bioorg. Med. Chem. 2000, 8, 1423–1432. [Google Scholar] [CrossRef]
- Nesměrák, K.; Toropov, A.A.; Toropova, A.P.; Ertan-Bolelli, T.; Yildiz, I. QSAR of antimycobacterial activity of benzoxazoles by optimal SMILES-based descriptors. Med. Chem. Res. 2017, 26, 3203–3208. [Google Scholar] [CrossRef]
- Ortega-Tenezaca, B.; Quevedo-Tumailli, V.; Bediaga, H.; Collados, J.; Arrasate, S.; Madariaga, G.; Munteanu, C.R.; Cordeiro, M.; Gonzalez-Diaz, H. PTML Multi-Label Algorithms: Models, Software, and Applications. Curr. Top. Med. Chem. 2020, 20, 2326–2337. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Multitasking models for quantitative structure-biological effect relationships: Current status and future perspectives to speed up drug discovery. Expert Opin. Drug Discov. 2015, 10, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Speck-Planche, A. The urgent need for pan-antiviral agents: From multitarget discovery to multiscale design. Future Med. Chem. 2021, 13, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Dominguez, E.; Armijos-Jaramillo, V.D.; Tejera, E.; Gonzalez-Diaz, H. Multioutput Perturbation-Theory Machine Learning (PTML) Model of ChEMBL Data for Antiretroviral Compounds. Mol. Pharm. 2019, 16, 4200–4212. [Google Scholar] [CrossRef]
- Nocedo-Mena, D.; Cornelio, C.; Camacho-Corona, M.D.R.; Garza-Gonzalez, E.; Waksman de Torres, N.; Arrasate, S.; Sotomayor, N.; Lete, E.; Gonzalez-Diaz, H. Modeling Antibacterial Activity with Machine Learning and Fusion of Chemical Structure Information with Microorganism Metabolic Networks. J. Chem. Inf. Model. 2019, 59, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Bediaga, H.; Arrasate, S.; Gonzalez-Diaz, H. PTML Combinatorial Model of ChEMBL Compounds Assays for Multiple Types of Cancer. ACS Comb. Sci. 2018, 20, 621–632. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Scotti, M.T.; Scotti, L.; Speck-Planche, A. Multi-Target Drug Discovery via PTML Modeling: Applications to the Design of Virtual Dual Inhibitors of CDK4 and HER2. Curr. Top. Med. Chem. 2021, 21, 661–675. [Google Scholar] [CrossRef]
- Diez-Alarcia, R.; Yanez-Perez, V.; Muneta-Arrate, I.; Arrasate, S.; Lete, E.; Meana, J.J.; Gonzalez-Diaz, H. Big Data Challenges Targeting Proteins in GPCR Signaling Pathways; Combining PTML-ChEMBL Models and [(35)S]GTPgammaS Binding Assays. ACS Chem. Neurosci. 2019, 10, 4476–4491. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Speck-Planche, A. PTML Modeling for Alzheimer’s Disease: Design and Prediction of Virtual Multi-Target Inhibitors of GSK3B, HDAC1, and HDAC6. Curr. Top. Med. Chem. 2020, 20, 1657–1672. [Google Scholar] [CrossRef] [PubMed]
- Ferreira da Costa, J.; Silva, D.; Caamano, O.; Brea, J.M.; Loza, M.I.; Munteanu, C.R.; Pazos, A.; Garcia-Mera, X.; Gonzalez-Diaz, H. Perturbation Theory/Machine Learning Model of ChEMBL Data for Dopamine Targets: Docking, Synthesis, and Assay of New l-Prolyl-l-leucyl-glycinamide Peptidomimetics. ACS Chem. Neurosci. 2018, 9, 2572–2587. [Google Scholar] [CrossRef] [PubMed]
- Abeijon, P.; Garcia-Mera, X.; Caamano, O.; Yanez, M.; Lopez-Castro, E.; Romero-Duran, F.J.; Gonzalez-Diaz, H. Multi-Target Mining of Alzheimer Disease Proteome with Hansch’s QSBR-Perturbation Theory and Experimental-Theoretic Study of New Thiophene Isosters of Rasagiline. Curr. Drug Targets 2017, 18, 511–521. [Google Scholar] [CrossRef]
- Concu, R.; MN, D.S.C.; Munteanu, C.R.; Gonzalez-Diaz, H. PTML Model of Enzyme Subclasses for Mining the Proteome of Biofuel Producing Microorganisms. J. Proteome Res. 2019, 18, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Dieguez-Santana, K.; Casanola-Martin, G.M.; Green, J.R.; Rasulev, B.; Gonzalez-Diaz, H. Predicting Metabolic Reaction Networks with Perturbation-Theory Machine Learning (PTML) Models. Curr. Top. Med. Chem. 2021, 21, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Santana, R.; Zuluaga, R.; Ganan, P.; Arrasate, S.; Onieva, E.; Gonzalez-Diaz, H. Designing nanoparticle release systems for drug-vitamin cancer co-therapy with multiplicative perturbation-theory machine learning (PTML) models. Nanoscale 2019, 11, 21811–21823. [Google Scholar] [CrossRef] [PubMed]
- Santana, R.; Zuluaga, R.; Ganan, P.; Arrasate, S.; Onieva, E.; Gonzalez-Diaz, H. Predicting coated-nanoparticle drug release systems with perturbation-theory machine learning (PTML) models. Nanoscale 2020, 12, 13471–13483. [Google Scholar] [CrossRef]
- Santana, R.; Zuluaga, R.; Ganan, P.; Arrasate, S.; Onieva, E.; Montemore, M.M.; Gonzalez-Diaz, H. PTML Model for Selection of Nanoparticles, Anticancer Drugs, and Vitamins in the Design of Drug-Vitamin Nanoparticle Release Systems for Cancer Cotherapy. Mol. Pharm. 2020, 17, 2612–2627. [Google Scholar] [CrossRef]
- Gonzalez-Durruthy, M.; Monserrat, J.M.; Viera de Oliveira, P.; Fagan, S.B.; Werhli, A.V.; Machado, K.; Melo, A.; Gonzalez-Diaz, H.; Concu, R.; MN, D.S.C. Computational MitoTarget Scanning Based on Topological Vacancies of Single-Walled Carbon Nanotubes with the Human Mitochondrial Voltage-Dependent Anion Channel (hVDAC1). Chem. Res. Toxicol. 2019, 32, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Luan, F.; Speck-Planche, A.; Cordeiro, M.N.D.S. In silico assessment of the acute toxicity of chemicals: Recent advances and new model for multitasking prediction of toxic effect. Mini Rev. Med. Chem. 2015, 15, 677–686. [Google Scholar] [CrossRef]
- Martinez-Arzate, S.G.; Tenorio-Borroto, E.; Barbabosa Pliego, A.; Diaz-Albiter, H.M.; Vazquez-Chagoyan, J.C.; Gonzalez-Diaz, H. PTML Model for Proteome Mining of B-Cell Epitopes and Theoretical-Experimental Study of Bm86 Protein Sequences from Colima, Mexico. J. Proteome Res. 2017, 16, 4093–4103. [Google Scholar] [CrossRef]
- Tenorio-Borroto, E.; Penuelas-Rivas, C.G.; Vasquez-Chagoyan, J.C.; Castanedo, N.; Prado-Prado, F.J.; Garcia-Mera, X.; Gonzalez-Diaz, H. Model for high-throughput screening of drug immunotoxicity—Study of the anti-microbial G1 over peritoneal macrophages using flow cytometry. Eur. J. Med. Chem. 2014, 72, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- Zhu, T.; Cao, S.; Su, P.C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; van der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Pasca, M.R.; Degiacomi, G.; Ribeiro, A.L.; Zara, F.; De Mori, P.; Heym, B.; Mirrione, M.; Brerra, R.; Pagani, L.; Pucillo, L.; et al. Clinical isolates of Mycobacterium tuberculosis in four European hospitals are uniformly susceptible to benzothiazinones. Antimicrob. Agents Chemother. 2010, 54, 1616–1618. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Shi, P.Y.; Li, H.; Zhou, J. Broad Spectrum Antiviral Agent Niclosamide and Its Therapeutic Potential. ACS Infect. Dis. 2020, 6, 909–915. [Google Scholar] [CrossRef]
- Fan, X.; Xu, J.; Files, M.; Cirillo, J.D.; Endsley, J.J.; Zhou, J.; Endsley, M.A. Dual activity of niclosamide to suppress replication of integrated HIV-1 and Mycobacterium tuberculosis (Beijing). Tuberculosis (Edinb) 2019, 116S, S28–S33. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, Y. Antituberculosis activity of certain antifungal and antihelmintic drugs. Tuber. Lung Dis. 1999, 79, 319–320. [Google Scholar] [CrossRef]
- Kaushik, A.; Ammerman, N.C.; Tasneen, R.; Story-Roller, E.; Dooley, K.E.; Dorman, S.E.; Nuermberger, E.L.; Lamichhane, G. In vitro and in vivo activity of biapenem against drug-susceptible and rifampicin-resistant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2017, 72, 2320–2325. [Google Scholar] [CrossRef]
- Upton, A.M.; Cho, S.; Yang, T.J.; Kim, Y.; Wang, Y.; Lu, Y.; Wang, B.; Xu, J.; Mdluli, K.; Ma, Z.; et al. In vitro and in vivo activities of the nitroimidazole TBA-354 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Truchon, J.F.; Bayly, C.I. Evaluating virtual screening methods: Good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B. mycoCSM: Using Graph-Based Signatures to Identify Safe Potent Hits against Mycobacteria. J. Chem. Inf. Model. 2020, 60, 3450–3456. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Mok, N.Y.; Brenk, R. Mining the ChEMBL database: An efficient chemoinformatics workflow for assembling an ion channel-focused screening library. J. Chem. Inf. Model. 2011, 51, 2449–2454. [Google Scholar] [CrossRef]
- Overington, J. ChEMBL. An interview with John Overington, team leader, chemogenomics at the European Bioinformatics Institute Outstation of the European Molecular Biology Laboratory (EMBL-EBI). Interview by Wendy A. Warr. J. Comput. Aided Mol. Des. 2009, 23, 195–198. [Google Scholar] [CrossRef]
- Ollinger, J.; Kumar, A.; Roberts, D.M.; Bailey, M.A.; Casey, A.; Parish, T. A high-throughput whole cell screen to identify inhibitors of Mycobacterium tuberculosis. PLoS ONE 2019, 14, e0205479. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Valdes-Martini, J.R.; Marrero-Ponce, Y.; Garcia-Jacas, C.R.; Martinez-Mayorga, K.; Barigye, S.J.; Vaz d’Almeida, Y.S.; Pham-The, H.; Perez-Gimenez, F.; Morell, C.A. QuBiLS-MAS, open source multi-platform software for atom- and bond-based topological (2D) and chiral (2.5D) algebraic molecular descriptors computations. J. Cheminform. 2017, 9, 35. [Google Scholar] [CrossRef]
- Valdés-Martini, J.R.; García-Jacas, C.R.; Marrero-Ponce, Y.; Silveira Vaz ‘d Almeida, Y.; Morell, C. QUBILs-MAS: Free Software for Molecular Descriptors Calculator from Quadratic, Bilinear and Linear Maps Based on Graph-Theoretic Electronic-Density Matrices and Atomic Weightings; v1.0; CAMD-BIR Unit, CENDA Registration Number: 2373-2012; Villa Clara, Cuba, 2012. Available online: http://tomocomd.com/ (accessed on 29 June 2021).
- Medina Marrero, R.; Marrero-Ponce, Y.; Barigye, S.J.; Echeverria Diaz, Y.; Acevedo-Barrios, R.; Casanola-Martin, G.M.; Garcia Bernal, M.; Torrens, F.; Perez-Gimenez, F. QuBiLs-MAS method in early drug discovery and rational drug identification of antifungal agents. SAR QSAR Environ. Res. 2015, 26, 943–958. [Google Scholar] [CrossRef]
- Marrero-Ponce, Y.; Siverio-Mota, D.; Galvez-Llompart, M.; Recio, M.C.; Giner, R.M.; Garcia-Domenech, R.; Torrens, F.; Aran, V.J.; Cordero-Maldonado, M.L.; Esguera, C.V.; et al. Discovery of novel anti-inflammatory drug-like compounds by aligning in silico and in vivo screening: The nitroindazolinone chemotype. Eur. J. Med. Chem. 2011, 46, 5736–5753. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. De novo computational design of compounds virtually displaying potent antibacterial activity and desirable in vitro ADMET profiles. Med. Chem. Res. 2017, 26, 2345–2356. [Google Scholar] [CrossRef]
- Urias, R.W.; Barigye, S.J.; Marrero-Ponce, Y.; Garcia-Jacas, C.R.; Valdes-Martini, J.R.; Perez-Gimenez, F. IMMAN: Free software for information theory-based chemometric analysis. Mol. Divers. 2015, 19, 305–319. [Google Scholar] [CrossRef]
- Godden, J.W.; Bajorath, J. Differential Shannon Entropy as a sensitive measure of differences in database variability of molecular descriptors. J. Chem. Inf. Comput. Sci. 2001, 41, 1060–1066. [Google Scholar] [CrossRef]
- Quinlan, J.R. Induction of decision trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar] [CrossRef] [Green Version]
- Pearson, K. Notes on regression and inheritance in the case of two parents. Proc. R. Soc. Lond. 1895, 58, 240–242. [Google Scholar] [CrossRef]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef]
- TIBCO-Software-Inc. STATISTICA (Data Analysis Software System); v13.5.0.17; TIBCO-Software-Inc.: Palo Alto, CA, USA, 2018; Available online: http://tibco.com (accessed on 2 July 2021).
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of different approaches to define the applicability domain of QSAR models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef] [Green Version]
- Netzeva, T.I.; Worth, A.; Aldenberg, T.; Benigni, R.; Cronin, M.T.; Gramatica, P.; Jaworska, J.S.; Kahn, S.; Klopman, G.; Marchant, C.A.; et al. Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships. The report and recommendations of ECVAM Workshop 52. Altern. Lab. Anim. 2005, 33, 155–173. [Google Scholar] [CrossRef]
- Speck-Planche, A. Combining Ensemble Learning with a Fragment-Based Topological Approach to Generate New Molecular Diversity in Drug Discovery: In Silico Design of Hsp90 Inhibitors. ACS Omega 2018, 3, 14704–14716. [Google Scholar] [CrossRef]
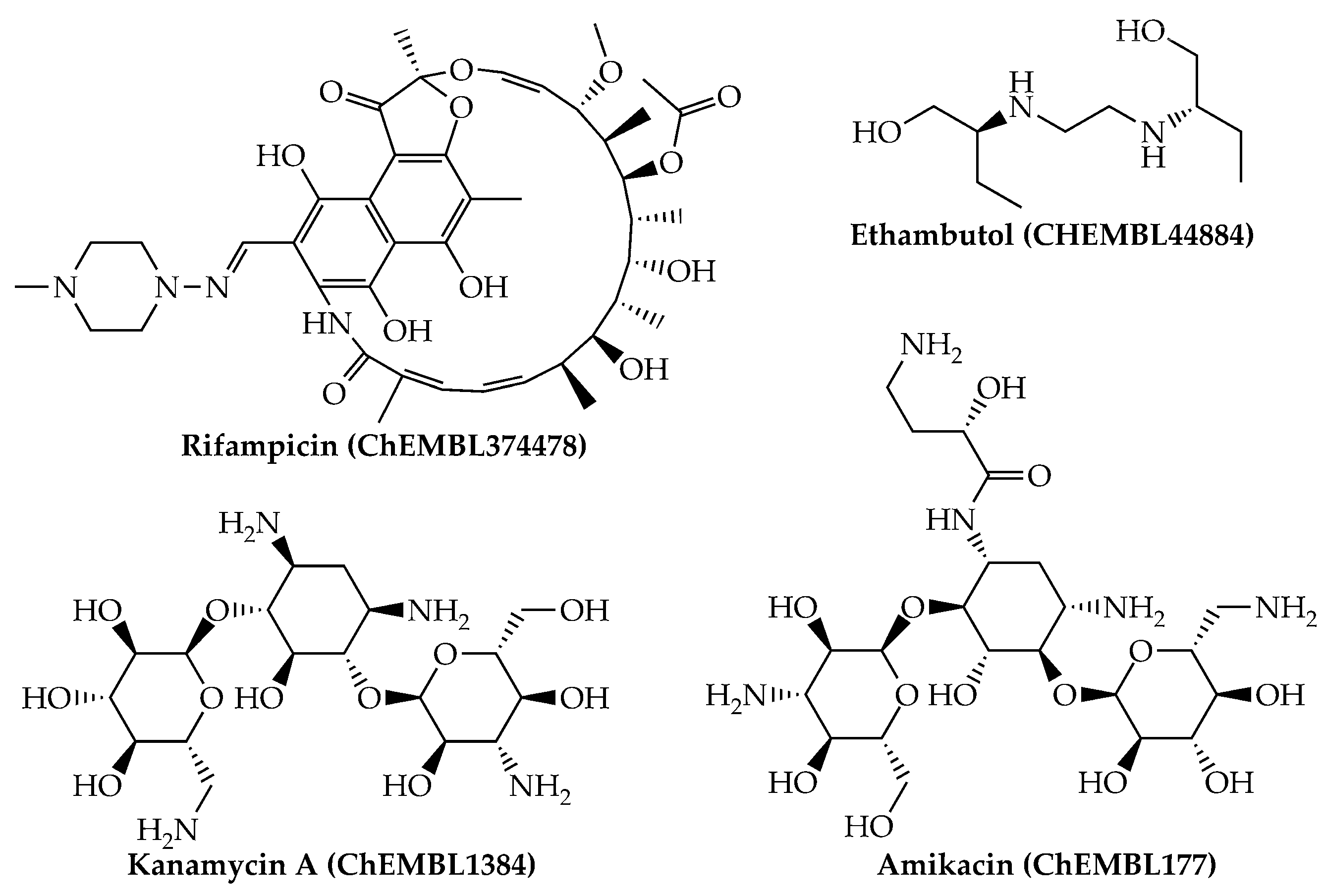
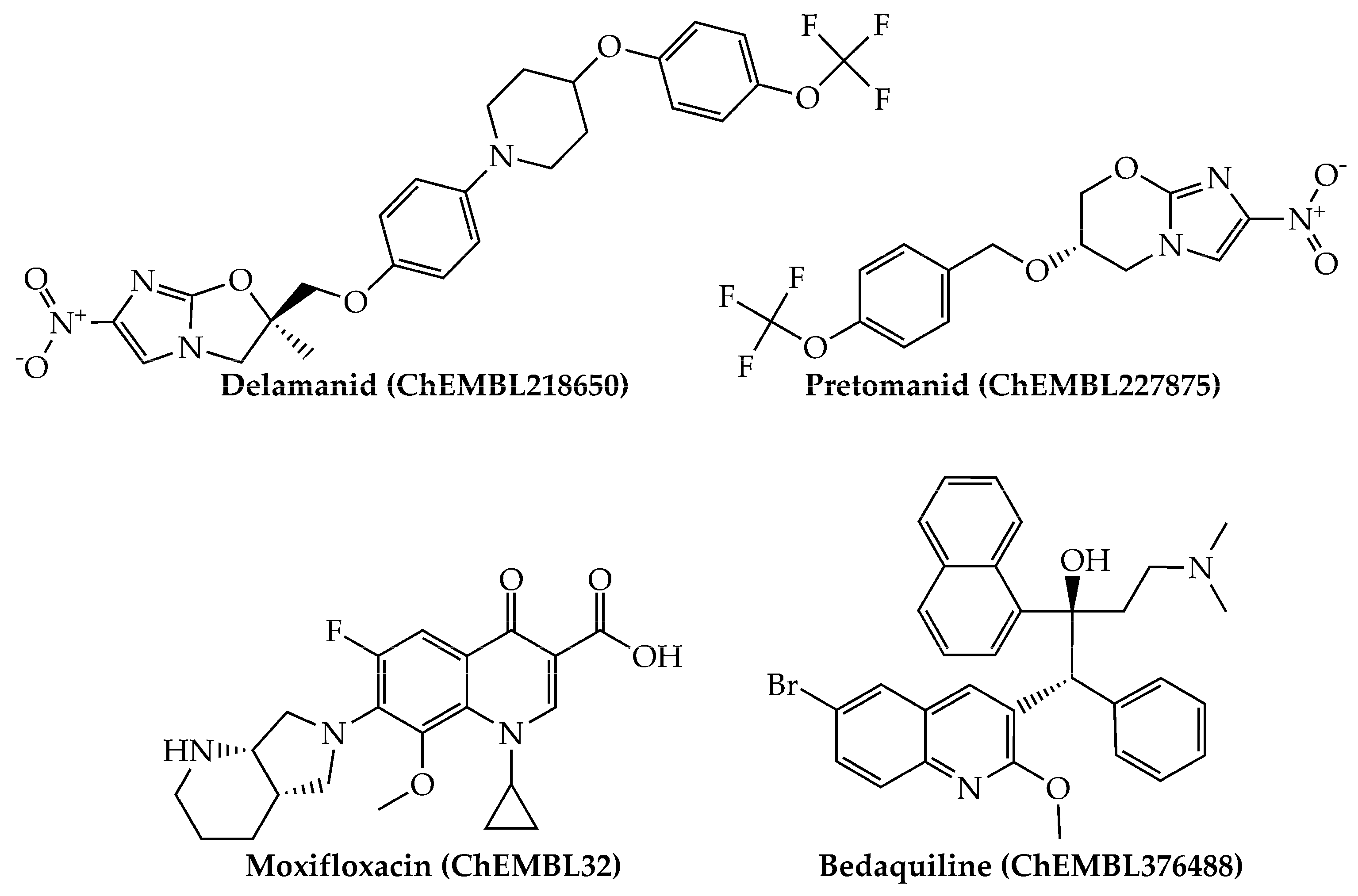

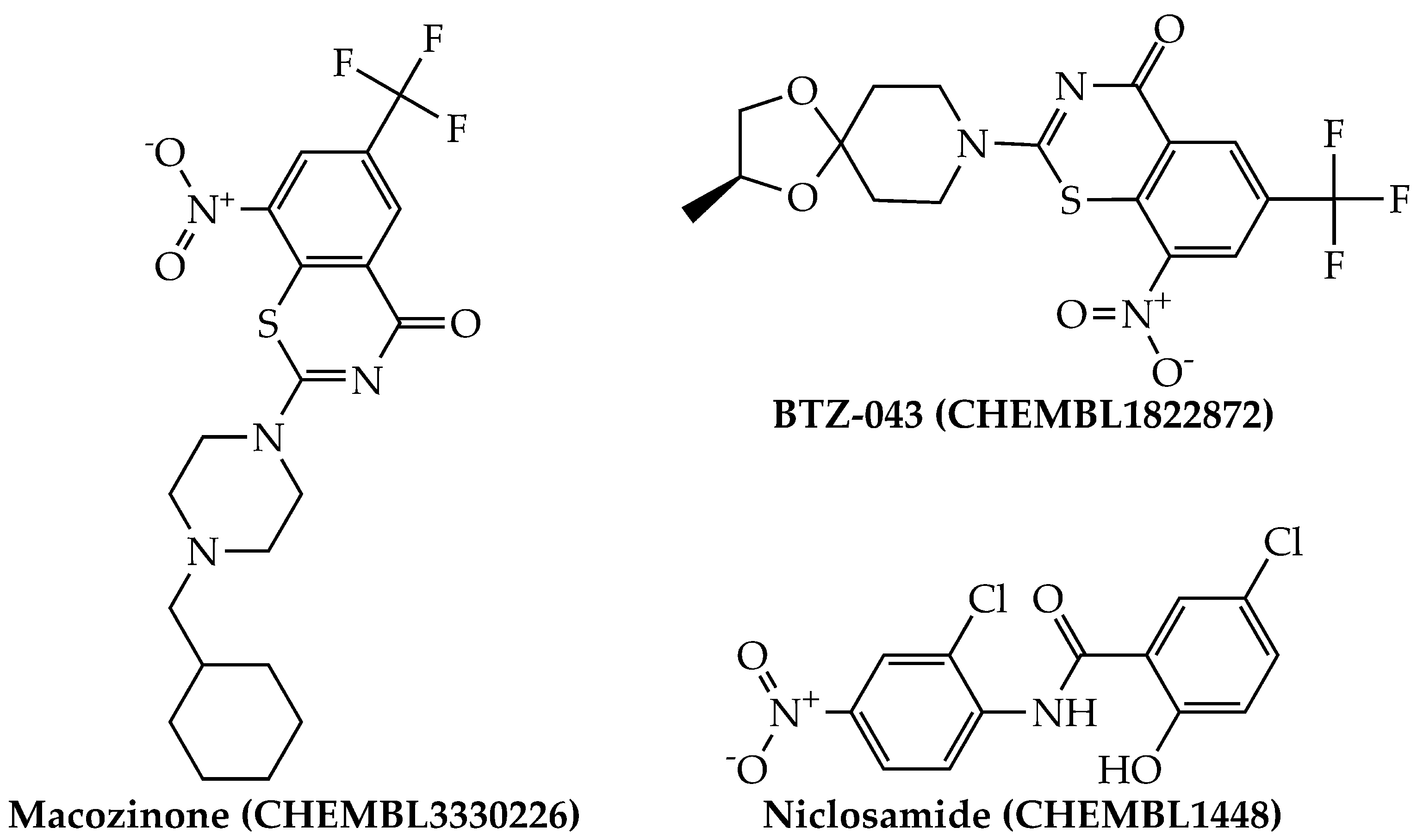

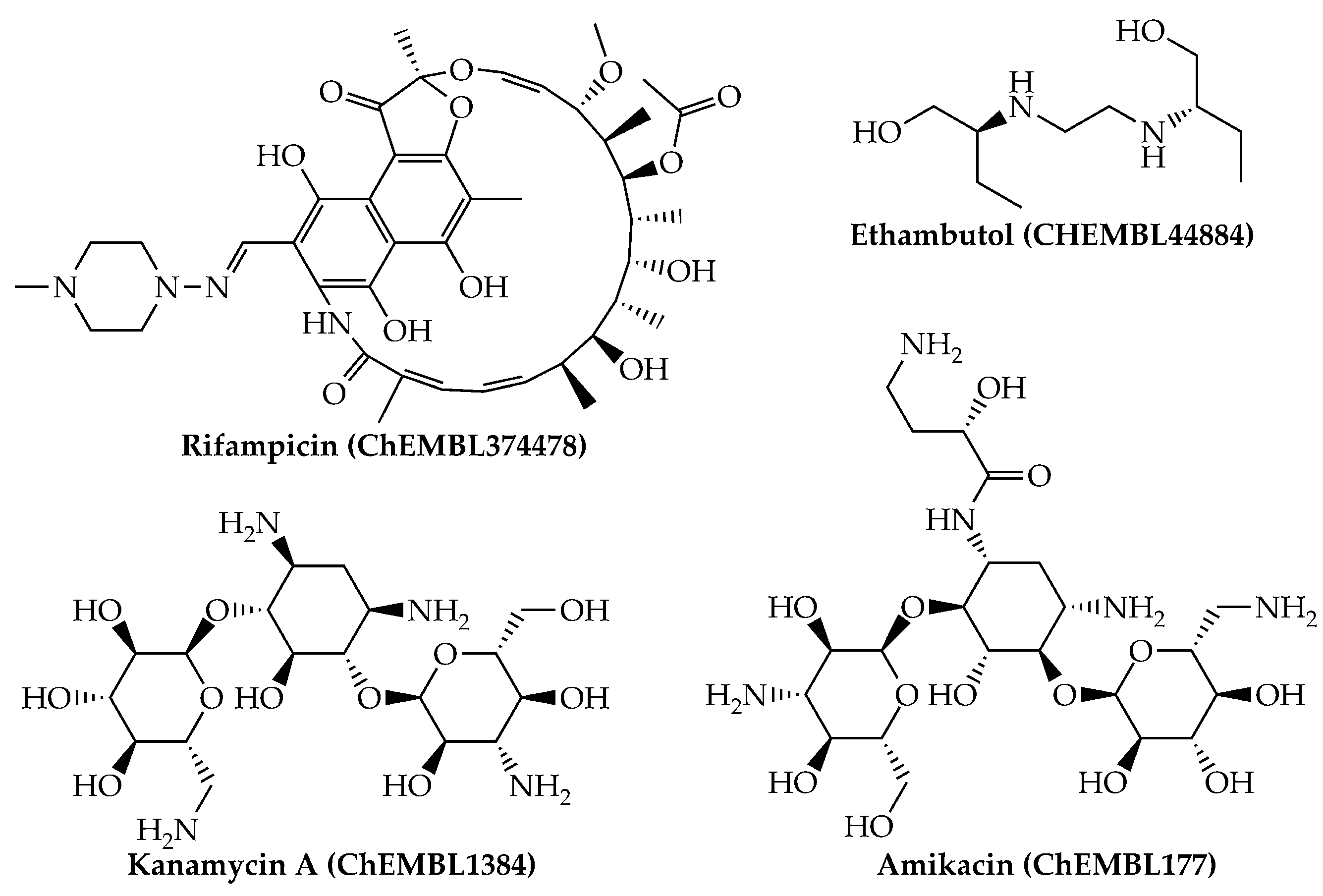{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Definition |
|---|---|
| D[LASSq3(HYD)G]ta | Deviation of the atom-based stochastic quadratic index of order 3, weighted by the hydrophobicity of the halogens and their chemical environments, and depending on the chemical structure and the assay time. |
| D[LASSq0(POL)G]ta | Deviation of the atom-based stochastic quadratic index of order 0, weighted by the polarizability of the halogens, and depending on the chemical structure and the assay time. |
| D[LASSq0(HYD)Y]ta | Deviation of the atom-based stochastic quadratic index of order 0, weighted by the hydrophobicity of the heteroatoms, and depending on the chemical structure and the assay time. |
| D[LASSq1(PSA)Y]ta | Deviation of the atom-based stochastic quadratic index of order 1, weighted by the polar surface area of the heteroatoms and their adjacent atoms, and depending on the chemical structure and the assay time. |
| D[LASSq0(HYD)M]bs | Deviation of the atom-based stochastic quadratic index of order 0, weighted by the hydrophobicity of the methyl groups, and depending on the chemical structure and the Mtb strain against which the molecule was tested. |
| D[LASSq0(AW)P]bs | Deviation of the atom-based stochastic quadratic index of order 0, weighted by the atomic weight of the aromatic carbons, and depending on the chemical structure and the Mtb strain against which the molecule was tested. |
| D[LASSq2(HYD)Y]bs | Deviation of the atom-based stochastic quadratic index of order 2, weighted by the hydrophobicity of the heteroatoms and their chemical environments, and depending on the chemical structure and the Mtb strain against which the molecule was tested. |
| D[LASSq2(HYD)G]ap | Deviation of the atom-based stochastic quadratic index of order 2, weighted by the hydrophobicity of the halogens and their chemical environments, and depending on the chemical structure and information regarding the assay protocol. |
| D[LASSq5(AW)G]ap | Deviation of the atom-based stochastic quadratic index of order 5, weighted by the atomic weight of the halogens and their chemical environments, and depending on the chemical structure and information regarding the assay protocol. |
| D[LASSq3(KU)G]ap | Deviation of the atom-based stochastic quadratic index of order 3, weighted by Kupchick’s vertex degrees of the halogens and their chemical environments, and depending on the chemical structure and information regarding the assay protocol. |
| D[LASSq2(HYD)M]ap | Deviation of the atom-based stochastic quadratic index of order 2, weighted by the hydrophobicity of the methyl groups and their chemical environments, and depending on the chemical structure and information regarding the assay protocol. |
| D[LASSq2(AW)P]ap | Deviation of the atom-based stochastic quadratic index of order 2, weighted by the atomic weight of the aromatic carbons and their chemical environments, and depending on the chemical structure and information regarding the assay protocol. |
| Network Notation a | Training Algorithm b | Error Function c | Hidden Activation d | Output Activation |
|---|---|---|---|---|
| MLP 12-45-2 | BFGS 116 | Entropy | Tanh | Softmax |
| MLP 12-33-2 | BFGS 138 | Entropy | Logistic | Softmax |
| MLP 12-41-2 | BFGS 104 | SOS | Tanh | Logistic |
| SYMBOLS a | Training Set | Test Set |
|---|---|---|
| NActive | 602 | 201 |
| CCActive | 572 | 172 |
| Sn(%) | 95.02% | 85.57% |
| NInactive | 582 | 186 |
| CCInactive | 534 | 160 |
| Sp(%) | 91.75% | 86.02% |
| MCC | 0.869 | 0.716 |
| Symbol | Class-Based Means | Tendency a | |
|---|---|---|---|
| Active | Inactive | ||
| D[LASSq3(HYD)G]ta | −1.1976 × 10−4 | −2.0514 × 10−2 | Increase |
| D[LASSq0(POL)G]ta | 5.0005 × 10−3 | 6.3674 × 10−3 | Decrease |
| D[LASSq0(HYD)Y]ta | 5.1000 × 10−3 | −4.4105 × 10−1 | Increase |
| D[LASSq1(PSA)Y]ta | 1.2460 × 10−3 | −1.6800 × 10−1 | Increase |
| D[LASSq0(HYD)M]bs | 1.4621 × 10−2 | −3.0065 × 10−1 | Increase |
| D[LASSq0(AW)P]bs | 8.1935 × 10−3 | −2.2074 × 10−1 | Increase |
| D[LASSq2(HYD)Y]bs | −4.0897 × 10−3 | −2.1845 × 10−1 | Increase |
| D[LASSq2(HYD)G]ap | 1.0793 × 10−2 | −2.4162 × 10−1 | Increase |
| D[LASSq5(AW)G]ap | 1.2911 × 10−2 | −1.8708 × 10−1 | Increase |
| D[LASSq3(KU)G]ap | 9.6900 × 10−3 | −1.1239 × 10−1 | Increase |
| D[LASSq2(HYD)M]ap | 1.6609 × 10−2 | −2.8205 × 10−1 | Increase |
| D[LASSq2(AW)P]ap | 1.1549 × 10−2 | −2.8297 × 10−1 | Increase |
| ID a | Name | FA(%) b | S(TSAD) c | log10(MIC) d | MIC (nM) e |
|---|---|---|---|---|---|
| CHEMBL78535 | Ancriviroc | 100.00 | 288 | −4.624 | 23768.40 |
| CHEMBL1450565 | Aklomide | 100.00 | 279 | −4.128 | 74473.20 |
| CHEMBL2104712 | Nisterime acetate | 100.00 | 279 | −4.673 | 21232.44 |
| CHEMBL2106822 | Nizofenone | 95.83 | 288 | −4.332 | 46558.61 |
| CHEMBL2104159 | Bromoxanide | 100.00 | 276 | −4.838 | 14521.12 |
| CHEMBL1199080 | Bretylium | 95.83 | 279 | −3.91 | 123026.88 |
| CHEMBL3330226 | Macozinone | 95.83 | 279 | −7.558 | 27.67 |
| CHEMBL1909324 | Pinaverium | 95.83 | 279 | −4.668 | 21478.30 |
| CHEMBL292702 | Maitansine | 95.83 | 279 | −4.776 | 16749.43 |
| CHEMBL2111120 | Nitralamine | 95.83 | 276 | −4.02 | 95499.26 |
| CHEMBL289832 | Licostinel | 95.83 | 276 | −3.826 | 149279.44 |
| CHEMBL1448 | Niclosamide | 95.83 | 276 | −5.249 | 5636.38 |
| CHEMBL2104616 | Clonitazene | 91.67 | 288 | −4.866 | 13614.45 |
| CHEMBL1269025 | Edoxaban | 91.67 | 288 | −5.731 | 1857.80 |
| CHEMBL2106056 | Cronidipine | 91.67 | 288 | −4.771 | 16943.38 |
| CHEMBL1241348 | Faldaprevir | 91.67 | 288 | −5.275 | 5308.84 |
| CHEMBL2013174 | Vedroprevir | 91.67 | 288 | −5.446 | 3580.96 |
| CHEMBL2104730 | Nitroxinil | 91.67 | 279 | −3.881 | 131522.48 |
| CHEMBL493636 | Sulfanitran | 91.67 | 279 | −4.678 | 20989.40 |
| CHEMBL9484 | Clofilium | 91.67 | 279 | −4.042 | 90782.05 |
| CHEMBL1822872 | BTZ-043 | 91.67 | 279 | −7.01 | 97.72 |
| CHEMBL1178725 | Nolpitantium | 91.67 | 279 | −4.876 | 13304.54 |
| CHEMBL2104390 | Ilatreotide | 91.67 | 279 | −5.123 | 7533.56 |
| CHEMBL56367 | Nimesulide | 87.50 | 288 | −4.907 | 12387.97 |
| CHEMBL2107448 | Loprazolam | 87.50 | 288 | −4.945 | 11350.11 |
| CHEMBL3670800 | ALK-4290 | 87.50 | 288 | −5.298 | 5035.01 |
| CHEMBL1181731 | Teglarinad | 87.50 | 288 | −5.154 | 7014.55 |
| CHEMBL1170047 | Iniparib | 87.50 | 279 | −4.117 | 76383.58 |
| CHEMBL491 | Hydroxyflutamide | 87.50 | 279 | −4.221 | 60117.37 |
| CHEMBL452 | Clonazepam | 87.50 | 279 | −4.223 | 59841.16 |
| CHEMBL1274 | Nilutamide | 87.50 | 279 | −4.559 | 27605.78 |
| CHEMBL2110930 | Fubrogonium | 87.50 | 279 | −4.125 | 74989.42 |
| CHEMBL2110825 | Dodeclonium | 87.50 | 279 | −4.19 | 64565.42 |
| CHEMBL397647 | JNJ-17166864 | 87.50 | 279 | −4.53 | 29512.09 |
| CHEMBL2105721 | Nivocasan | 87.50 | 279 | −4.644 | 22698.65 |
| CHEMBL2107326 | Dasantafil | 87.50 | 279 | −5.015 | 9660.51 |
| CHEMBL1908326 | Meclonazepam | 83.33 | 288 | −4.281 | 52360.04 |
| CHEMBL1823817 | CE-224535 | 83.33 | 288 | −4.736 | 18365.38 |
| CHEMBL1276663 | Cefozopran | 83.33 | 288 | −5.143 | 7194.49 |
| CHEMBL2110800 | Ciclonium | 83.33 | 279 | −4.48 | 33113.11 |
| CHEMBL1337 | Nitisinone | 83.33 | 279 | −4.569 | 26977.39 |
| CHEMBL512306 | [18F]D | 83.33 | 279 | −4.532 | 29376.50 |
| CHEMBL435191 | Edotecarin | 83.33 | 279 | −4.521 | 30130.06 |
| CHEMBL2074922 | Efonidipine | 83.33 | 279 | −4.894 | 12764.39 |
| MIC90 Cutoff Value (nM) a | tab | bsc,d | ape |
|---|---|---|---|
| <1146.75 | 10d | Mtb (H37Rv_NRF) | LORA method |
| 10d | Mtb (H37Rv) | Spectrophotometric assay (OD580-OD600) | |
| ≤1500 | 14d | Mtb (H37Rv) | Broth dilution method |
| <7622.22 | 3d | Mtb (H37Rv_NRF) | Spectrophotometric assay (OD580-OD600) |
| 3d | Mtb (MC2 6220_NRF) | Spectrophotometric assay (OD580-OD600) | |
| 3d | Mtb (MC2 6220_RF) | Spectrophotometric assay (OD580-OD600) | |
| <5829.77 | 4d | Mtb (H37Rv) | AlamarBlue/Resazurin/MABA method |
| ≤5300 | 5d | Mtb (H37Rv) | AlamarBlue/Resazurin/MABA method |
| 5d | Mtb (H37Rv_ATCC 25618) | Spectrophotometric assay (OD580-OD600) | |
| 5d | Mtb (H37Rv) | Spectrophotometric assay (OD580-OD600) | |
| 5d | Mtb (INH-R) | AlamarBlue/Resazurin/MABA method | |
| 5d | Mtb (H37Rv_ATCC 27294) | Broth dilution method | |
| ≤5000 | 6d | Mtb (H37Rv_ATCC 25618) | AlamarBlue/Resazurin/MABA method |
| 6d | Mtb (H37Rv) | AlamarBlue/Resazurin/MABA method | |
| 6d | Mtb (MC2 6220_NRF) | Spectrophotometric assay (OD580-OD600) | |
| ≤4940 | 7d | Mtb (H37Rv) | AlamarBlue/Resazurin/MABA method |
| 7d | Mtb (H37Rv_ATCC 27294) | AlamarBlue/Resazurin/MABA method | |
| 7d | Mtb (INH-R) | AlamarBlue/Resazurin/MABA method | |
| 7d | Mtb (H37Rv_ATCC 27294) | Broth dilution method | |
| 7d | Mtb (H37Rv) | Broth dilution method | |
| 7d | Mtb (RMP-R) | AlamarBlue/Resazurin/MABA method | |
| 7d | Mtb (H37Rv) | Spectrophotometric assay (OD580-OD600) | |
| 7d | Mtb (MC2 6220_RF) | Spectrophotometric assay (OD580-OD600) | |
| 7d | Mtb (MC2 6220_NRF) | Spectrophotometric assay (OD580-OD600) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleandrova, V.V.; Scotti, M.T.; Speck-Planche, A. Computational Drug Repurposing for Antituberculosis Therapy: Discovery of Multi-Strain Inhibitors. Antibiotics 2021, 10, 1005. https://doi.org/10.3390/antibiotics10081005
Kleandrova VV, Scotti MT, Speck-Planche A. Computational Drug Repurposing for Antituberculosis Therapy: Discovery of Multi-Strain Inhibitors. Antibiotics. 2021; 10(8):1005. https://doi.org/10.3390/antibiotics10081005
Chicago/Turabian StyleKleandrova, Valeria V., Marcus T. Scotti, and Alejandro Speck-Planche. 2021. "Computational Drug Repurposing for Antituberculosis Therapy: Discovery of Multi-Strain Inhibitors" Antibiotics 10, no. 8: 1005. https://doi.org/10.3390/antibiotics10081005
APA StyleKleandrova, V. V., Scotti, M. T., & Speck-Planche, A. (2021). Computational Drug Repurposing for Antituberculosis Therapy: Discovery of Multi-Strain Inhibitors. Antibiotics, 10(8), 1005. https://doi.org/10.3390/antibiotics10081005