
In Vitro Evaluation of the Potential Pharmacological Activity and Molecular Targets of New Benzimidazole-Based Schiff Base Metal Complexes

 ,
,  , , ,
, , ,  ,
,  and
and
Abstract

1. Introduction
2. Results and Discussion
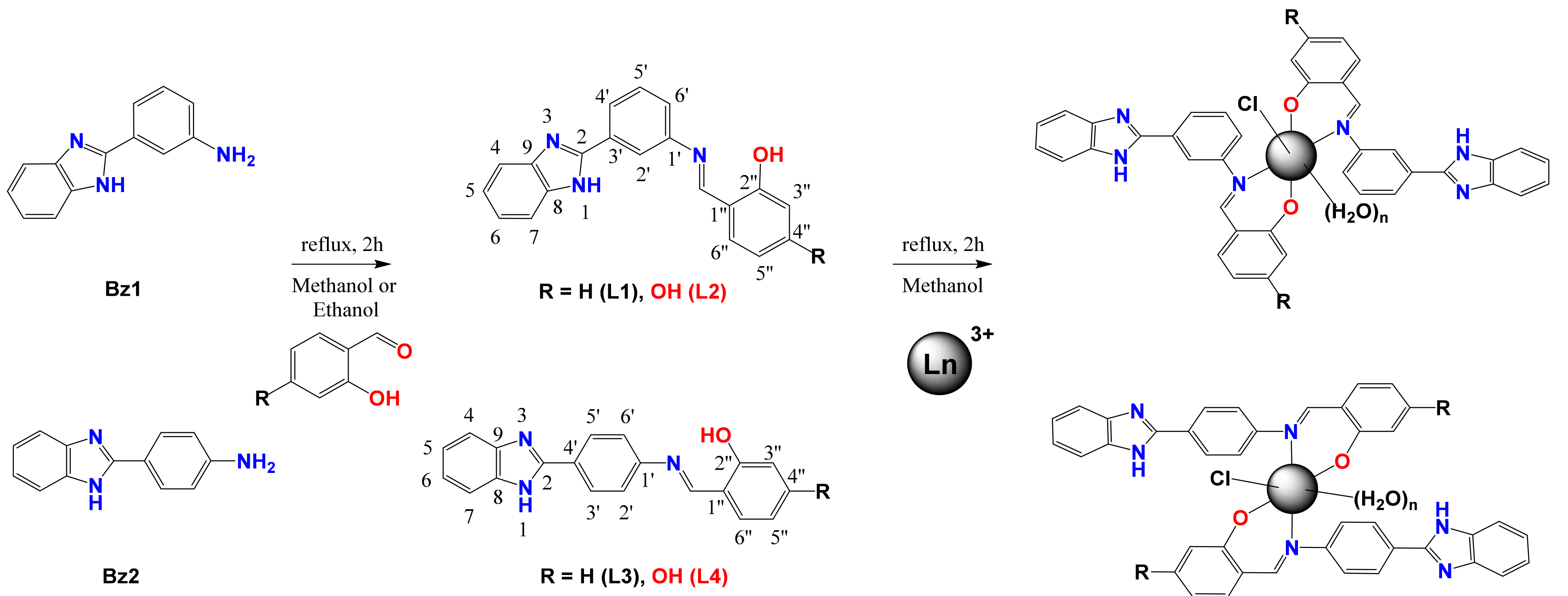
2.1. Chemistry
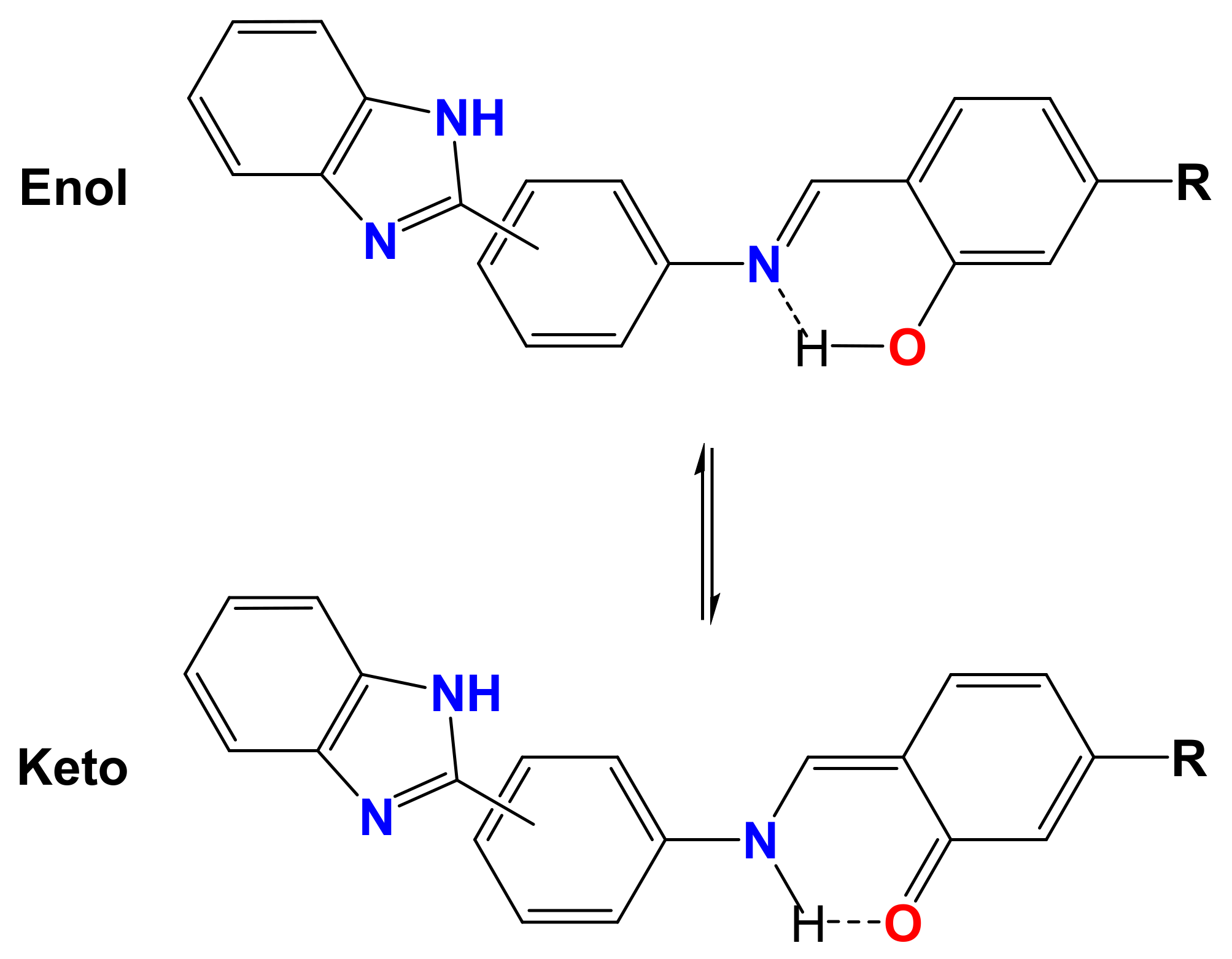
Spectral Characteristics
2.2. Biological Studies
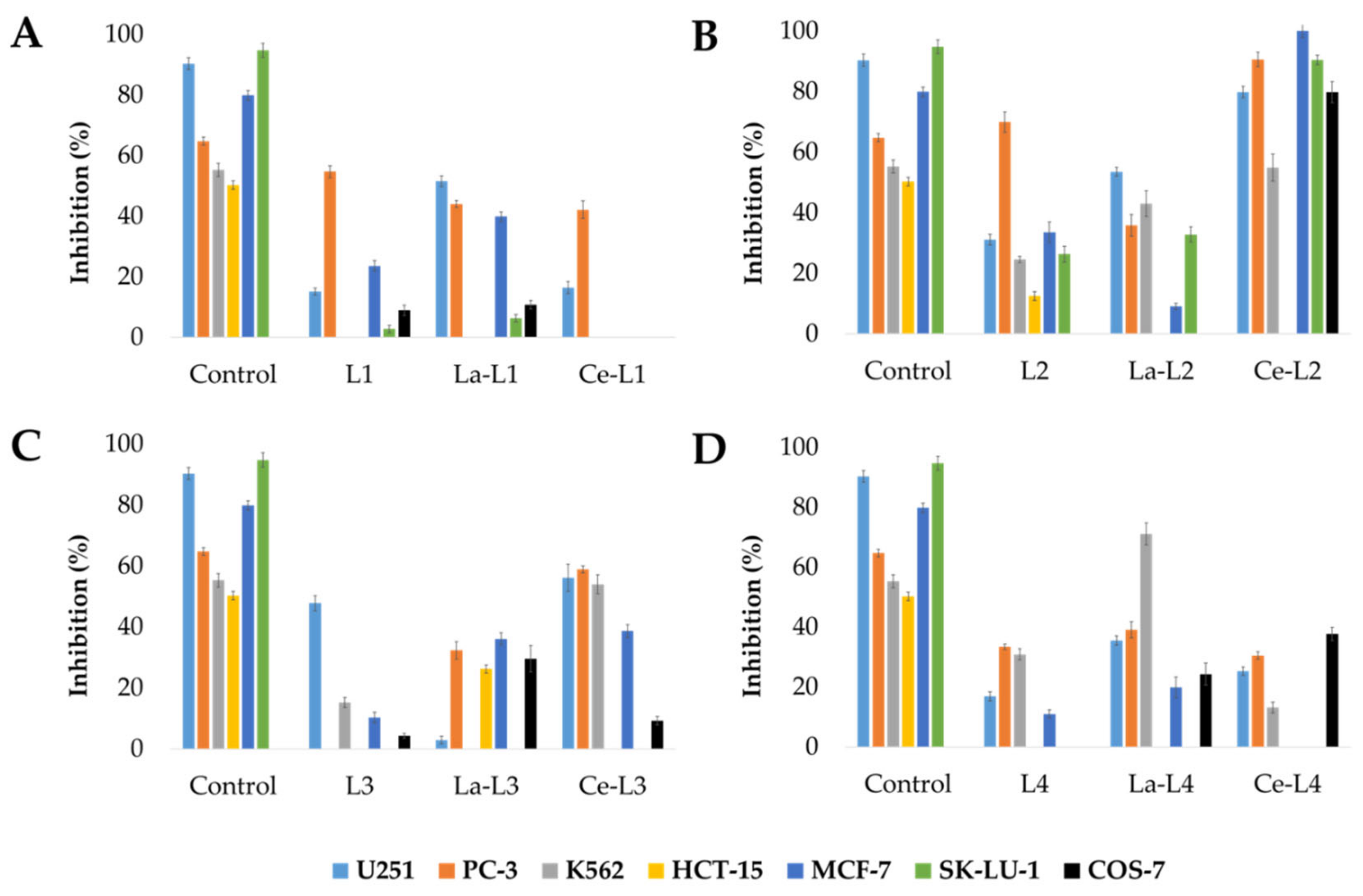
2.2.1. Antiproliferative Properties
2.2.2. Antiparasitic Properties
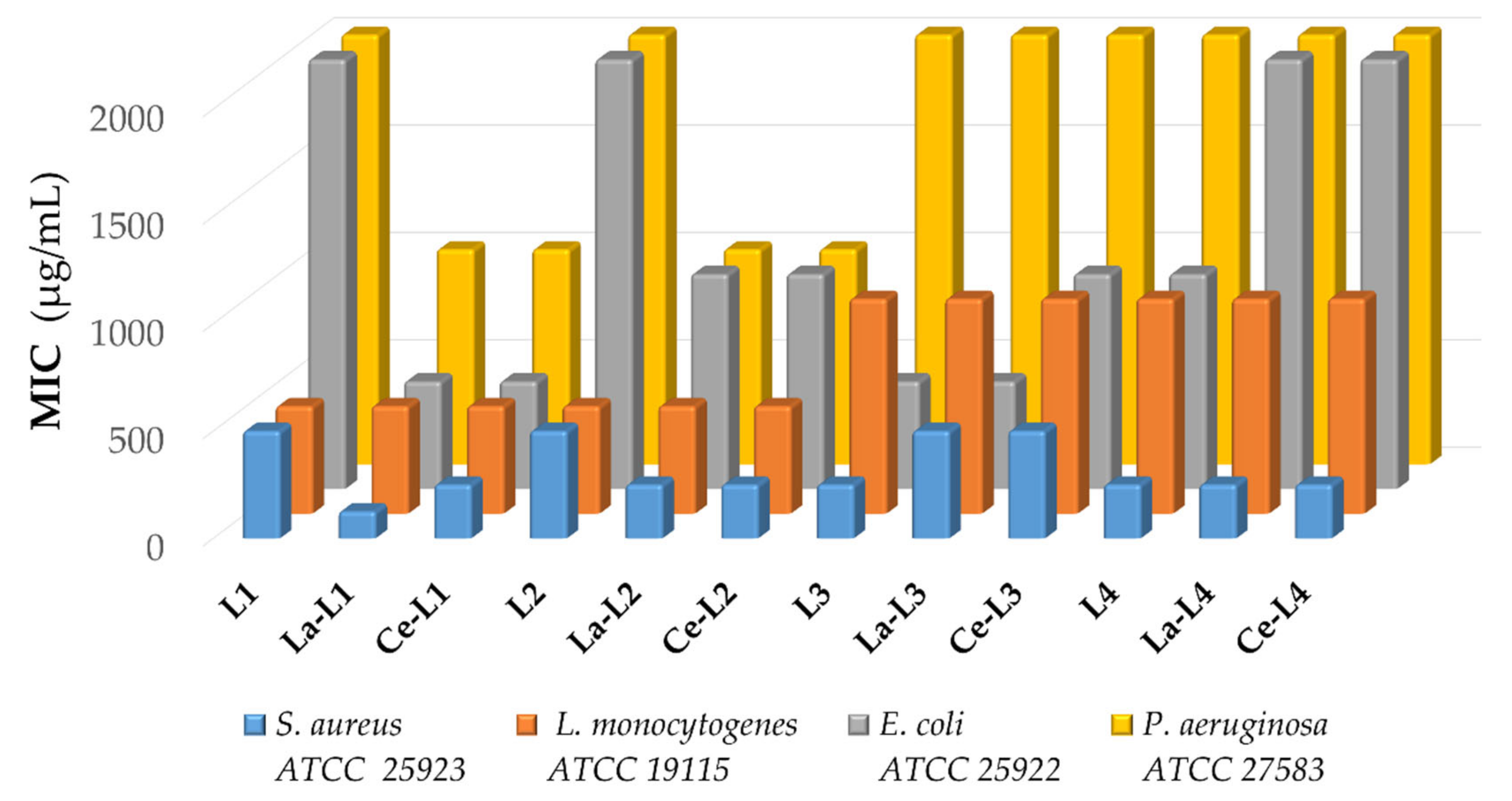
2.2.3. Antibacterial Properties
2.3. Interaction with Potential Molecular Targets
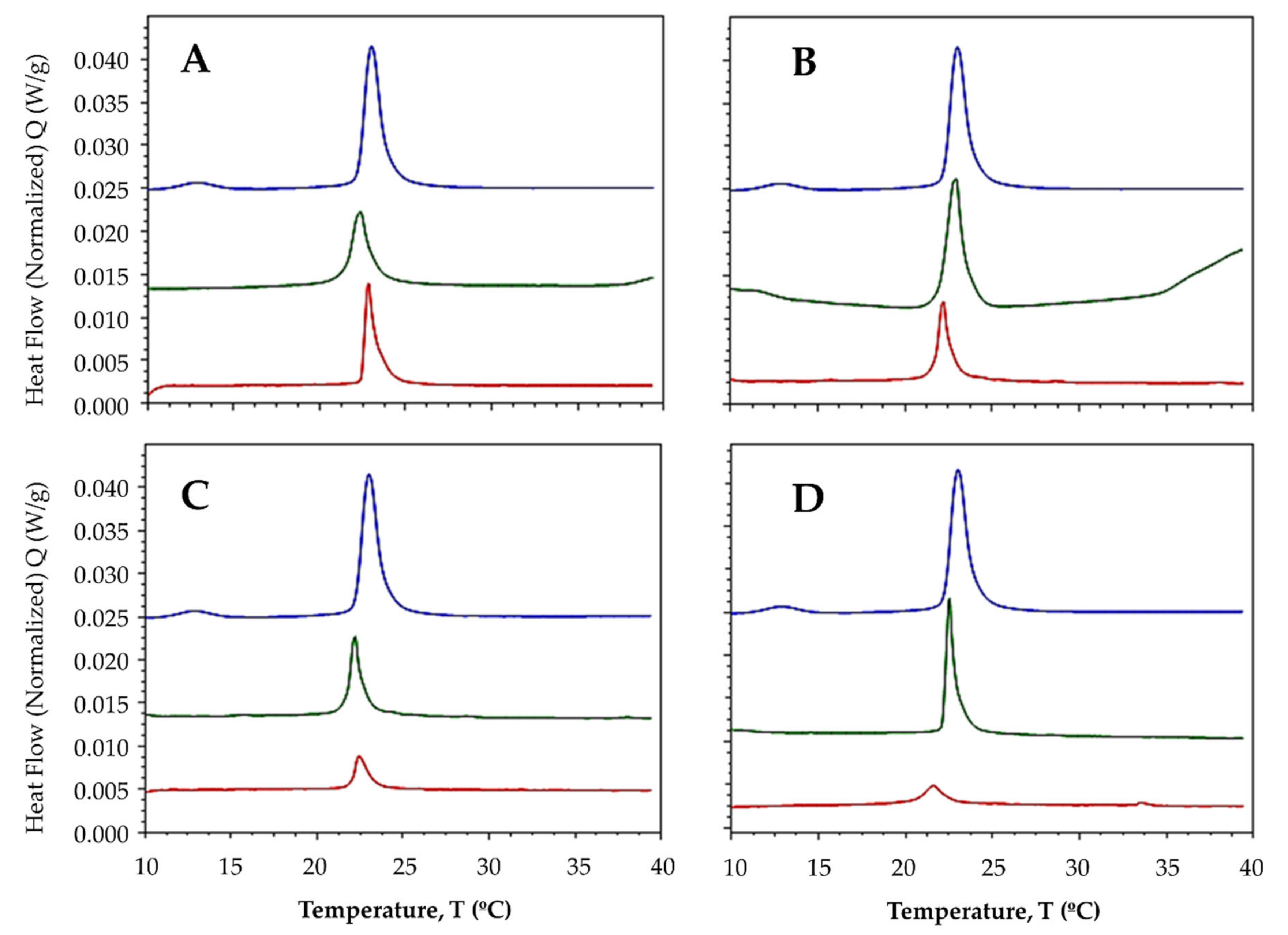
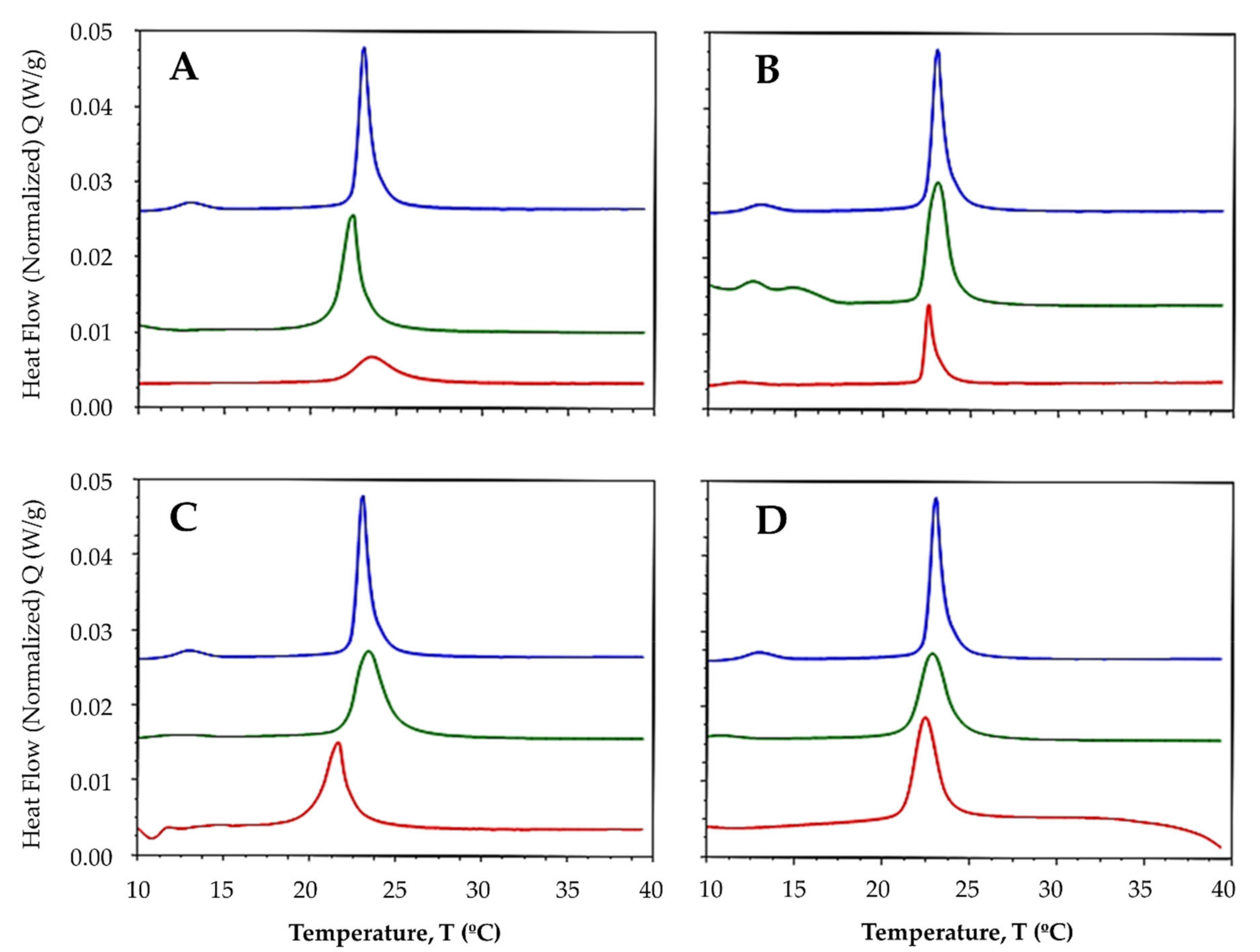
2.3.1. Interaction with Synthetic Model Membranes
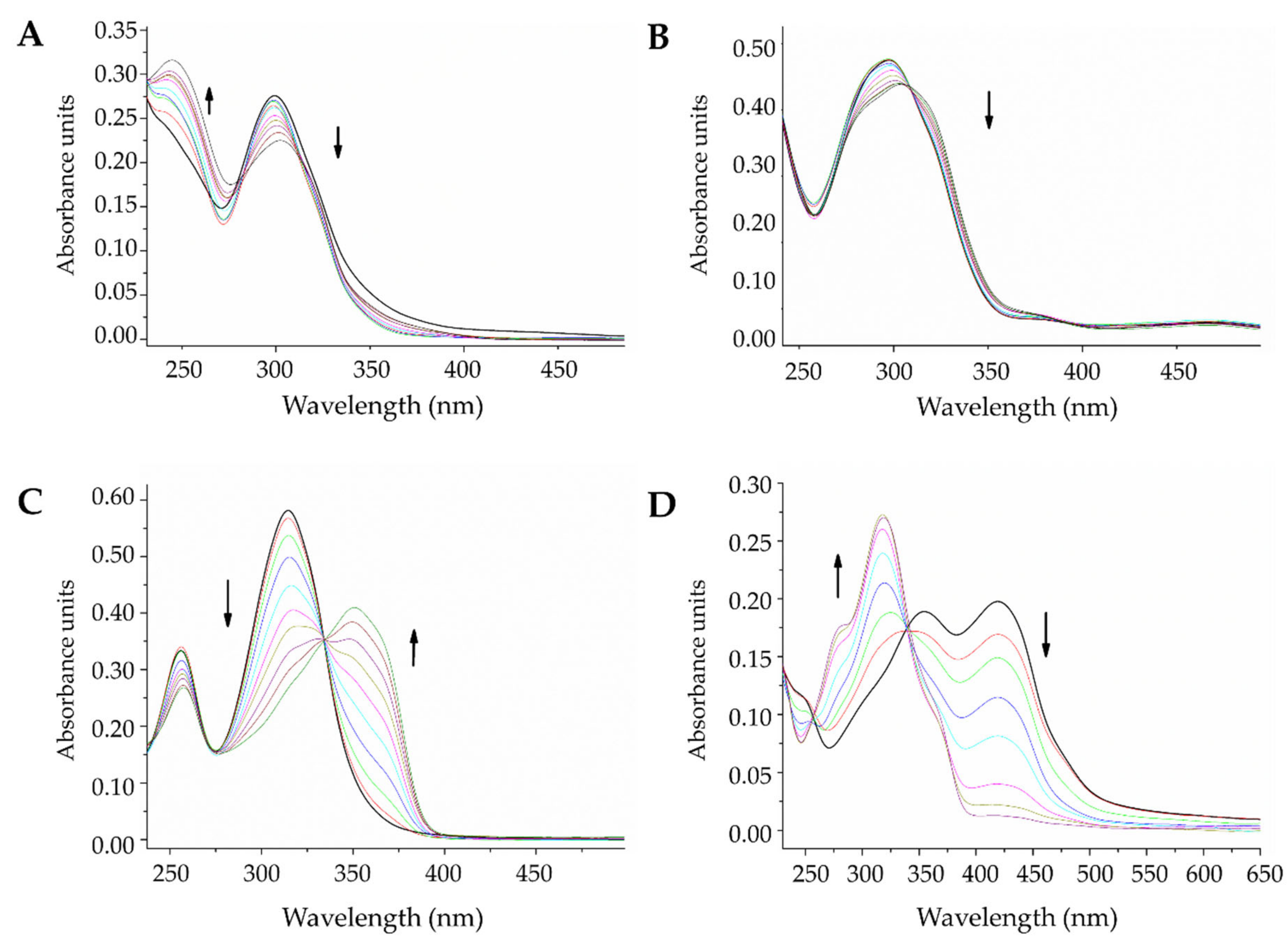
2.3.2. DNA Interaction by Electronic Absorption Monitoring
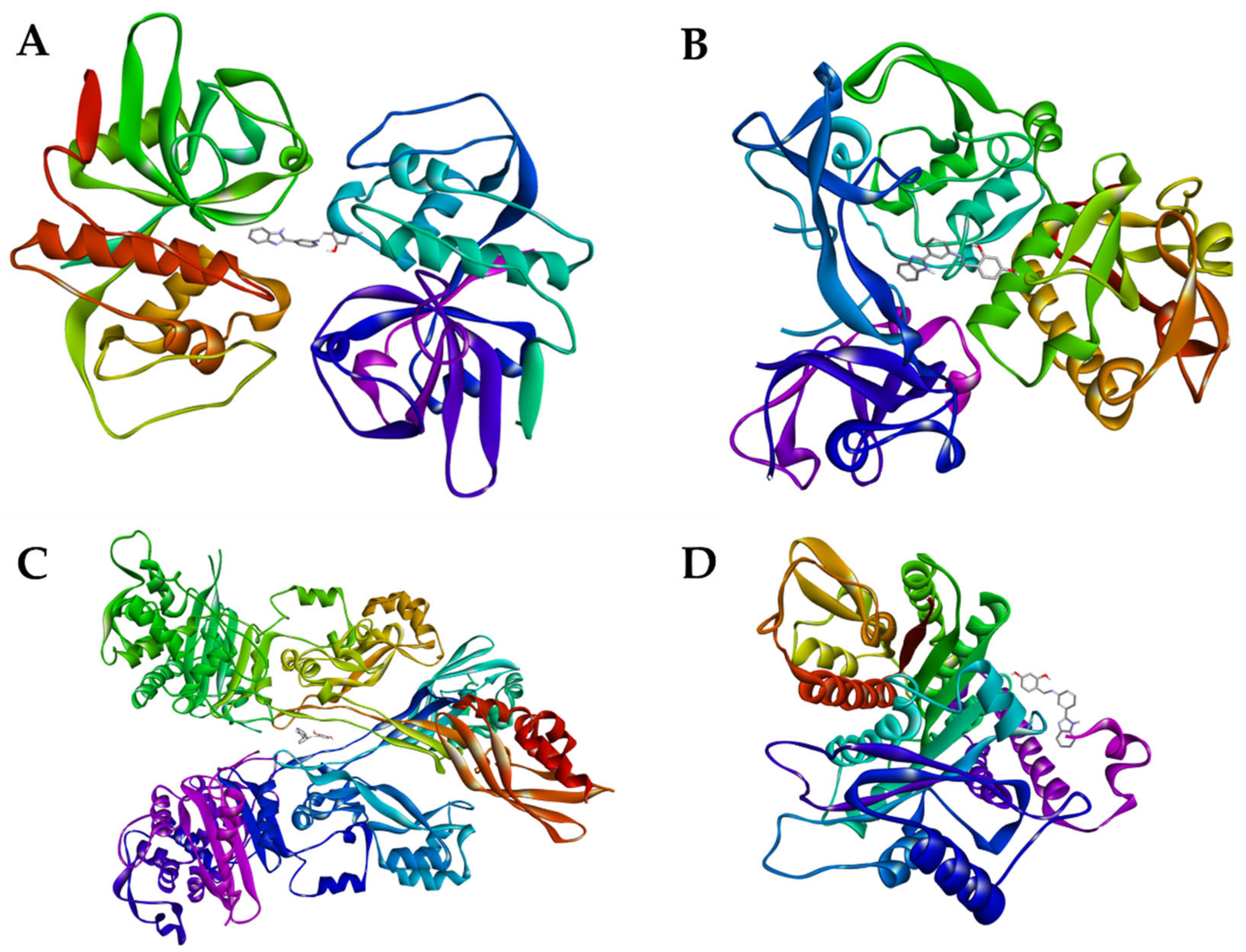
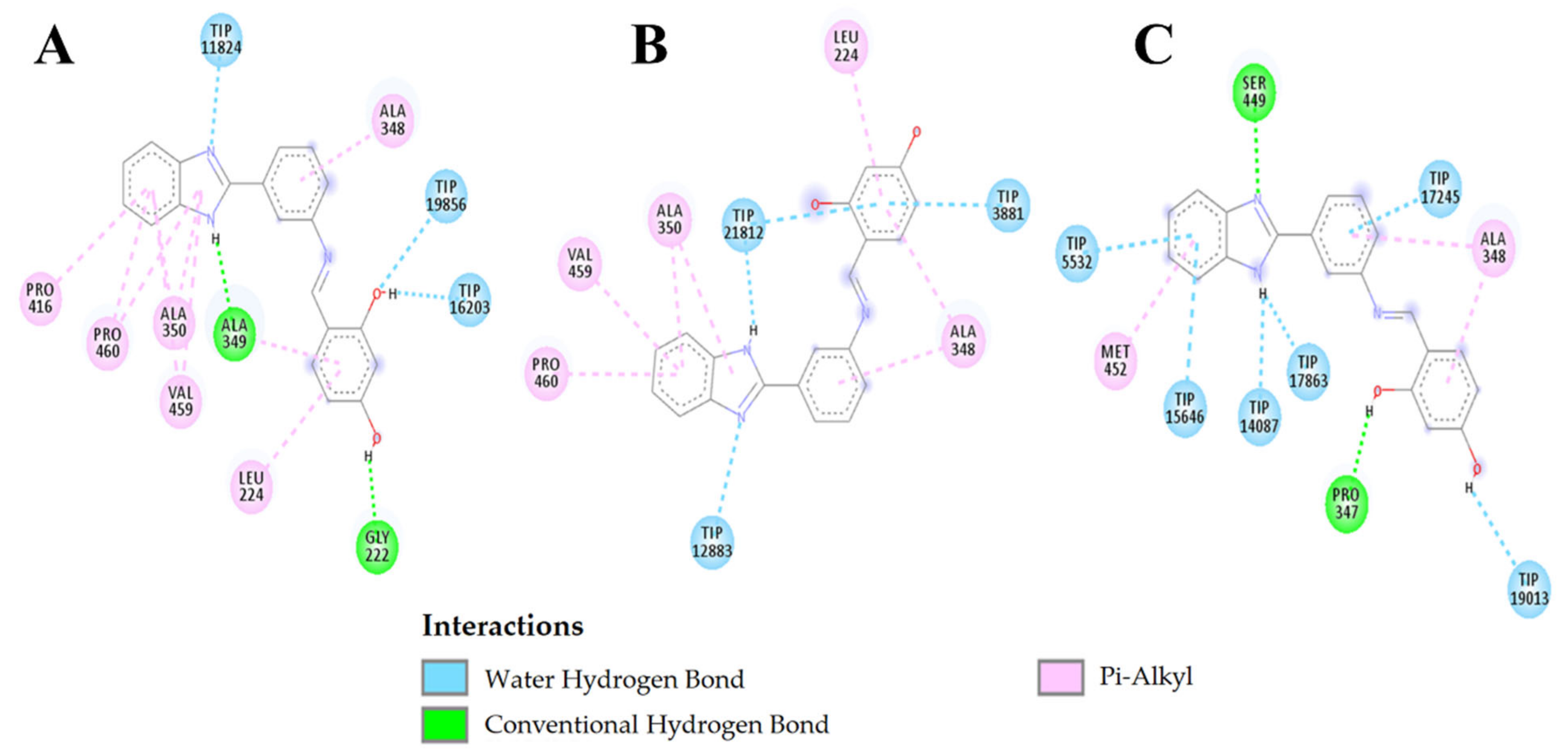
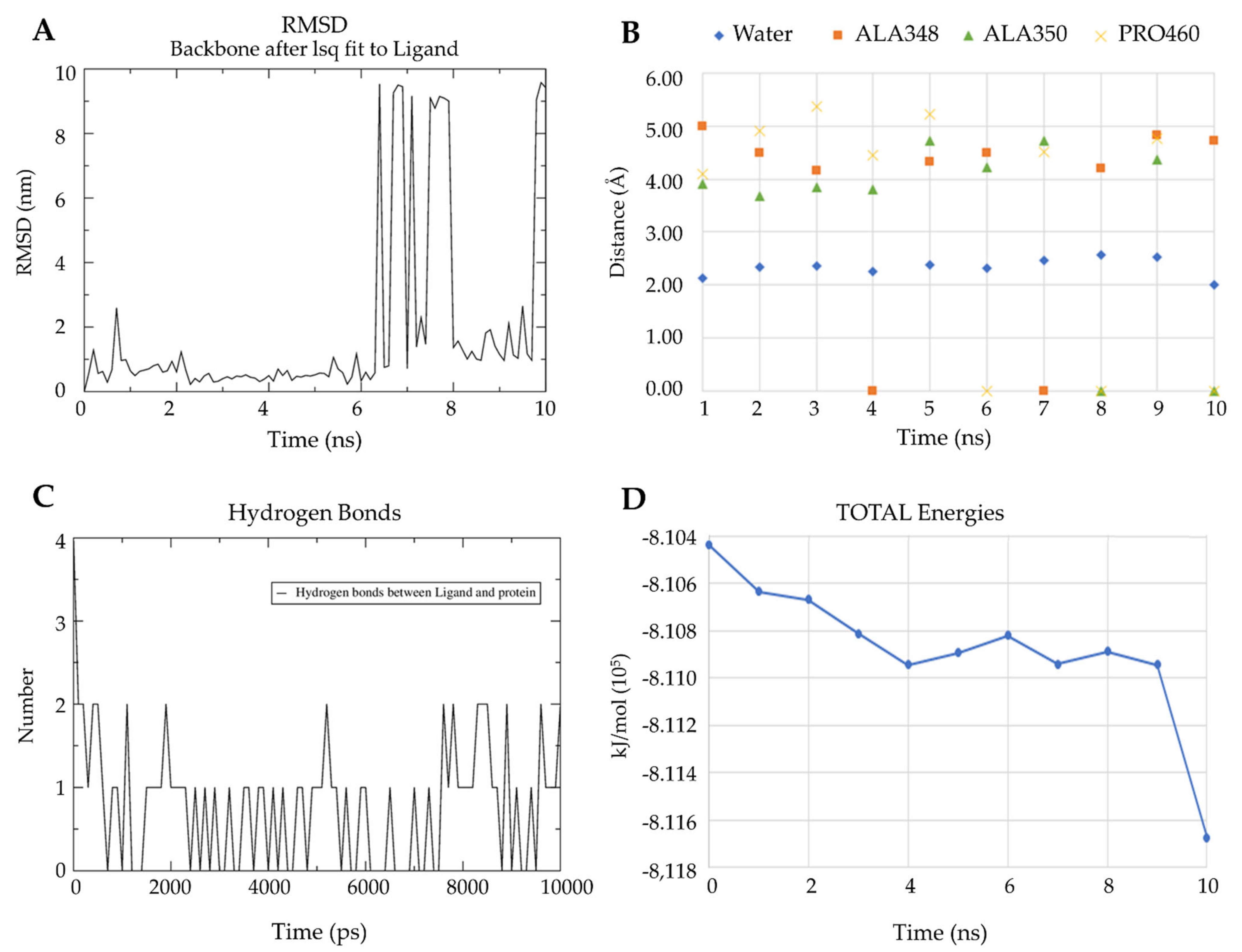
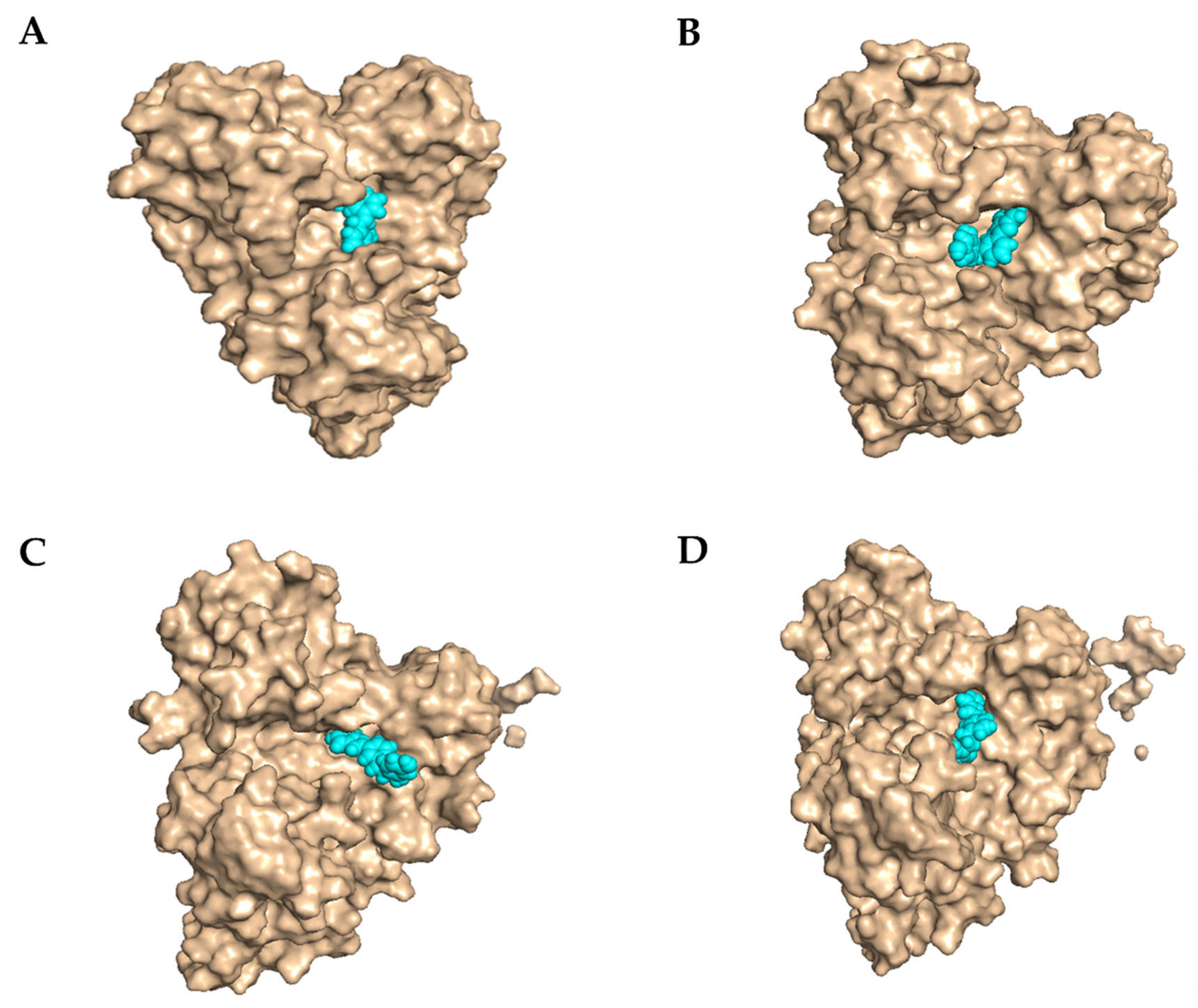
2.3.3. Molecular Docking
3. Materials and Methods
3.1. Materials and Physical Measurements
3.2. Synthesis of Compounds
3.2.1. Synthesis of Benzimidazoles
3.2.2. Synthesis of Schiff Bases
3.2.3. Synthesis of Lanthanide Complexes
3.3. Biological Assays
3.3.1. Cytotoxicity Studies
3.3.2. In Vitro Leishmanicidal Activity
3.3.3. In Vitro Trypanocidal Activity
3.3.4. In Vitro Anti-Plasmodial Activity
3.3.5. In Vitro Antibacterial Activity
3.4. Interaction with Model Membranes
3.4.1. Membrane Preparation
3.4.2. Differential Scanning Calorimetry
3.5. DNA Interaction Studies
Electronic Absorption Monitoring Assays
3.6. Molecular Docking
3.6.1. Construction of the Ligands and Complexes 3D Model
3.6.2. Obtaining the Receptors
3.6.3. Controls
3.6.4. Validation of Docking between L2 and Leishmanin with Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McQuitty, R.J. Metal-based drugs. Sci. Prog. 2014, 97, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, R.K. Metal Complexes in Medicine: An Overview and Update from Drug Design Perspective. Cancer Ther. Oncol. Int. J. 2019, 14. [Google Scholar] [CrossRef]
- Gasser, G. Metal complexes and medicine: A successful combination. Chimia 2015, 69, 442–446. [Google Scholar] [CrossRef]
- Kellert, M.; Sárosi, I.; Rajaratnam, R.; Meggers, E.; Lönnecke, P.; Hey-Hawkins, E. Ruthenacarborane–phenanthroline derivatives as potential metallodrugs. Molecules 2020, 25, 2322. [Google Scholar] [CrossRef]
- Naureen, B.; Miana, G.A.; Shahid, K.; Asghar, M.; Tanveer, S.; Sarwar, A. Iron (III) and zinc (II) monodentate Schiff base metal complexes: Synthesis, characterisation and biological activities. J. Mol. Struct. 2021, 1231. [Google Scholar] [CrossRef]
- Liu, X.; Hamon, J.R. Recent developments in penta-, hexa- and heptadentate Schiff base ligands and their metal complexes. Coord. Chem. Rev. 2019, 389, 94–118. [Google Scholar] [CrossRef]
- Kumar, J.; Rai, A.; Raj, V. A Comprehensive Review on the Pharmacological Activity of Schiff Base Containing Derivatives. Org. Med. Chem. 2017, 1, 1–15. [Google Scholar] [CrossRef]
- Hranjec, M.; Starčević, K.; Pavelić, S.K.; Lučin, P.; Pavelić, K.; Karminski Zamola, G. Synthesis, spectroscopic characterization and antiproliferative evaluation in vitro of novel Schiff bases related to benzimidazoles. Eur. J. Med. Chem. 2011, 46, 2274–2279. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorganic Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef]
- Andrasiak, I.; Rybka, J.; Knopinska-Posluszny, W.; Wrobel, T. Efficacy and Safety of Bendamustine and Ibrutinib in Previously Untreated Patients With Chronic Lymphocytic Leukemia: Indirect Comparison. Clin. Lymphoma, Myeloma Leuk. 2017, 17, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.S.E.P.W.; Kviecinski, M.R.; Ourique, F.; Parisotto, E.B.; Grinevicius, V.M.A.S.; Correia, J.F.G.; Wilhelm Filho, D.; Pedrosa, R.C. Albendazole as a promising molecule for tumor control. Redox Biol. 2016, 10, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.J.; Jeyakkumar, P.; Avula, S.R.; Zhou, Q.; Zhou, C.H. Design, synthesis and biological evaluation of 5-fluorouracil-derived benzimidazoles as novel type of potential antimicrobial agents. Bioorganic Med. Chem. Lett. 2016, 26, 2584–2588. [Google Scholar] [CrossRef]
- Robinson, M.W.; McFerran, N.; Trudgett, A.; Hoey, L.; Fairweather, I. A possible model of benzimidazole binding to β-tubulin disclosed by invoking an inter-domain movement. J. Mol. Graph. Model. 2004, 23, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Qin, Y.J.; Yang, N.; Zhang, Y.L.; Liu, C.H.; Zhu, H.L. Synthesis, biological evaluation, and molecular docking studies of novel 1-benzene acyl-2-(1-methylindol-3-yl)-benzimidazole derivatives as potential tubulin polymerization inhibitors. Eur. J. Med. Chem. 2015, 99, 125–137. [Google Scholar] [CrossRef]
- Singhal, S.; Khanna, P.; Khanna, L. Synthesis, DFT studies, molecular docking, antimicrobial screening and UV fluorescence studies on ct-DNA for novel Schiff bases of 2-(1-aminobenzyl) benzimidazole. Heliyon 2019, 5, e02596. [Google Scholar] [CrossRef]
- Wang, Y.N.; Bheemanaboina, R.R.Y.; Cai, G.X.; Zhou, C.H. Novel purine benzimidazoles as antimicrobial agents by regulating ROS generation and targeting clinically resistant Staphylococcus aureus DNA groove. Bioorganic Med. Chem. Lett. 2018, 28, 1621–1628. [Google Scholar] [CrossRef]
- Ranjan, N.; Story, S.; Fulcrand, G.; Leng, F.; Ahmad, M.; King, A.; Sur, S.; Wang, W.; Tse-Dinh, Y.C.; Arya, D.P. Selective Inhibition of Escherichia coli RNA and DNA Topoisomerase i by Hoechst 33258 Derived Mono-and Bisbenzimidazoles. J. Med. Chem. 2017, 60, 4904–4922. [Google Scholar] [CrossRef]
- Miana, G.E.; Ribone, S.R.; Vera, D.M.A.; Sánchez-Moreno, M.; Mazzieri, M.R.; Quevedo, M.A. Design, synthesis and molecular docking studies of novel N-arylsulfonyl-benzimidazoles with anti Trypanosoma cruzi activity. Eur. J. Med. Chem. 2019, 165, 1–10. [Google Scholar] [CrossRef]
- Kumaravel, G.; Raman, N. A treatise on benzimidazole based Schiff base metal(II) complexes accentuating their biological efficacy: Spectroscopic evaluation of DNA interactions, DNA cleavage and antimicrobial screening. Mater. Sci. Eng. C 2017, 70, 184–194. [Google Scholar] [CrossRef]
- Sukkur Saleem, S.; Sankarganesh, M.; Adwin Jose, P.; Dhaveethu Raja, J. Design, synthesis, antioxidant, antimicrobial, DNA binding and molecular docking studies of morpholine based Schiff base ligand and its metal(II) complexes. Inorg. Chem. Commun. 2021, 124, 108396. [Google Scholar] [CrossRef]
- Mahmood, K.; Hashmi, W.; Ismail, H.; Mirza, B.; Twamley, B.; Akhter, Z.; Rozas, I.; Baker, R.J. Synthesis, DNA binding and antibacterial activity of metal(II) complexes of a benzimidazole Schiff base. Polyhedron 2019, 157, 326–334. [Google Scholar] [CrossRef]
- Kalarani, R.; Sankarganesh, M.; Kumar, G.G.V.; Kalanithi, M. Synthesis, spectral, DFT calculation, sensor, antimicrobial and DNA binding studies of Co(II), Cu(II) and Zn(II) metal complexes with 2-amino benzimidazole Schiff base. J. Mol. Struct. 2020, 1206, 127725. [Google Scholar] [CrossRef]
- Andiappan, K.; Sanmugam, A.; Deivanayagam, E.; Karuppasamy, K.; Kim, H.S.; Vikraman, D. In vitro cytotoxicity activity of novel Schiff base ligand-lanthanide complexes. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, M.T.; Zabiszak, M.; Nowak, M.; Jastrzab, R. Lanthanides: Schiff base complexes, applications in cancer diagnosis, therapy, and antibacterial activity. Coord. Chem. Rev. 2018, 370, 42–54. [Google Scholar] [CrossRef]
- Siddiqi, Z.A.; Shahid, A.M.; Khalid, M.; Sharma, P.K.; Siddique, A. Spectroscopic, luminescence, electrochemical and antimicrobial studies of lanthanide complexes of bis-benzimidazole derived ligands. J. Mol. Struct. 2013, 1037, 402–411. [Google Scholar] [CrossRef]
- Aragón-Muriel, A.; Camprubí-Robles, M.; González-Rey, E.; Salinas-Castillo, A.; Rodríguez-Diéguez, A.; Gómez-Ruiz, S.; Polo-Cerón, D. Dual investigation of lanthanide complexes with cinnamate and phenylacetate ligands: Study of the cytotoxic properties and the catalytic oxidation of styrene. Polyhedron 2014, 80, 117–128. [Google Scholar] [CrossRef]
- Váquiro-Reyes, I.Y.; Aragón-Muriel, A.; Polo-Cerón, D. Síntesis, caracterización y evaluación farmacológica de nuevos complejos metálicos derivados de híbridos heteroaromáticos (benzimidazol/oxadiazol). Rev. Colomb. Ciencias Químico-Farmacéuticas 2019, 48, 557–588. [Google Scholar] [CrossRef]
- Aragón-Muriel, A.; Upegui, Y.; Muñoz, J.A.; Robledo, S.M.; Polo-Cerón, D. Synthesis, characterization and biological evaluation of rare earth complexes against tropical diseases Leishmaniasis, Malaria and Trypanosomiasis. Av. Quim. 2016, 11, 53–61. [Google Scholar]
- Magd-El-Din, A.A.; Mousa, H.A.; Labib, A.A.; Hassan, A.S.; Abd El-All, A.S.; Ali, M.M.; El-Rashedy, A.A.; El-Desoky, A.H. Benzimidazole-Schiff bases and their complexes: Synthesis, anticancer activity and molecular modeling as Aurora kinase inhibitor. Zeitschrift Naturforsch. Sect. C J. Biosci. 2018, 73, 465–478. [Google Scholar] [CrossRef]
- Geary, W.J. The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Chandrakala, M.; Sheshadri, B.S.; Nanje Gowda, N.M.; Murthy, K.G.S.; Nagasundara, K.R. Synthesis and spectral studies of 2-salicylidine-4-aminophenyl benzimidazole and its reaction with divalent Zn, Cd and Hg: Crystal structure of the cadmium bromide complex. J. Chem. Res. 2010, 576–580. [Google Scholar] [CrossRef]
- Roopashree, B.; Gayathri, V.; Gopi, A.; Devaraju, K.S. Syntheses, characterizations, and antimicrobial activities of binuclear ruthenium(III) complexes containing 2-substituted benzimidazole derivatives. J. Coord. Chem. 2012, 65, 4023–4040. [Google Scholar] [CrossRef]
- Chandrakala, M.; Nanje Gowda, N.M.; Murthy, K.G.S.; Nagasundara, K.R. Activation of - N=CH - bond in a Schiff base by divalent nickel monitored by NMR evidence. Magn. Reson. Chem. 2012, 50, 335–340. [Google Scholar] [CrossRef]
- Suman, G.R.; Bubbly, S.G.; Gudennavar, S.B.; Muthu, S.; Roopashree, B.; Gayatri, V.; Nanje Gowda, N.M. Structural investigation, spectroscopic and energy level studies of Schiff base: 2-[(3′-N-salicylidenephenyl)benzimidazole] using experimental and DFT methods. J. Mol. Struct. 2017, 1139, 247–254. [Google Scholar] [CrossRef]
- Chandrakala, M. Hydrolysis of -N=CH- Bond in 2-Salicylidene-4-aminophenyl benzimidazole by Palladium(II). Asian J. Chem. 2019, 31, 287–292. [Google Scholar] [CrossRef]
- Dutta Gupta, S.; Revathi, B.; Mazaira, G.I.; Galigniana, M.D.; Subrahmanyam, C.V.S.; Gowrishankar, N.L.; Raghavendra, N.M. 2,4-dihydroxy benzaldehyde derived Schiff bases as small molecule Hsp90 inhibitors: Rational identification of a new anticancer lead. Bioorg. Chem. 2015, 59, 97–105. [Google Scholar] [CrossRef][Green Version]
- Alam, S.A.M.F.; Ahmad, T.; Nazmuzzaman, M.; Ray, S.K.; Sharifuzzaman, M.; Karim, M.R.; Alam, M.G.; Ajam, M.M.; Maitra, P.; Mandol, D.; et al. Synthesis of Benzimidazole Derivatives Containing Schiff Base Exhibiting Antimicrobial Activities. Int. J. Res. Stud. Biosci. 2017, 5, 18–24. [Google Scholar] [CrossRef]
- Singh, A.K.; Goswami, B. Synthesis, Characterization and Biological Evaluation of Some Mannich Schiff Base Derivatives of Substituted Benzimidazole. Int. J. Pharm. Phytopharm. Res. 2013, 2, 302–305. [Google Scholar]
- Fonkui, T.Y.; Ikhile, M.I.; Njobeh, P.B.; Ndinteh, D.T. Benzimidazole schiff base derivatives: Synthesis, characterization and antimicrobial activity. BMC Chem. 2019, 13, 1–11. [Google Scholar] [CrossRef]
- Aragón-Muriel, A.; Liscano-Martínez, Y.; Rufino-Felipe, E.; Morales-Morales, D.; Oñate-Garzón, J.; Polo-Cerón, D. Synthesis, biological evaluation and model membrane studies on metal complexes containing aromatic N,O-chelate ligands. Heliyon 2020, 6. [Google Scholar] [CrossRef]
- Blaszczak-Światkiewicz, K.; Mirowski, M.; Kaplinska, K.; Kruszynśki, R.; Trzesowska-Kruszyńska, A.; Mikiciuk-Olasik, E. New benzimidazole derivatives with potential cytotoxic activity - study of their stability by RP-HPLC. Acta Biochim. Pol. 2012, 59, 279–288. [Google Scholar] [CrossRef]
- Palacios, G.; Parodi, A.; Upegui, Y.A.; Montoya, A.; Pulido, S.; Vélez, I.D.; Robledo, S.M. Studies in vitro on infectivity and sensitivity to antileishmanial drugs in New World Leishmania species transfected with the green fluorescent protein [pIR3(-)-eGFP]. Parasitology 2017, 144, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Fonkui, T.Y.; Ikhile, M.I.; Ndinteh, D.T.; Njobeh, P.B. Microbial activity of some heterocyclic schiff bases and metal complexes: A review. Trop. J. Pharm. Res. 2018, 17, 2507–2518. [Google Scholar] [CrossRef]
- Touafri, L.; Hellal, A.; Chafaa, S.; Khelifa, A.; Kadri, A. Synthesis, characterisation and DFT studies of three Schiff bases derived from histamine. J. Mol. Struct. 2017, 1149, 750–760. [Google Scholar] [CrossRef]
- Arzanlou, M.; Chai, W.C.; Venter, H. Intrinsic, adaptive and acquired antimicrobial resistance in Gram-negative bacteria. Essays Biochem. 2017, 60. [Google Scholar] [CrossRef]
- Pachori, P.; Gothalwal, R.; Gandhi, P. Emergence of antibiotic resistance Pseudomonas aeruginosa in intensive care unit; a critical review. Genes Dis. 2019, 6, 109–119. [Google Scholar] [CrossRef]
- Schenone, M.; Wagner, B.K.; Clemons, P.A.; Program, B. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013, 9, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Aragón-Muriel, A.; Ausili, A.; Sánchez, K.; Rojas, A.O.E.; Londoño Mosquera, J.; Polo-Cerón, D.; Oñate-Garzón, J. Studies on the Interaction of Alyteserin 1c Peptide and Its Cationic Analogue with Model Membranes Imitating Mammalian and Bacterial Membranes. Biomolecules 2019, 9, 527. [Google Scholar] [CrossRef]
- Bilge, D.; Kazanci, N.; Severcan, F. Acyl chain length and charge effect on Tamoxifen-lipid model membrane interactions. J. Mol. Struct. 2013, 1040, 75–82. [Google Scholar] [CrossRef]
- Kaddah, S.; Khreich, N.; Kaddah, F.; Khrouz, L.; Charcosset, C.; Greige-Gerges, H. Corticoids modulate liposome membrane fluidity and permeability depending on membrane composition and experimental protocol design. Biochimie 2018, 153, 33–45. [Google Scholar] [CrossRef]
- Ihlenfeldt, M.; Gantner, G.; Harrer, M.; Puschendorf, B.; Putzer, H.; Grunicke, H. Interaction of the Alkylating Antitumor Agent 2,3,5-Tris(ethyleneimino)-benzoquinone with the Plasma Membrane of Ehrlich Ascites Tumor Cells. Cancer Res. 1981, 41, 289–293. [Google Scholar] [PubMed]
- Severcan, F.; Kazanci, N.; Zorlu, F. Tamoxifen increases membrane fluidity at high concentrations. Biosci. Rep. 2000, 20, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Zschörnig, O. The effect of metal cations on the phase behavior and hydration characteristics of phospholipid membranes. Chem. Phys. Lipids 2002, 115, 39–61. [Google Scholar] [CrossRef]
- Le, C.T.M.; Houri, A.; Balage, N.; Smith, B.J.; Mechler, A. Interaction of small ionic species with phospholipid membranes: The role of metal coordination. Front. Mater. 2019, 5, 1–9. [Google Scholar] [CrossRef]
- Oñate-Garzón, J.; Ausili, A.; Manrique-Moreno, M.; Torrecillas, A.; Aranda, F.J.; Patiño, E.; Gomez-Fernández, J.C. The increase in positively charged residues in cecropin D-like Galleria mellonella favors its interaction with membrane models that imitate bacterial membranes. Arch. Biochem. Biophys. 2017, 629, 54–62. [Google Scholar] [CrossRef]
- Ninham, B.W.; Larsson, K.; Lo Nostro, P. Two sides of the coin. Part 1. Lipid and surfactant self-assembly revisited. Colloids Surfaces B Biointerfaces 2017, 152, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Gil, E.S.; da Silva, C.B.; Nogara, P.A.; da Silveira, C.H.; da Rocha, J.B.T.; Iglesias, B.A.; Lüdtke, D.S.; Gonçalves, P.F.B.; Rodembusch, F.S. Synthesis, photophysical characterization, CASSCF/CASPT2 calculations and CT-DNA interaction study of amino and azido benzazole analogues. J. Mol. Liq. 2020, 297, 111938. [Google Scholar] [CrossRef]
- Vamsikrishna, N.; Daravath, S.; Ganji, N.; Pasha, N.; Shivaraj. Synthesis, structural characterization, DNA interaction, antibacterial and cytotoxicity studies of bivalent transition metal complexes of 6-aminobenzothiazole Schiff base. Inorg. Chem. Commun. 2020, 113, 107767. [Google Scholar] [CrossRef]
- Chatziefthimiou, S.D.; Lazarou, Y.G.; Hadjoudis, E.; Dziembowska, T.; Mavridis, I.M. Keto forms of salicylaldehyde schiff bases: Structural and theoretical aspects. J. Phys. Chem. B 2006, 110, 23701–23709. [Google Scholar] [CrossRef]
- Abu-Dief, A.M.; Abdel-Rahman, L.H.; Abdel-Mawgoud, A.A.H. A robust in vitro Anticancer, Antioxidant and Antimicrobial Agents Based on New Metal-Azomethine Chelates Incorporating Ag(I), Pd (II) and VO (II) Cations: Probing the Aspects of DNA Interaction. Appl. Organomet. Chem. 2020, 34, 1–20. [Google Scholar] [CrossRef]
- Biver, T. Use of UV-vis spectrometry to gain information on the mode of binding of small molecules to DNAs and RNAs. Appl. Spectrosc. Rev. 2012, 47, 272–325. [Google Scholar] [CrossRef]
- Zhang, L.; Addla, D.; Ponmani, J.; Wang, A.; Xie, D.; Wang, Y.N.; Zhang, S.L.; Geng, R.X.; Cai, G.X.; Li, S.; et al. Discovery of membrane active benzimidazole quinolones-based topoisomerase inhibitors as potential DNA-binding antimicrobial agents. Eur. J. Med. Chem. 2016, 111, 160–182. [Google Scholar] [CrossRef]
- Nimesh, H.; Sur, S.; Sinha, D.; Yadav, P.; Anand, P.; Bajaj, P.; Virdi, J.S.; Tandon, V. Synthesis and biological evaluation of novel bisbenzimidazoles as Escherichia coli topoisomerase IA inhibitors and potential antibacterial agents. J. Med. Chem. 2014, 57, 5238–5257. [Google Scholar] [CrossRef] [PubMed]
- Žgur-Bertok, D. DNA Damage Repair and Bacterial Pathogens. PLoS Pathog. 2013, 9, 1–5. [Google Scholar] [CrossRef]
- Liu, Y.; Grimm, M.; Dai, W.T.; Hou, M.C.; Xiao, Z.X.; Cao, Y. CB-Dock: A web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacol. Sin. 2020, 41, 138–144. [Google Scholar] [CrossRef]
- Braña, M.F.; Castellano, J.M.; Yunta, M.J.R. Synthesis of benzimidazo-substituted 3-quinolinecarboxylic acids as antibacterial agents. J. Heterocycl. Chem. 1990, 27, 1177–1180. [Google Scholar] [CrossRef]
- Cernatescu, C.; ComaniŢă, E. Benzazole derivatives. IX. 2-(4′-aminophenyl)- and 2-(4′-aminophenyl)-1-methyl-benzimidazoles Reactions with phenolic aldehydes. Roum. Biotechnol. Lett. 2006, 11, 2845–2850. [Google Scholar]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Otero, E.; Vergara, S.; Robledo, S.M.; Cardona, W.; Carda, M.; Vélez, I.D.; Rojas, C.; Otálvaro, F. Synthesis, leishmanicidal and Cytotoxic activity of triclosan-chalcone, triclosan-chromone and triclosan-coumarin hybrids. Molecules 2014, 19, 13251–13266. [Google Scholar] [CrossRef] [PubMed]
- Pulido, S.A.; Muñoz, D.L.; Restrepo, A.M.; Mesa, C.V.; Alzate, J.F.; Vélez, I.D.; Robledo, S.M. Improvement of the green fluorescent protein reporter system in Leishmania spp. for the in vitro and in vivo screening of antileishmanial drugs. Acta Trop. 2012, 122, 36–45. [Google Scholar] [CrossRef]
- Buckner, F.S.; Verlinde, C.L.M.J.; La Flamme, A.C.; Van Voorhis, W.C. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing β-galactosidase. Antimicrob. Agents Chemother. 1996, 40, 2592–2597. [Google Scholar] [CrossRef] [PubMed]
- Trager, W.; Jensen, J. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Insuasty, B.; Montoya, A.; Becerra, D.; Quiroga, J.; Abonia, R.; Robledo, S.; Vélez, I.D.; Upegui, Y.; Nogueras, M.; Cobo, J. Synthesis of novel analogs of 2-pyrazoline obtained from [(7-chloroquinolin-4-yl)amino]chalcones and hydrazine as potential antitumor and antimalarial agents. Eur. J. Med. Chem. 2013, 67, 252–262. [Google Scholar] [CrossRef]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2002, 49, 1049. [Google Scholar] [CrossRef]
- Oñate-Garzón, J.; Manrique-Moreno, M.; Trier, S.; Leidy, C.; Torres, R.; Patiño, E. Antimicrobial activity and interactions of cationic peptides derived from Galleria mellonella cecropin D-like peptide with model membranes. J. Antibiot. 2017, 70, 238–245. [Google Scholar] [CrossRef]
- Londoño-Mosquera, J.-D.; Aragón-Muriel, A.; Polo Cerón, D. Synthesis, antibacterial activity and DNA interactions of lanthanide(III) complexes of N(4)-substituted thiosemicarbazones. Univ. Sci. 2018, 23, 141–169. [Google Scholar] [CrossRef]
- Chen, H.L.; Su, P.Y.; Chang, Y.S.; Wu, S.Y.; Liao, Y.D.; Yu, H.M.; Lauderdale, T.L.; Chang, K.; Shih, C. Identification of a Novel Antimicrobial Peptide from Human Hepatitis B Virus Core Protein Arginine-Rich Domain (ARD). PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef]
- Saugar, J.M.; Rodríguez-Hernández, M.J.; De La Torre, B.G.; Pachón-Ibañez, M.E.; Fernández-Reyes, M.; Andreu, D.; Pachón, J.; Rivas, L. Activity of cecropin A-melittin hybrid peptides against colistin-resistant clinical strains of Acinetobacter baumannii: Molecular basis for the differential mechanisms of action. Antimicrob. Agents Chemother. 2006, 50, 1251–1256. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cytotoxicity | Antiparasitic Activity | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| U-937 Cell Line | Erythrocytes | Anti-L. braziliensis a | Anti-T. cruzi b | Anti-P. falciparum c | |||||||||
| LC50 | SD | LC50 | SD | EC50 | SD | SI | EC50 | SD | SI | EC50 | SD | SI | |
| L1 | 7.77 | 0.46 | >200 | NA | >4 | NA | <1.94 | 17.03 | 2.13 | 0.46 | 14.04 | 1.01z | >14.24 |
| La-L1 | 53.65 | 8.06 | >200 | NA | 106.09 | 61.07 | 0.51 | 62.24 | 8.25 | 0.86 | 13.80 | 1.06 | >14.49 |
| Ce-L1 | 44.64 | 10.68 | >200 | NA | >25 | NA | <1.78 | 178.99 | 28.94 | 0.25 | 15.16 | 0.26 | >13.19 |
| L2 | 17.09 | 4.74 | >200 | NA | >8 | NA | <2.13 | 14.97 | 0.05 | 1.14 | 15.57 | 0.92 | >12.84 |
| La-L2 | 18.17 | 3.94 | >200 | NA | 12.67 | 2.47 | 1.43 | 29.27 | 4.62 | 0.62 | 12.81 | 0.54 | >15.63 |
| Ce-L2 | 45.37 | 10.12 | >200 | NA | 26.17 | 1.46 | 1.73 | 56.39 | 4.18 | 0.80 | 13.73 | 0.63 | >14.56 |
| L3 | 20.40 | 5.62 | >200 | NA | >10 | NA | <2.04 | 30.53 | 2.72 | 0.67 | 15.54 | 0.99 | >12.87 |
| La-L3 | 28.83 | 6.55 | >200 | NA | >14 | NA | <2.05 | >15 | NA | <1.92 | 12.79 | 1.81 | >15.63 |
| Ce-L3 | 61.73 | 3.81 | >200 | NA | >30 | NA | <2.06 | 130.58 | 14.63 | 0.47 | 16.05 | 0.56 | >12.46 |
| L4 | 14.27 | 1.34 | >200 | NA | >7 | NA | <2.03 | 13.64 | 1.72 | 1.05 | 13.98 | 0.44 | >14.30 |
| La-L4 | 122.39 | 18.06 | >200 | NA | >50 | NA | <2.44 | >50 | NA | <2.44 | 18.35 | 0.49 | >10.89 |
| Ce-L4 | 41.77 | 7.41 | >200 | NA | >20 | NA | <2.09 | >20 | NA | <2.09 | 15.54 | 0.45 | >12.87 |
| Amphotericin B | 36.5 | 2.8 | ND | ND | 0.31 | 0.08 | 456.3 | ND | ND | ND | ND | ND | ND |
| Benznidazole | >200 | NA | ND | ND | ND | ND | ND | 11.89 | 3.30 | >16.8 | ND | ND | ND |
| Chloroquine | ND | ND | >200 | NA | ND | ND | ND | ND | ND | ND | 0.01 | 0.003 | >200 |
| Compound | Gram-Positive Strains | Gram-Negative Strains | ||
|---|---|---|---|---|
| S. aureus ATCC® 25923 | L. monocytogenes ATCC® 19115 | E. coli ATCC® 25922 | P. aeruginosa ATCC® 27583 | |
| L1 | 500 | 500 | 2000 | 2000 |
| La-L1 | 125 | 500 | 500 | 1000 |
| Ce-L1 | 250 | 500 | 500 | 1000 |
| L2 | 500 | 500 | 2000 | 2000 |
| La-L2 | 250 | 500 | 1000 | 1000 |
| Ce-L2 | 250 | 500 | 1000 | 1000 |
| L3 | 250 | 1000 | 500 | 2000 |
| La-L3 | 500 | 1000 | 500 | 2000 |
| Ce-L3 | 500 | 1000 | 1000 | 2000 |
| L4 | 250 | 1000 | 1000 | 2000 |
| La-L4 | 250 | 1000 | 2000 | 2000 |
| Ce-L4 | 250 | 1000 | 2000 | 2000 |
| CP a | 0.50 | 0.50 | <0.008 | >4.00 |
| ÂgNO3 b | <100 | <100 | <100 | <100 |
| MLV | Compound | Pretransition Temperature (°C) | Tm (°C) | DH (J·g−1) |
|---|---|---|---|---|
| DMPC | - | 12.94 | 23.02 | 1.48 |
| L1 | - | 22.39 | 0.82 | |
| La-L1 | - | 22.85 | 0.62 | |
| L2 | - | 22.94 | 1.05 | |
| La-L2 | - | 22.22 | 0.57 | |
| L3 | - | 22.22 | 0.54 | |
| La-L3 | - | 22.46 | 0.25 | |
| L4 | - | 22.53 | 0.70 | |
| La-L4 | - | 21.60 | 0.21 | |
| DMPC/DMPG (3:1) | - | 12.71 | 23.02 | 1.72 |
| L1 | - | 22.41 | 1.48 | |
| La-L1 | - | 23.55 | 0.61 | |
| L2 | - | 23.06 | 1.46 | |
| La-L2 | - | 22.58 | 0.51 | |
| L3 | - | 23.39 | 1.50 | |
| La-L3 | - | 21.64 | 1.31 | |
| L4 | - | 22.86 | 1.41 | |
| La-L4 | - | 22.47 | 1.38 |
| Compound | Band Followed (nm) | Estimated Constant (104 M−1) |
|---|---|---|
| L1 | 300 | 0.3 |
| L2 | 297 | 1.1 |
| L3 | 314 | 1.3 |
| L4 | 420 | 0.7 |
| La-L1 | 299 | 1.6 |
| La-L2 | 299 | 1.3 |
| La-L3 | 318 | 1.6 |
| La-L4 | 323 | 1.4 |
| Ce-L1 | 295 | 0.6 |
| Ce-L2 | 294 | 1.1 |
| Ce-L3 | 329 | 2.2 |
| Ce-L4 | 324 | 1.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aragón-Muriel, A.; Liscano, Y.; Upegui, Y.; Robledo, S.M.; Ramírez-Apan, M.T.; Morales-Morales, D.; Oñate-Garzón, J.; Polo-Cerón, D. In Vitro Evaluation of the Potential Pharmacological Activity and Molecular Targets of New Benzimidazole-Based Schiff Base Metal Complexes. Antibiotics 2021, 10, 728. https://doi.org/10.3390/antibiotics10060728
Aragón-Muriel A, Liscano Y, Upegui Y, Robledo SM, Ramírez-Apan MT, Morales-Morales D, Oñate-Garzón J, Polo-Cerón D. In Vitro Evaluation of the Potential Pharmacological Activity and Molecular Targets of New Benzimidazole-Based Schiff Base Metal Complexes. Antibiotics. 2021; 10(6):728. https://doi.org/10.3390/antibiotics10060728
Chicago/Turabian StyleAragón-Muriel, Alberto, Yamil Liscano, Yulieth Upegui, Sara M. Robledo, María Teresa Ramírez-Apan, David Morales-Morales, Jose Oñate-Garzón, and Dorian Polo-Cerón. 2021. "In Vitro Evaluation of the Potential Pharmacological Activity and Molecular Targets of New Benzimidazole-Based Schiff Base Metal Complexes" Antibiotics 10, no. 6: 728. https://doi.org/10.3390/antibiotics10060728
APA StyleAragón-Muriel, A., Liscano, Y., Upegui, Y., Robledo, S. M., Ramírez-Apan, M. T., Morales-Morales, D., Oñate-Garzón, J., & Polo-Cerón, D. (2021). In Vitro Evaluation of the Potential Pharmacological Activity and Molecular Targets of New Benzimidazole-Based Schiff Base Metal Complexes. Antibiotics, 10(6), 728. https://doi.org/10.3390/antibiotics10060728