
The Potential of Human Peptide LL-37 as an Antimicrobial and Anti-Biofilm Agent


Abstract
1. Introduction
2. Challenges Associated with Using LL-37 as an Antimicrobial Agent
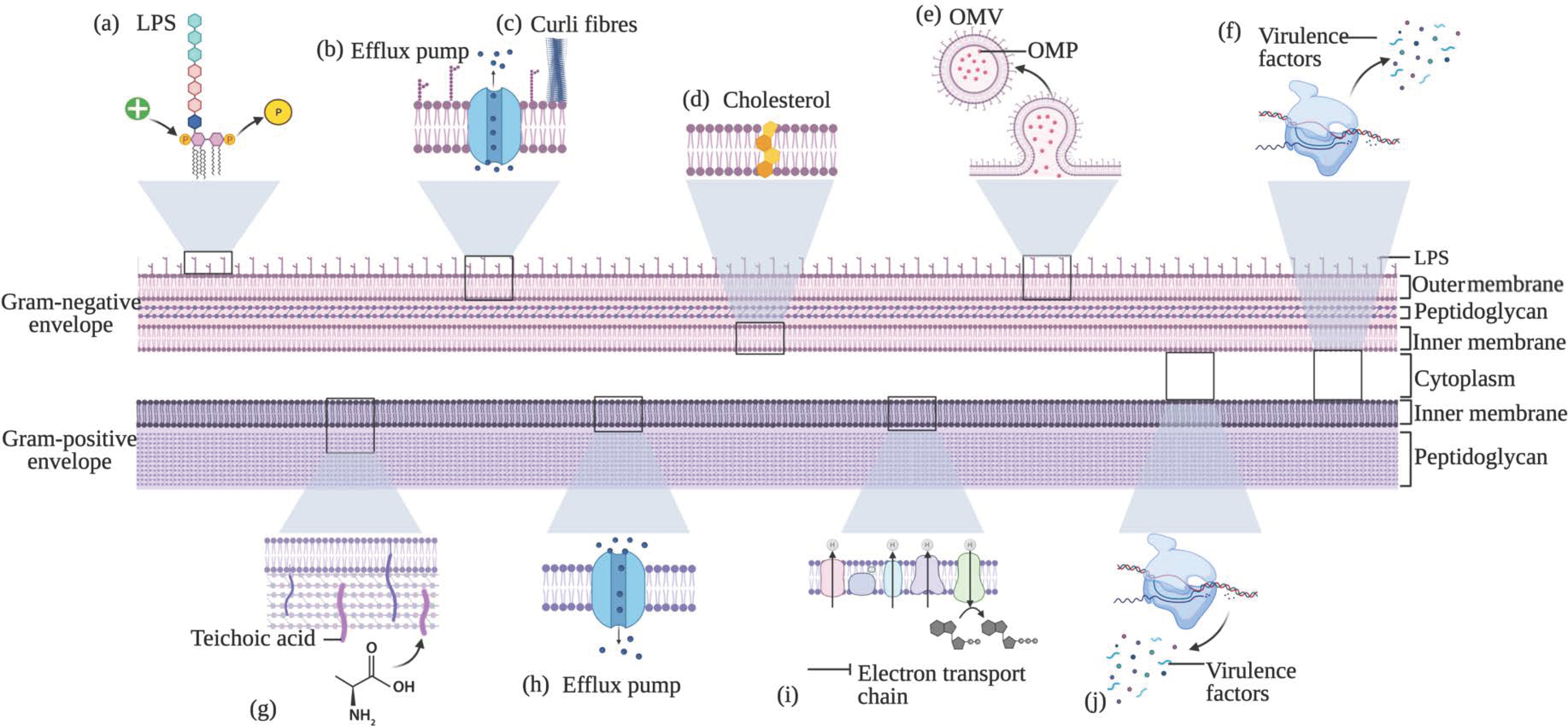
2.1. Resistance
2.1.1. Cell Membrane and Charge Modifications
2.1.2. Efflux Pumps
2.1.3. Incorporation of Exogenous Molecules into the Bacterial Membrane
2.1.4. Outer Membrane Proteins and Vesicles
2.1.5. Proteases
2.1.6. Regulation of Gene Expression, Virulence and Phenotypic Changes
2.1.7. Metabolic Changes
2.2. Cross-Resistance
2.2.1. Colistin
2.2.2. Polymyxin B
2.2.3. Chlorhexidine
2.3. Mutations
2.4. Biofilms
3. Methods to Improve LL-37 as a Therapeutic Agent
3.1. Immobilization Techniques and LL-37 Delivery Systems
3.2. LL-37 Derivatives
3.2.1. Sequence
3.2.2. Helicity
3.2.3. Hydrophobicity
3.2.4. Charge
3.2.5. Configuration
3.3. Synergy
3.3.1. Combinations with LL-37
Cell Wall Inhibitors
Nucleic Acid Synthesis Inhibitors
Protein Synthesis Inhibitors
Other Combinations
3.3.2. Synergy with LL-37 Derivatives
3.3.3. Synergy against Biofilms
3.3.4. Combinations That Enhance LL-37
4. Final Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- IACG. No Time to Wait: Infections from Drug-Resistant Securing the Future from Drug-Resistant Infections. 2019. Available online: https://www.who.int/docs/default-source/documents/no-time-to-wait-securing-the-future-from-drug-resistant-infections-en.pdf?sfvrsn=5b424d7_6 (accessed on 1 October 2020).
- Mobarki, N.; Almerabi, B.; Hattan, A. Antibiotic Resistance Crisis. Int. J. Med. Dev. Ctries. 2019, 40, 561–564. [Google Scholar] [CrossRef]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the host defense peptide landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Fotina, H.; Wang, L.; Hu, J. Antimicrobial peptides as novel alternatives to antibiotics. Sci. Messenger LNU Vet. Med. Biotechnol. 2020, 22. [Google Scholar] [CrossRef]
- Lei, J.; Sun, L.C.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q.Y. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919. [Google Scholar] [PubMed]
- Joo, H.S.; Fu, C.I.; Otto, M. Bacterial strategies of resistance to antimicrobial peptides. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150292. [Google Scholar] [CrossRef]
- Iacob, S.A.; Iacob, D.G. Antibacterial function of the human cathelicidin-18 peptide (LL-37) between theory and practice. Protein Pept. Lett. 2014, 21, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Bandurska, K.; Berdowska, A.; Barczyńska-Felusiak, R.; Krupa, P. Unique features of human cathelicidin LL-37. BioFactors 2015, 41, 289–300. [Google Scholar] [CrossRef]
- Malhotra, S.; Hayes, D.; Wozniak, D.J. Cystic fibrosis and pseudomonas aeruginosa: The host-microbe interface. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef]
- Wang, G. Human antimicrobial peptides and proteins. Pharmaceuticals 2014, 7, 545–594. [Google Scholar] [CrossRef]
- Neshani, A.; Zare, H.; Akbari Eidgahi, M.R.; Kamali Kakhki, R.; Safdari, H.; Khaledi, A.; Ghazvini, K. LL-37: Review of antimicrobial profile against sensitive and antibiotic-resistant human bacterial pathogens. Gene Rep. 2019, 17, 100519. [Google Scholar] [CrossRef]
- Bruce, K.E.; Rued, B.E.; Tsui, H.C.T.; Winkler, M.E. The Opp (AmiACDEF) oligopeptide transporter mediates resistance of serotype 2 Streptococcus pneumoniae D39 to killing by chemokine CXCL10 and other antimicrobial peptides. J. Bacteriol. 2018, 200. [Google Scholar] [CrossRef]
- Chung, M.C.; Dean, S.N.; Van Hoek, M.L. Acyl carrier protein is a bacterial cytoplasmic target of cationic antimicrobial peptide LL-37. Biochem. J. 2015, 470, 243–253. [Google Scholar] [CrossRef]
- Yang, B.; Good, D.; Mosaiab, T.; Liu, W.; Ni, G.; Kaur, J.; Liu, X.; Jessop, C.; Yang, L.; Fadhil, R.; et al. Significance of LL-37 on Immunomodulation and Disease Outcome. Biomed. Res. Int. 2020, 2020, 8349712. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Wuerth, K.; Hancock, R.E.W. Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Chem. Biol. 2013, 9, 761. [Google Scholar] [CrossRef]
- Van der Does, A.M.; Hiemstra, P.S.; Mookherjee, N. Antimicrobial host defence peptides: Immunomodulatory functions and translational prospects. Adv. Exp. Med. Biol. 2019, 1117, 149–171. [Google Scholar]
- Maria-Neto, S.; De Almeida, K.C.; Macedo, M.L.R.; Franco, O.L. Understanding bacterial resistance to antimicrobial peptides: From the surface to deep inside. Biochim. Biophys. Acta Biomembr. 2015, 1848, 3078–3088. [Google Scholar] [CrossRef]
- Kubicek-Sutherland, J.Z.; Lofton, H.; Vestergaard, M.; Hjort, K.; Ingmer, H.; Andersson, D.I. Antimicrobial peptide exposure selects for Staphylococcus aureus resistance to human defence peptides. J. Antimicrob. Chemother. 2017, 72, 115–127. [Google Scholar] [CrossRef]
- Kang, J.; Dietz, M.J.; Li, B. Antimicrobial peptide LL-37 is bactericidal against Staphylococcus aureus biofilms. PLoS ONE 2019, 14, e0216676. [Google Scholar] [CrossRef]
- McQuade, R.; Roxas, B.; Viswanathan, V.K.; Vedantam, G. Clostridium difficile clinical isolates exhibit variable susceptibility and proteome alterations upon exposure to mammalian cationic antimicrobial peptides. Anaerobe 2012, 18, 614–620. [Google Scholar] [CrossRef]
- Leszczyńska, K.; Namiot, D.; Byfield, F.J.; Cruz, K.; Zendzian-Piotrowska, M.; Fein, D.E.; Savage, P.B.; Diamond, S.; Mcculloch, C.A.; Janmey, P.A.; et al. Antibacterial activity of the human host defence peptide LL-37 and selected synthetic cationic lipids against bacteria associated with oral and upper respiratory tract infections. J. Antimicrob. Chemother. 2013, 68, 610–618. [Google Scholar] [CrossRef]
- Lofton, H.; Pränting, M.; Thulin, E.; Andersson, D.I. Mechanisms and Fitness Costs of Resistance to Antimicrobial Peptides LL-37, CNY100HL and Wheat Germ Histones. PLoS ONE 2013, 8, e68875. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.Y.; Otto, M. Do antimicrobial peptides and antimicrobial-peptide resistance play important roles during bacterial infection? Future Microbiol. 2018, 13, 1073–1075. [Google Scholar] [CrossRef]
- Brannon, J.R.; Thomassin, J.L.; Desloges, I.; Gruenheid, S.; Le Moual, H. Role of uropathogenic Escherichia coli OmpT in the resistance against human cathelicidin LL-37. FEMS Microbiol. Lett. 2013, 345, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Martynowycz, M.W.; Rice, A.; Andreev, K.; Nobre, T.M.; Kuzmenko, I.; Wereszczynski, J.; Gidalevitz, D. Salmonella Membrane Structural Remodeling Increases Resistance to Antimicrobial Peptide LL-37. ACS Infect. Dis. 2019, 5, 1214–1222. [Google Scholar] [CrossRef]
- Duperthuy, M.; Sjöström, A.E.; Sabharwal, D.; Damghani, F.; Uhlin, B.E.; Wai, S.N. Role of the Vibrio cholerae Matrix Protein Bap1 in Cross-Resistance to Antimicrobial Peptides. PLoS Pathog. 2013, 9, e1003620. [Google Scholar] [CrossRef]
- Uhlmann, J.; Rohde, M.; Siemens, N.; Kreikemeyer, B.; Bergman, P.; Johansson, L.; Norrby-Teglund, A. LL-37 triggers formation of streptococcus pyogenes extracellular vesicle-like structures with immune stimulatory properties. J. Innate Immun. 2016, 8, 243–257. [Google Scholar] [CrossRef]
- Thwaite, J.E.; Hibbs, S.; Titball, R.W.; Atkins, T.P. Proteolytic degradation of human antimicrobial peptide LL-37 by Bacillus anthracis may contribute to virulence. Antimicrob. Agents Chemother. 2006, 50, 2316–2322. [Google Scholar] [CrossRef]
- Khan, A.; Davlieva, M.; Panesso, D.; Rincon, S.; Miller, W.R.; Diaz, L.; Reyes, J.; Cruz, M.R.; Pemberton, O.; Nguyen, A.H.; et al. Antimicrobial sensing coupled with cell membrane remodeling mediates antibiotic resistance and virulence in Enterococcus faecalis. Proc. Natl. Acad. Sci. USA 2019, 116, 26925–26932. [Google Scholar] [CrossRef]
- Zhu, Y.; Mohapatra, S.; Weisshaar, J.C. Rigidification of the Escherichia coli cytoplasm by the human antimicrobial peptide LL-37 revealed by superresolution fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2019, 116, 1017–1026. [Google Scholar] [CrossRef]
- LaRock, C.N.; Nizet, V. Cationic antimicrobial peptide resistance mechanisms of streptococcal pathogens. Biochim. Biophys. Acta Biomembr. 2015, 1848, 3047–3054. [Google Scholar] [CrossRef]
- Golla, R.M.; Mishra, B.; Dang, X.; Lakshmaiah Narayana, J.; Li, A.; Xu, L.; Wang, G. Resistome of Staphylococcus aureus in Response to Human Cathelicidin LL-37 and Its Engineered Antimicrobial Peptides. ACS Infect. Dis. 2020, 6, 1866–1881. [Google Scholar] [CrossRef] [PubMed]
- Motamedi, N.; Danelishvili, L.; Bermudez, L.E. Identification of Mycobacterium avium genes associated with resistance to host antimicrobial peptides. J. Med. Microbiol. 2014, 63, 923. [Google Scholar] [CrossRef]
- Mücke, P.A.; Maaß, S.; Kohler, T.P.; Hammerschmidt, S.; Becher, D. Proteomic adaptation of streptococcus pneumoniae to the human antimicrobial peptide LL-37. Microorganisms 2020, 8, 413. [Google Scholar] [CrossRef] [PubMed]
- Papasergi, S.; Brega, S.; Mistou, M.Y.; Firon, A.; Oxaran, V.; Dover, R.; Teti, G.; Shai, Y.; Trieu-Cuot, P.; Dramsi, S. The gbs pi-2a pilus is required for virulence in mice neonates. PLoS ONE 2011, 6, e18747. [Google Scholar] [CrossRef]
- Mazda, Y.; Kawada-Matsuo, M.; Kanbara, K.; Oogai, Y.; Shibata, Y.; Yamashita, Y.; Miyawaki, S.; Komatsuzawa, H. Association of CiaRH with resistance of Streptococcus mutans to antimicrobial peptides in biofilms. Mol. Oral Microbiol. 2012, 27, 124–135. [Google Scholar] [CrossRef]
- Koppen, B.C.; Mulder, P.P.G.; de Boer, L.; Riool, M.; Drijfhout, J.W.; Zaat, S.A.J. Synergistic microbicidal effect of cationic antimicrobial peptides and teicoplanin against planktonic and biofilm-encased Staphylococcus aureus. Int. J. Antimicrob. Agents 2019, 53, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Van Sorge, N.M.; Cole, J.N.; Kuipers, K.; Henningham, A.; Aziz, R.K.; Kasirer-Friede, A.; Lin, L.; Berends, E.T.M.; Davies, M.R.; Dougan, G.; et al. The classical lancefield antigen of group A Streptococcus is a virulence determinant with implications for vaccine design. Cell Host Microbe 2014, 15, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Cullen, T.W.; Giles, D.K.; Wolf, L.N.; Ecobichon, C.; Boneca, I.G.; Trent, M.S. Helicobacter pylori versus the host: Remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog. 2011, 7, e1002454. [Google Scholar] [CrossRef]
- Gooderham, W.J.; Gellatly, S.L.; Sanschagrin, F.; McPhee, J.B.; Bains, M.; Cosseau, C.; Levesque, R.C.; Hancock, R.E.W. The sensor kinase PhoQ mediates virulence in Pseudomonas aeruginosa. Microbiology 2009, 155, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.R.; Hancock, R.E.W.; Fernandez, R.C. Bordetella pertussis lipid a glucosamine modification confers resistance to cationic antimicrobial peptides and increases resistance to outer membrane perturbation. Antimicrob. Agents Chemother. 2014, 58, 4931–4934. [Google Scholar] [CrossRef]
- De Majumdar, S.; Yu, J.; Fookes, M.; McAteer, S.P.; Llobet, E.; Finn, S.; Spence, S.; Monahan, A.; Kissenpfennig, A.; Ingram, R.J.; et al. Elucidation of the RamA Regulon in Klebsiella pneumoniae Reveals a Role in LPS Regulation. PLoS Pathog. 2015, 11, e1004627. [Google Scholar] [CrossRef]
- Bociek, K.; Ferluga, S.; Mardirossian, M.; Benincasa, M.; Tossi, A.; Gennaro, R.; Scocchi, M. Lipopolysaccharide phosphorylation by the WaaY kinase affects the susceptibility of Escherichia coli to the human antimicrobial peptide LL-37. J. Biol. Chem. 2015, 290, 19933–19941. [Google Scholar] [CrossRef]
- Trombley, M.P.; Post, D.M.B.; Rinker, S.D.; Reinders, L.M.; Fortney, K.R.; Zwickl, B.W.; Janowicz, D.M.; Baye, F.M.; Katz, B.P.; Spinola, S.M.; et al. Phosphoethanolamine transferase LptA in haemophilus ducreyi modifies lipid a and contributes to human defensin resistance in vitro. PLoS ONE 2015, 10, e124373. [Google Scholar] [CrossRef]
- Webber, M.A.; Piddock, L.J.V. The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 2003, 51, 9–11. [Google Scholar] [CrossRef]
- Peschel, A.; Sahl, H.G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 2006, 4, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Staroń, A.; Finkeisen, D.E.; Mascher, T. Peptide antibiotic sensing and detoxification modules of Bacillus subtilis. Antimicrob. Agents Chemother. 2011, 55, 515–525. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, M.; Zhou, H.; Li, C.; Luk, A.; Zhao, G.P.; Fung, K.; Ip, M. Role of two-component system response regulator bceR in the antimicrobial resistance, virulence, biofilm formation, and stress response of Group B Streptococcus. Front. Microbiol. 2019, 10, 10. [Google Scholar] [CrossRef]
- Tian, X.L.; Salim, H.; Dong, G.; Parcells, M.; Li, Y.H. The BceABRS four-component system that is essential for cell envelope stress response is involved in sensing and response to host defence peptides and is required for the biofilm formation and fitness of streptococcus mutans. J. Med. Microbiol. 2018, 67, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Mount, K.L.B.; Townsend, C.A.; Rinker, S.D.; Gu, X.; Fortney, K.R.; Zwickl, B.W.; Janowicz, D.M.; Spinola, S.M.; Katz, B.P.; Bauer, M.E. Haemophilus ducreyi SapA contributes to cathelicidin resistance and virulence in humans. Infect. Immun. 2010, 78, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Rinker, S.D.; Gu, X.; Fortney, K.R.; Zwickl, B.W.; Katz, B.P.; Janowicz, D.M.; Spinola, S.M.; Bauer, M.E. Permeases of the sap transporter are required for cathelicidin resistance and virulence of haemophilus ducreyi in humans. J. Infect. Dis. 2012, 206, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Rinker, S.D.; Trombley, M.P.; Gu, X.; Fortney, K.R.; Bauer, M.E. Deletion of mtrC in Haemophilus ducreyi increases sensitivity to human antimicrobial peptides and activates the CpxRA regulon. Infect. Immun. 2011, 79, 2324–2334. [Google Scholar] [CrossRef] [PubMed]
- Shelton, C.L.; Raffel, F.K.; Beatty, W.L.; Johnson, S.M.; Mason, K.M. Sap transporter mediated import and subsequent degradation of antimicrobial peptides in Haemophilus. PLoS Pathog. 2011, 7, e1002360. [Google Scholar] [CrossRef]
- Blodkamp, S.; Kadlec, K.; Gutsmann, T.; Quiblier, C.; Naim, H.Y.; Schwarz, S.; von Köckritz-Blickwede, M. Effects of SecDF on the antimicrobial functions of cathelicidins against Staphylococcus aureus. Vet. Microbiol. 2017, 200, 52–58. [Google Scholar] [CrossRef]
- Geörg, M.; Maudsdotter, L.; Tavares, R.; Jonsson, A.B. Meningococcal resistance to antimicrobial peptides is mediated by bacterial adhesion and host cell RhoA and Cdc42 signalling. Cell. Microbiol. 2013, 15, 1938–1954. [Google Scholar] [CrossRef]
- Zähner, D.; Zhou, X.; Chancey, S.T.; Pohl, J.; Shafer, W.M.; Stephens, D.S. Human antimicrobial peptide LL-37 induces MefE/Mel-mediated macrolide resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2010, 54, 3516–3519. [Google Scholar] [CrossRef]
- McGee, D.J.; George, A.E.; Trainor, E.A.; Horton, K.E.; Hildebrandt, E.; Testerman, T.L. Cholesterol enhances Helicobacter pylori resistance to antibiotics and LL-37. Antimicrob. Agents Chemother. 2011, 55, 2897–2904. [Google Scholar] [CrossRef] [PubMed]
- Goytia, M.; Shafer, W.M. Polyamines can increase resistance of Neisseria gonorrhoeae to mediators of the innate human host defense. Infect. Immun. 2010, 78, 3187–3195. [Google Scholar] [CrossRef]
- Rompikuntal, P.K.; Vdovikova, S.; Duperthuy, M.; Johnson, T.L.; Åhlund, M.; Lundmark, R.; Oscarsson, J.; Sandkvist, M.; Uhlin, B.E.; Wai, S.N. Outer membrane vesicle-mediated export of processed PrtV protease from Vibrio cholerae. PLoS ONE 2015, 10, e134098. [Google Scholar] [CrossRef] [PubMed]
- Urashima, A.; Sanou, A.; Yen, H.; Tobe, T. Enterohaemorrhagic Escherichia coli produces outer membrane vesicles as an active defence system against antimicrobial peptide LL-37. Cell. Microbiol. 2017, 19, e12758. [Google Scholar] [CrossRef]
- Jan, A.T. Outer Membrane Vesicles (OMVs) of gram-negative bacteria: A perspective update. Front. Microbiol. 2017, 8, 1053. [Google Scholar] [CrossRef]
- Lin, M.F.; Tsai, P.W.; Chen, J.Y.; Lin, Y.Y.; Lan, C.Y. OmpA binding mediates the effect of antimicrobial peptide LL-37 on Acinetobacter baumannii. PLoS ONE 2015, 10, e141107. [Google Scholar] [CrossRef]
- Horie, T.; Inomata, M.; Into, T. OmpA-Like Proteins of Porphyromonas gingivalis Mediate Resistance to the Antimicrobial Peptide LL-37. J. Pathog. 2018, 2018, 2068435. [Google Scholar] [CrossRef]
- Thomassin, J.L.; Brannon, J.R.; Gibbs, B.F.; Gruenheid, S.; Le Moual, H. OmpT outer membrane proteases of enterohemorrhagic and enteropathogenic Escherichia coli contribute differently to the degradation of human LL-37. Infect. Immun. 2012, 80, 483–492. [Google Scholar] [CrossRef]
- Desloges, I.; Taylor, J.A.; Leclerc, J.M.; Brannon, J.R.; Portt, A.; Spencer, J.D.; Dewar, K.; Marczynski, G.T.; Manges, A.; Gruenheid, S.; et al. Identification and characterization of OmpT-like proteases in uropathogenic Escherichia coli clinical isolates. Microbiologyopen 2019, 8, e915. [Google Scholar] [CrossRef] [PubMed]
- McPhee, J.B.; Small, C.L.; Reid-Yu, S.A.; Brannon, J.R.; Le Moual, H.; Coombes, B.K. Host defense peptide resistance contributes to colonization and maximal intestinal pathology by Crohn’s disease-associated adherent-invasive Escherichia coli. Infect. Immun. 2014, 82, 3383–3393. [Google Scholar] [CrossRef]
- Chang, T.W.; Lin, Y.M.; Wang, C.F.; Liaos, Y. Di Outer membrane lipoprotein Lpp is gram-negative bacterial cell surface receptor for cationic antimicrobial peptides. J. Biol. Chem. 2012, 287, 418–428. [Google Scholar] [CrossRef]
- Lin, Y.M.; Wu, S.J.; Chang, T.W.; Wang, C.F.; Suen, C.S.; Hwang, M.J.; Chang, M.D.T.; Chen, Y.T.; Liao, Y. Di Outer membrane protein I of pseudomonas aeruginosa is a target of cationic antimicrobial peptide/protein. J. Biol. Chem. 2010, 285, 8985–8994. [Google Scholar] [CrossRef]
- Culp, E.; Wright, G.D. Bacterial proteases, untapped antimicrobial drug targets. J. Antibiot. 2017, 70, 366–377. [Google Scholar] [CrossRef]
- Hollands, A.; Gonzalez, D.; Leire, E.; Donald, C.; Gallo, R.L.; Sanderson-Smith, M.; Dorrestein, P.C.; Nizet, V. A bacterial pathogen co-opts host plasmin to resist killing by cathelicidin antimicrobial peptides. J. Biol. Chem. 2012, 287, 40891–40897. [Google Scholar] [CrossRef]
- Kany, A.M.; Sikandar, A.; Yahiaoui, S.; Haupenthal, J.; Walter, I.; Empting, M.; Köhnke, J.; Hartmann, R.W. Tackling Pseudomonas aeruginosa Virulence by a Hydroxamic Acid-Based LasB Inhibitor. ACS Chem. Biol. 2018, 13, 2449–2455. [Google Scholar] [CrossRef] [PubMed]
- Koziel, J.; Karim, A.Y.; Przybyszewska, K.; Ksiazek, M.; Rapala-Kozik, M.; Nguyen, K.A.; Potempa, J. Proteolytic inactivation of LL-37 by karilysin, a novel virulence mechanism of Tannerella forsythia. J. Innate Immun. 2010, 2, 288–293. [Google Scholar] [CrossRef]
- Claunch, K.M.; Bush, M.; Evans, C.R.; Malmquist, J.A.; Hale, M.C.; McGillivray, S.M. Transcriptional profiling of the clpX mutant in Bacillus anthracis reveals regulatory connection with the lrgAB operon. Microbiology 2018, 164, 659–669. [Google Scholar] [CrossRef]
- Sonesson, A.; Przybyszewska, K.; Eriksson, S.; Mörgelin, M.; Kjellström, S.; Davies, J.; Potempa, J.; Schmidtchen, A. Identification of bacterial biofilm and the Staphylococcus aureus derived protease, staphopain, on the skin surface of patients with atopic dermatitis. Sci. Rep. 2017, 7, 8689. [Google Scholar] [CrossRef]
- Strempe, N.; Neidig, A.; Nusser, M.; Geffers, R.; Vieillard, J.; Lesouhaitier, O.; Brenner-Weiss, G.; Overhage, J. Human host defense peptide LL-37 Stimulates virulence factor production and adaptive resistance in Pseudomonas aeruginosa. PLoS ONE 2013, 8, e82240. [Google Scholar] [CrossRef]
- Velarde, J.J.; Ashbaugh, M.; Wessels, M.R. The human antimicrobial peptide LL-37 binds directly to CsrS, a sensor histidine kinase of group a streptococcus, to activate expression of virulence factors. J. Biol. Chem. 2014, 289, 36315–36324. [Google Scholar] [CrossRef]
- Tran-Winkler, H.J.; Love, J.F.; Gryllos, I.; Wessels, M.R. Signal transduction through CsrRS confers an invasive phenotype in group a Streptococcus. PLoS Pathog. 2011, 7, e1002361. [Google Scholar] [CrossRef] [PubMed]
- Wessels, M.R.; Love, J.F.; Tran-Winkler, H.J. Vitamin D and the human antimicrobial peptide LL-37 enhance group a streptococcus resistance to killing by human cells. MBio 2012, 3. [Google Scholar] [CrossRef]
- Cole, J.N.; Pence, M.A.; von Köckritz-Blickwede, M.; Hollands, A.; Gallo, R.L.; Walker, M.J.; Nizeta, V. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. MBio 2010, 1. [Google Scholar] [CrossRef]
- Mairpady Shambat, S.; Siemens, N.; Monk, I.R.; Mohan, D.B.; Mukundan, S.; Krishnan, K.C.; Prabhakara, S.; Snäll, J.; Kearns, A.; Vandenesch, F.; et al. A point mutation in AgrC determines cytotoxic or colonizing properties associated with phenotypic variants of ST22 MRSA strains. Sci. Rep. 2016, 6, 31360. [Google Scholar] [CrossRef]
- LaRock, C.N.; Döhrmann, S.; Todd, J.; Corriden, R.; Olson, J.; Johannssen, T.; Lepenies, B.; Gallo, R.L.; Ghosh, P.; Nizet, V. Group A Streptococcal M1 Protein Sequesters Cathelicidin to Evade Innate Immune Killing. Cell Host Microbe 2015, 18, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Tandhavanant, S.; Thanwisai, A.; Limmathurotsakul, D.; Korbsrisate, S.; Day, N.P.; Peacock, S.J.; Chantratita, N. Effect of colony morphology variation of Burkholderia pseudomallei on intracellular survival and resistance to antimicrobial environments in human macrophages in vitro. BMC Microbiol. 2010, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Wuersching, S.N.; Huth, K.C.; Hickel, R.; Kollmuss, M. Inhibitory effect of LL-37 and human lactoferricin on growth and biofilm formation of anaerobes associated with oral diseases. Anaerobe 2021, 67, 102301. [Google Scholar] [CrossRef]
- Eini, A.; Sol, A.; Coppenhagen-Glazer, S.; Skvirsky, Y.; Zini, A.; Bachrach, G. Oxygen deprivation affects the antimicrobial action of LL-37 as determined by microplate real-time kinetic measurements under anaerobic conditions. Anaerobe 2013, 22, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Al-Farsi, H.M.; Al-Adwani, S.; Ahmed, S.; Vogt, C.; Ambikan, A.T.; Leber, A.; Al-Jardani, A.; Al-Azri, S.; Al-Muharmi, Z.; Toprak, M.S.; et al. Effects of the Antimicrobial Peptide LL-37 and Innate Effector Mechanisms in Colistin-Resistant Klebsiella pneumoniae with mgrB Insertions. Front. Microbiol. 2019, 10, 2632. [Google Scholar] [CrossRef]
- Crémieux, A.C.; Dinh, A.; Nordmann, P.; Mouton, W.; Tattevin, P.; Ghout, I.; Jayol, A.; Aimer, O.; Gatin, L.; Verdier, M.C.; et al. Efficacy of colistin alone and in various combinations for the treatment of experimental osteomyelitis due to carbapenemase-producing Klebsiella pneumoniae. J. Antimicrob. Chemother. 2019, 74, 2666–2675. [Google Scholar] [CrossRef]
- Napier, B.A.; Burd, E.M.; Satola, S.W.; Cagle, S.M.; Ray, S.M.; Mcgann, P.; Pohl, J.; Lesho, E.P.; Weiss, D.S. Clinical use of colistin induces cross-resistance to host antimicrobials in Acinetobacter baumannii. MBio 2013, 4. [Google Scholar] [CrossRef]
- Tzeng, Y.L.; Berman, Z.; Toh, E.; Bazan, J.A.; Turner, A.N.; Retchless, A.C.; Wang, X.; Nelson, D.E.; Stephens, D.S. Heteroresistance to the model antimicrobial peptide polymyxin B in the emerging Neisseria meningitidis lineage 11.2 urethritis clade: Mutations in the pilMNOPQ operon. Mol. Microbiol. 2019, 111, 254–268. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist. Updates 2016, 26, 43–57. [Google Scholar] [CrossRef]
- Fleitas, O.; Franco, O.L. Induced bacterial cross-resistance toward host antimicrobial peptides: A worrying phenomenon. Front. Microbiol. 2016, 7, 381. [Google Scholar] [CrossRef] [PubMed]
- Jayol, A.; Poirel, L.; Brink, A.; Villegas, M.V.; Yilmaz, M.; Nordmann, P. Resistance to colistin associated with a single amino acid change in protein PmrB among Klebsiella pneumoniae isolates of worldwide origin. Antimicrob. Agents Chemother. 2014, 58, 4762–4766. [Google Scholar] [CrossRef]
- García-Quintanilla, M.; Pulido, M.R.; Moreno-Martínez, P.; Martín-Peña, R.; López-Rojas, R.; Pachón, J.; McConnell, M.J. Activity of host antimicrobials against multidrug-resistant acinetobacter baumannii acquiring colistin resistance through loss of lipopolysaccharide. Antimicrob. Agents Chemother. 2014, 58, 2972–2975. [Google Scholar] [CrossRef]
- Dobias, J.; Poirel, L.; Nordmann, P. Cross-resistance to human cationic antimicrobial peptides and to polymyxins mediated by the plasmid-encoded MCR-1? Clin. Microbiol. Infect. 2017, 23, 676.e1–676.e5. [Google Scholar] [CrossRef] [PubMed]
- Poirel, L.; Jayol, A.; Nordmanna, P. Polymyxins: Antibacterial activity, susceptibility testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef] [PubMed]
- Wand, M.E.; Bock, L.J.; Bonney, L.C.; Sutton, J.M. Mechanisms of increased resistance to chlorhexidine and cross-resistance to colistin following exposure of Klebsiella pneumoniae clinical isolates to chlorhexidine. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Hashemi, M.M.; Holden, B.S.; Coburn, J.; Taylor, M.F.; Weber, S.; Hilton, B.; Zaugg, A.L.; McEwan, C.; Carson, R.; Andersen, J.L.; et al. Proteomic analysis of resistance of gram-negative bacteria to chlorhexidine and impacts on susceptibility to colistin, antimicrobial peptides, and ceragenins. Front. Microbiol. 2019, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rojas, A.; Makarova, O.; Müller, U.; Rolff, J. Cationic Peptides Facilitate Iron-induced Mutagenesis in Bacteria. PLoS Genet. 2015, 11, 5546. [Google Scholar] [CrossRef]
- Limoli, D.H.; Wozniak, D.J. Mutagenesis by host antimicrobial peptides: Insights into microbial evolution during chronic infections. Microb. Cell 2014, 1, 247. [Google Scholar] [CrossRef]
- Limoli, D.H.; Rockel, A.B.; Host, K.M.; Jha, A.; Kopp, B.T.; Hollis, T.; Wozniak, D.J. Cationic Antimicrobial Peptides Promote Microbial Mutagenesis and Pathoadaptation in Chronic Infections. PLoS Pathog. 2014, 10, e1004083. [Google Scholar] [CrossRef]
- Malhotra, S.; Limoli, D.H.; English, A.E.; Parsek, M.R.; Wozniak, D.J. Mixed communities of mucoid and nonmucoid Pseudomonas aeruginosa exhibit enhanced resistance to host antimicrobials. MBio 2018, 9. [Google Scholar] [CrossRef]
- Pence, M.A.; Haste, N.M.; Meharena, H.S.; Olson, J.; Gallo, R.L.; Nizet, V.; Kristian, S.A. Beta-lactamase repressor blai modulates staphylococcus aureus cathelicidin antimicrobial peptide resistance and virulence. PLoS ONE 2015, 10, e136605. [Google Scholar] [CrossRef]
- Matson, J.S.; Yoo, H.J.; Hakansson, K.; Dirita, V.J. Polymyxin B resistance in el tor vibrio cholerae requires lipid acylation catalyzed by MsbB. J. Bacteriol. 2010, 192, 2044–2052. [Google Scholar] [CrossRef] [PubMed]
- Nhu, N.T.K.; Phan, M.D.; Peters, K.M.; Lo, A.W.; Forde, B.M.; Chong, T.M.; Yin, W.F.; Chan, K.G.; Chromek, M.; Brauner, A.; et al. Discovery of new genes involved in curli production by a uropathogenic Escherichia coli strain from the highly virulent O45:K1:H7 lineage. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Woods, E.C.; Edwards, A.N.; Childress, K.O.; Jones, J.B.; McBride, S.M. The C. difficile clnRAB operon initiates adaptations to the host environment in response to LL-37. PLoS Pathog. 2018, 14, e1007153. [Google Scholar] [CrossRef] [PubMed]
- Stohl, E.A.; Dale, E.M.; Criss, A.K.; Seifert, H.S. Neisseria gonorrhoeae metalloprotease NGO1686 is required for full piliation, and piliation is required for resistance to H2O2- and neutrophil-mediated killing. MBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Oguri, T.; Yeo, W.S.; Bae, T.; Lee, H. Identification of envC and its cognate amidases as novel determinants of intrinsic resistance to Cationic antimicrobial peptides. Antimicrob. Agents Chemother. 2016, 60, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Munguia, J.; LaRock, D.L.; Tsunemoto, H.; Olson, J.; Cornax, I.; Pogliano, J.; Nizet, V. The Mla pathway is critical for Pseudomonas aeruginosa resistance to outer membrane permeabilization and host innate immune clearance. J. Mol. Med. 2017, 95, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; De Sessions, P.F.; Teoh, G.H.K.; Mohamed, A.N.N.; Zhu, Y.O.; Koh, V.H.Q.; Ang, M.L.T.; Dedon, P.C.; Hibberd, M.L.; Alonsoa, S. Transcriptional profiling of Mycobacterium tuberculosis exposed to in vitro lysosomal stress. Infect. Immun. 2016, 84, 2505–2523. [Google Scholar] [CrossRef]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial biofilm and associated infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef]
- Duplantier, A.J.; van Hoek, M.L. The human cathelicidin antimicrobial peptide LL-37 as a potential treatment for polymicrobial infected wounds. Front. Immunol. 2013, 4, 143. [Google Scholar] [CrossRef]
- Kanthawong, S.; Bolscher, J.G.M.; Veerman, E.C.I.; Van Marle, J.; De Soet, H.J.J.; Nazmi, K.; Wongratanacheewin, S.; Taweechaisupapong, S. Antimicrobial and antibiofilm activity of LL-37 and its truncated variants against Burkholderia pseudomallei. Int. J. Antimicrob. Agents 2012, 39, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Wnorowska, U.; Piktel, E.; Durnaś, B.; Fiedoruk, K.; Savage, P.B.; Bucki, R. Use of ceragenins as a potential treatment for urinary tract infections. BMC Infect. Dis. 2019, 19, 369. [Google Scholar] [CrossRef]
- Kai-Larsen, Y.; Lüthje, P.; Chromek, M.; Peters, V.; Wang, X.; Holm, Å.; Kádas, L.; Hedlund, K.O.; Johansson, J.; Chapman, M.R.; et al. Uropathogenic Escherichia coli modulates immune responses and its curli fimbriae interact with the antimicrobial peptide LL-37. PLoS Pathog. 2010, 6, e1001010. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Y.; Wu, Y.; Cui, Y.; Liu, Y.; Shi, X.; Zhang, Q.; Chen, Q.; Sun, Q.; Hu, Q. Phenotypic and genetic changes in the life cycle of small colony variants of Salmonella enterica serotype Typhimurium induced by streptomycin. Ann. Clin. Microbiol. Antimicrob. 2016, 15, 37. [Google Scholar] [CrossRef]
- De Breij, A.; Riool, M.; Kwakman, P.H.S.; De Boer, L.; Cordfunke, R.A.; Drijfhout, J.W.; Cohen, O.; Emanuel, N.; Zaat, S.A.J.; Nibbering, P.H.; et al. Prevention of Staphylococcus aureus biomaterial-associated infections using a polymer-lipid coating containing the antimicrobial peptide OP-145. J. Control. Release 2016, 222, 1–8. [Google Scholar] [CrossRef]
- Pestrak, M.J.; Chaney, S.B.; Eggleston, H.C.; Dellos-Nolan, S.; Dixit, S.; Mathew-Steiner, S.S.; Roy, S.; Parsek, M.R.; Sen, C.K.; Wozniak, D.J. Pseudomonas aeruginosa rugose small-colony variants evade host clearance, are hyper-inflammatory, and persist in multiple host environments. PLoS Pathog. 2018, 14, e1006842. [Google Scholar] [CrossRef]
- Lin, Q.; Deslouches, B.; Montelaro, R.C.; Di, Y.P. Prevention of ESKAPE pathogen biofilm formation by antimicrobial peptides WLBU2 and LL37. Int. J. Antimicrob. Agents 2018, 52, 667–672. [Google Scholar] [CrossRef]
- Amer, L.S.; Bishop, B.M.; van Hoek, M.L. Antimicrobial and antibiofilm activity of cathelicidins and short, synthetic peptides against Francisella. Biochem. Biophys. Res. Commun. 2010, 396. [Google Scholar] [CrossRef]
- Xie, F.; Zan, Y.; Zhang, X.; Zhang, H.; Jin, M.; Zhang, W.; Zhang, Y.; Liu, S. Differential abilities of Mammalian cathelicidins to inhibit bacterial biofilm formation and promote multifaceted immune functions of neutrophils. Int. J. Mol. Sci. 2020, 21, 1871. [Google Scholar] [CrossRef]
- Dean, S.N.; Bishop, B.M.; van Hoek, M.L. Natural and synthetic cathelicidin peptides with anti-microbial and anti-biofilm activity against Staphylococcus aureus. BMC Microbiol. 2011, 11, 114. [Google Scholar] [CrossRef]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.W.; Rehm, B.H.A.; Hancock, R.E.W. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–7182. [Google Scholar] [CrossRef]
- Sol, A.; Ginesin, O.; Chaushu, S.; Karra, L.; Coppenhagen-Glazer, S.; Ginsburg, I.; Bachracha, G. LL-37 Opsonizes and inhibits biofilm formation of aggregatibacter actinomycetemcomitans at subbactericidal concentrations. Infect. Immun. 2013, 81, 3577–3585. [Google Scholar] [CrossRef]
- Dean, S.N.; Bishop, B.M.; Van Hoek, M.L. Susceptibility of Pseudomonas aeruginosa biofilm to alpha-helical peptides: D-enantiomer of LL-37. Front. Microbiol. 2011, 2, 128. [Google Scholar] [CrossRef]
- Hell, É.; Giske, C.G.; Nelson, A.; Römling, U.; Marchini, G. Human cathelicidin peptide LL37 inhibits both attachment capability and biofilm formation of Staphylococcus epidermidis. Lett. Appl. Microbiol. 2010, 50, 211–215. [Google Scholar] [CrossRef]
- Dosler, S.; Karaaslan, E. Inhibition and destruction of Pseudomonas aeruginosa biofilms by antibiotics and antimicrobial peptides. Peptides 2014, 62, 32–37. [Google Scholar] [CrossRef]
- Blower, R.J.; Barksdale, S.M.; van Hoek, M.L. Snake cathelicidin NA-CATH and smaller helical antimicrobial peptides are effective against burkholderia thailandensis. PLoS Negl. Trop. Dis. 2015, 9, e3862. [Google Scholar] [CrossRef]
- Dean, S.N.; Walsh, C.; Goodman, H.; Van Hoek, M.L. Analysis of mixed biofilm (Staphylococcus aureus and Pseudomonas aeruginosa) by laser ablation electrospray ionization mass spectrometry. Biofouling 2015, 31. [Google Scholar] [CrossRef]
- Wang, G.; Narayana, J.L.; Mishra, B.; Zhang, Y.; Wang, F.; Wang, C.; Zarena, D.; Lushnikova, T.; Wang, X. Design of antimicrobial peptides: Progress made with human cathelicidin LL-37. Adv. Exp. Med. Biol. 2019, 1117, 215–240. [Google Scholar]
- Jaśkiewicz, M.; Neubauer, D.; Kazor, K.; Bartoszewska, S.; Kamysz, W. Antimicrobial Activity of Selected Antimicrobial Peptides Against Planktonic Culture and Biofilm of Acinetobacter baumannii. Probiotics Antimicrob. Proteins 2019, 11, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Song, D.W.; Kim, S.H.; Kim, H.H.; Lee, K.H.; Ki, C.S.; Park, Y.H. Multi-biofunction of antimicrobial peptide-immobilized silk fibroin nanofiber membrane: Implications for wound healing. Acta Biomater. 2016, 39, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Wang, G. Titanium surfaces immobilized with the major antimicrobial fragment FK-16 of human cathelicidin LL-37 are potent against multiple antibiotic-resistant bacteria. Biofouling 2017, 33, 544–555. [Google Scholar] [CrossRef]
- Sorrentino, I.; Gargano, M.; Ricciardelli, A.; Parrilli, E.; Buonocore, C.; de Pascale, D.; Giardina, P.; Piscitelli, A. Development of anti-bacterial surfaces using a hydrophobin chimeric protein. Int. J. Biol. Macromol. 2020, 164, 2293–2300. [Google Scholar] [CrossRef]
- Mishra, B.; Wang, G. Individual and combined effects of engineered peptides and antibiotics on Pseudomonas aeruginosa biofilms. Pharmaceuticals 2017, 10, 58. [Google Scholar] [CrossRef]
- De Breij, A.; Riool, M.; Cordfunke, R.A.; Malanovic, N.; De Boer, L.; Koning, R.I.; Ravensbergen, E.; Franken, M.; Van Der Heijde, T.; Boekema, B.K.; et al. The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci. Transl. Med. 2018, 10, eaan4044. [Google Scholar] [CrossRef]
- Lozeau, L.D.; Grosha, J.; Kole, D.; Prifti, F.; Dominko, T.; Camesano, T.A.; Rolle, M.W. Collagen tethering of synthetic human antimicrobial peptides cathelicidin LL37 and its effects on antimicrobial activity and cytotoxicity. Acta Biomater. 2017, 52, 9–20. [Google Scholar] [CrossRef]
- Nordström, R.; Nyström, L.; Andrén, O.C.J.; Malkoch, M.; Umerska, A.; Davoudi, M.; Schmidtchen, A.; Malmsten, M. Membrane interactions of microgels as carriers of antimicrobial peptides. J. Colloid Interface Sci. 2018, 513, 141–150. [Google Scholar] [CrossRef]
- Fumakia, M.; Ho, E.A. Nanoparticles encapsulated with LL37 and serpin A1 promotes wound healing and synergistically enhances antibacterial activity. Mol. Pharm. 2016, 13, 2318–2331. [Google Scholar] [CrossRef]
- Sadeghi, S.; Bakhshandeh, H.; Cohan, R.A.; Peirovi, A.; Ehsani, P.; Norouzian, D. Synergistic anti-staphylococcal activity of niosomal recombinant lysostaphin-LL-37. Int. J. Nanomed. 2019, 14, 9777. [Google Scholar] [CrossRef] [PubMed]
- Cassin, M.E.; Ford, A.J.; Orbach, S.M.; Saverot, S.E.; Rajagopalan, P. The design of antimicrobial LL37-modified collagen-hyaluronic acid detachable multilayers. Acta Biomater. 2016, 40, 119–129. [Google Scholar] [CrossRef]
- Teixeira, M.C.; Carbone, C.; Sousa, M.C.; Espina, M.; Garcia, M.L.; Sanchez-Lopez, E.; Souto, E.B. Nanomedicines for the delivery of antimicrobial peptides (Amps). Nanomaterials 2020, 10, 560. [Google Scholar] [CrossRef]
- Lin, X.; Wang, R.; Mai, S. Advances in delivery systems for the therapeutic application of LL37. J. Drug Deliv. Sci. Technol. 2020, 60, 102016. [Google Scholar] [CrossRef]
- Nagant, C.; Pitts, B.; Nazmi, K.; Vandenbranden, M.; Bolscher, J.G.; Stewart, P.S.; Dehaye, J.P. Identification of peptides derived from the human antimicrobial peptide LL-37 active against biofilms formed by Pseudomonas aeruginosa using a library of truncated fragments. Antimicrob. Agents Chemother. 2012, 56, 5698–5708. [Google Scholar] [CrossRef]
- Dong, N.; Li, X.R.; Xu, X.Y.; Lv, Y.F.; Li, Z.Y.; Shan, A.S.; Wang, J.L. Characterization of bactericidal efficiency, cell selectivity, and mechanism of short interspecific hybrid peptides. Amino Acids 2018, 50, 453–468. [Google Scholar] [CrossRef]
- Rajasekaran, G.; Kim, E.Y.; Shin, S.Y. LL-37-derived membrane-active FK-13 analogs possessing cell selectivity, anti-biofilm activity and synergy with chloramphenicol and anti-inflammatory activity. Biochim. Biophys. Acta Biomembr. 2017, 1859, 722–733. [Google Scholar] [CrossRef]
- Saporito, P.; Vang Mouritzen, M.; Løbner-Olesen, A.; Jenssen, H. LL-37 fragments have antimicrobial activity against Staphylococcus epidermidis biofilms and wound healing potential in HaCaT cell line. J. Pept. Sci. 2018, 24, e3080. [Google Scholar] [CrossRef]
- Thennarasu, S.; Tan, A.; Penumatchu, R.; Shelburne, C.E.; Heyl, D.L.; Ramamoorthy, A. Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide ll37. Biophys. J. 2010, 98, 248–257. [Google Scholar] [CrossRef]
- Al Tall, Y.; Abualhaijaa, A.; Alsaggar, M.; Almaaytah, A.; Masadeh, M.; Alzoubi, K.H. Design and characterization of a new hybrid peptide from LL-37 and BMAP-27. Infect. Drug Resist. 2019, 12, 1035. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, S.; Jafari, M.; Arjomand, M.; Mehrnejad, F. Molecular insights into the interactions of GF-17 with the gram-negative and gram-positive bacterial lipid bilayers. J. Cell. Biochem. 2018, 119, 9205–9216. [Google Scholar] [CrossRef]
- Shurko, J.F.; Galega, R.S.; Li, C.; Lee, G.C. Evaluation of LL-37 antimicrobial peptide derivatives alone and in combination with vancomycin against S. aureus. J. Antibiot. 2018, 71, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Haisma, E.M.; De Breij, A.; Chan, H.; Van Dissel, J.T.; Drijfhout, J.W.; Hiemstra, P.S.; El Ghalbzouri, A.; Nibbering, P.H. LL-37-derived peptides eradicate multidrug-resistant Staphylococcus aureus from thermally wounded human skin equivalents. Antimicrob. Agents Chemother. 2014, 58, 4411–4419. [Google Scholar] [CrossRef]
- Wanmakok, M.; Orrapin, S.; Intorasoot, A.; Intorasoot, S. Expression in Escherichia coli of novel recombinant hybrid antimicrobial peptide AL32-P113 with enhanced antimicrobial activity in vitro. Gene 2018, 671, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; McLean, D.T.F.; Linden, G.J.; McAuley, D.F.; McMullan, R.; Lundy, F.T. The naturally occurring host defense peptide, LL-37, and its truncated mimetics KE-18 and KR-12 have selected biocidal and antibiofilm activities against Candida albicans, Staphylococcus aureus, and Escherichia coli in vitro. Front. Microbiol. 2017, 8, 544. [Google Scholar] [CrossRef]
- Jacob, B.; Park, I.S.; Bang, J.K.; Shin, S.Y. Short KR-12 analogs designed from human cathelicidin LL-37 possessing both antimicrobial and antiendotoxic activities without mammalian cell toxicity. J. Pept. Sci. 2013, 19, 700–707. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. Compound Summary for CID 16198951, Cathelicidin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cathelicidin (accessed on 4 March 2021).
- Dannehl, C.; Gutsmann, T.; Brezesinski, G. Surface activity and structures of two fragments of the human antimicrobial LL-37. Colloids Surfaces B Biointerfaces 2013, 109, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Kanthawong, S.; Bolscher, J.G.M.; Veerman, E.C.I.; van Marle, J.; Nazmi, K.; Wongratanacheewin, S.; Taweechaisupapong, S. Antimicrobial activities of LL-37 and its truncated variants against Burkholderia thailandensis. Int. J. Antimicrob. Agents 2010, 36, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Sambanthamoorthy, K.; Palys, T.; Paranavitana, C. The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides 2013, 49, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yang, G.; Lu, S.; Chen, D.; Fan, S.; Xu, J.; Wu, B.; He, J. Design and antimicrobial activities of LL-37 derivatives inhibiting the formation of Streptococcus mutans biofilm. Chem. Biol. Drug Des. 2019, 93, 1175–1185. [Google Scholar] [CrossRef]
- Kim, E.Y.; Rajasekaran, G.; Shin, S.Y. LL-37-derived short antimicrobial peptide KR-12-a5 and its D-amino acid substituted analogs with cell selectivity, anti-biofilm activity, synergistic effect with conventional antibiotics, and anti-inflammatory activity. Eur. J. Med. Chem. 2017, 136, 428–441. [Google Scholar] [CrossRef]
- Caiaffa, K.S.; Massunari, L.; Danelon, M.; Abuna, G.F.; Bedran, T.B.L.; Santos-Filho, N.A.; Spolidorio, D.M.P.; Vizoto, N.L.; Cilli, E.M.; Duque, C. KR-12-a5 is a non-cytotoxic agent with potent antimicrobial effects against oral pathogens. Biofouling 2017, 33, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, Q.; Zheng, Z.; Zhao, L.; Shang, Y.; Wei, X.; Liao, X.; Zhang, R. Design, characterization and expression of a novel hybrid peptides melittin (1-13)-LL37 (17-30). Mol. Biol. Rep. 2014, 41, 4163–4169. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.B.; Wu, R.J.; Si, D.Y.; Liao, X.D.; Zhang, L.L.; Zhang, R.J. Novel hybrid peptide cecropin A (1-8)-LL37 (17-30) with potential antibacterial activity. Int. J. Mol. Sci. 2016, 17, 983. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.; Wu, D.; Li, W.; Zheng, X.; Li, W.; Shan, A. High specific selectivity and membrane-active mechanism of synthetic cationic hybrid antimicrobial peptides based on the peptide FV7. Int. J. Mol. Sci. 2017, 18, 339. [Google Scholar] [CrossRef] [PubMed]
- Chingaté, S.; Delgado, G.; Salazar, L.M.; Soto, C.Y. The ATPase activity of the mycobacterial plasma membrane is inhibited by the LL37-analogous peptide LLAP. Peptides 2015, 71, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh, H.; Memariani, H.; Ranjbar, R.; Pooshang Bagheri, K. The activity and action mechanism of novel short selective LL-37-derived anticancer peptides against clinical isolates of Escherichia coli. Chem. Biol. Drug Des. 2019, 93, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Woodburn, K.W.; Jaynes, J.M.; Clemens, L.E. Evaluation of the Antimicrobial Peptide, RP557, for the Broad-Spectrum Treatment of Wound Pathogens and Biofilm. Front. Microbiol. 2019, 10, 1688. [Google Scholar] [CrossRef]
- Mishra, B.; Golla, R.M.; Lau, K.; Lushnikova, T.; Wang, G. Anti-Staphylococcal Biofilm Effects of Human Cathelicidin Peptides. ACS Med. Chem. Lett. 2016, 7, 117–121. [Google Scholar] [CrossRef]
- Ming, L.; Huang, J.A. The antibacterial effects of antimicrobial peptides OP-145 against clinically isolated multi-resistant strains. Jpn. J. Infect. Dis. 2017, 70, JJID.2017. [Google Scholar] [CrossRef]
- Scheper, H.; Wubbolts, J.M.; Verhagen, J.A.M.; de Visser, A.W.; van der Wal, R.J.P.; Visser, L.G.; de Boer, M.G.J.; Nibbering, P.H. SAAP-148 Eradicates MRSA Persisters Within Mature Biofilm Models Simulating Prosthetic Joint Infection. Front. Microbiol. 2021, 12, 99. [Google Scholar] [CrossRef]
- Wongkaewkhiaw, S.; Taweechaisupapong, S.; Anutrakunchai, C.; Nazmi, K.; Bolscher, J.G.M.; Wongratanacheewin, S.; Kanthawong, S. D-LL-31 in combination with ceftazidime synergistically enhances bactericidal activity and biofilm destruction in Burkholderia pseudomallei. Biofouling 2019, 35, 573–584. [Google Scholar] [CrossRef]
- Wongkaewkhiaw, S.; Taweechaisupapong, S.; Thanaviratananich, S.; Bolscher, J.G.M.; Nazmi, K.; Anutrakunchai, C.; Chareonsudjai, S.; Kanthawong, S. D-LL-31 enhances biofilm-eradicating effect of currently used antibiotics for chronic rhinosinusitis and its immunomodulatory activity on human lung epithelial cells. PLoS ONE 2020, 15, e243315. [Google Scholar] [CrossRef]
- Narayana, J.L.; Mishra, B.; Lushnikova, T.; Golla, R.M.; Wang, G. Modulation of antimicrobial potency of human cathelicidin peptides against the ESKAPE pathogens and in vivo efficacy in a murine catheter-associated biofilm model. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Hanke, M.L.; Mishra, B.; Lushnikova, T.; Heim, C.E.; Thomas, V.C.; Bayles, K.W.; Kielian, T. Transformation of human cathelicidin LL-37 into selective, stable, and potent antimicrobial compounds. ACS Chem. Biol. 2014, 9, 1997–2002. [Google Scholar] [CrossRef]
- Gunasekera, S.; Muhammad, T.; Strömstedt, A.A.; Rosengren, K.J.; Göransson, U. Backbone Cyclization and Dimerization of LL-37-Derived Peptides Enhance Antimicrobial Activity and Proteolytic Stability. Front. Microbiol. 2020, 11, 168. [Google Scholar] [CrossRef]
- Wang, G.; Mishra, B.; Epand, R.F.; Epand, R.M. High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2160–2172. [Google Scholar] [CrossRef]
- Leszczyńska, K.; Namiot, A.; Janmey, P.A.; Bucki, R. Modulation of exogenous antibiotic activity by host cathelicidin LL-37. Apmis 2010, 118, 830–836. [Google Scholar] [CrossRef]
- Kamysz, E.; Sikorska, E.; Jaśkiewicz, M.; Bauer, M.; Neubauer, D.; Bartoszewska, S.; Barańska-Rybak, W.; Kamysz, W. Lipidated analogs of the ll-37-derived peptide fragment KR12—structural analysis, surface-active properties and antimicrobial activity. Int. J. Mol. Sci. 2020, 21, 887. [Google Scholar] [CrossRef]
- Wang, X.; Mishra, B.; Lushnikova, T.; Narayana, J.L.; Wang, G. Amino Acid Composition Determines Peptide Activity Spectrum and Hot-Spot-Based Design of Merecidin. Adv. Biosyst. 2018, 2, 1700259. [Google Scholar] [CrossRef] [PubMed]
- Shahmiri, M.; Enciso, M.; Adda, C.G.; Smith, B.J.; Perugini, M.A.; Mechler, A. Membrane Core-Specific Antimicrobial Action of Cathelicidin LL-37 Peptide Switches between Pore and Nanofibre Formation. Sci. Rep. 2016, 6, 38184. [Google Scholar] [CrossRef] [PubMed]
- Ulaeto, D.O.; Morris, C.J.; Fox, M.A.; Gumbleton, M.; Beck, K. Destabilization of α-helical structure in solution improves bactericidal activity of antimicrobial peptides: Opposite effects on bacterial and viral targets. Antimicrob. Agents Chemother. 2016, 60, 1984–1991. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.P.; Higgins, M.; Whitehurst, J.; Kisich, K.O.; Voskuil, M.I.; Hodges, R.S. Anti-Tuberculosis Activity of α-Helical Antimicrobial Peptides: De Novo Designed L- and D-Enantiomers Versus L- and D-LL37. Protein Pept. Lett. 2011, 18, 241–252. [Google Scholar] [CrossRef]
- Silva, T.; Magalhães, B.; Maia, S.; Gomes, P.; Nazmi, K.; Bolscher, J.G.M.; Rodrigues, P.N.; Bastos, M.; Gomes, M.S. Killing of mycobacterium avium by lactoferricin peptides: Improved activity of arginine- and d-Amino-Acid-containing molecules. Antimicrob. Agents Chemother. 2014, 58, 2461–2467. [Google Scholar] [CrossRef]
- De La Fuente-Núñez, C.; Reffuveille, F.; Mansour, S.C.; Reckseidler-Zenteno, S.L.; Hernández, D.; Brackman, G.; Coenye, T.; Hancock, R.E.W. D-Enantiomeric Peptides that Eradicate Wild-Type and Multidrug-Resistant Biofilms and Protect against Lethal Pseudomonas aeruginosa Infections. Chem. Biol. 2015, 22, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Zharkova, M.S.; Orlov, D.S.; Golubeva, O.Y.; Chakchir, O.B.; Eliseev, I.E.; Grinchuk, T.M.; Shamova, O.V. Application of antimicrobial peptides of the innate immune system in combination with conventional antibiotics-a novel way to combat antibiotic resistance? Front. Cell. Infect. Microbiol. 2019, 9, 128. [Google Scholar] [CrossRef]
- Chauhan, V.M.; Zhang, H.; Dalby, P.A.; Aylott, J.W. Advancements in the co-formulation of biologic therapeutics. J. Control. Release 2020, 327, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Geitani, R.; Ayoub Moubareck, C.; Touqui, L.; Karam Sarkis, D. Cationic antimicrobial peptides: Alternatives and/or adjuvants to antibiotics active against methicillin-resistant Staphylococcus aureus and multidrug-resistant Pseudomonas aeruginosa. BMC Microbiol. 2019, 19, 54. [Google Scholar] [CrossRef]
- Lin, L.; Nonejuie, P.; Munguia, J.; Hollands, A.; Olson, J.; Dam, Q.; Kumaraswamy, M.; Rivera, H.; Corriden, R.; Rohde, M.; et al. Azithromycin Synergizes with Cationic Antimicrobial Peptides to Exert Bactericidal and Therapeutic Activity Against Highly Multidrug-Resistant Gram-Negative Bacterial Pathogens. EBioMedicine 2015, 2, 690–698. [Google Scholar] [CrossRef]
- Payne, J.E.; Dubois, A.V.; Ingram, R.J.; Weldon, S.; Taggart, C.C.; Elborn, J.S.; Tunney, M.M. Activity of innate antimicrobial peptides and ivacaftor against clinical cystic fibrosis respiratory pathogens. Int. J. Antimicrob. Agents 2017, 50, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Sakoulas, G.; Rose, W.; Berti, A.; Olson, J.; Munguia, J.; Nonejuie, P.; Sakoulas, E.; Rybak, M.J.; Pogliano, J.; Nizet, V. Classical β-lactamase inhibitors potentiate the activity of daptomycin against methicillin-resistant Staphylococcus aureus and colistin against Acinetobacter baumannii. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Le, J.; Dam, Q.; Schweizer, M.; Thienphrapa, W.; Nizet, V.; Sakoulas, G. Effects of vancomycin versus nafcillin in enhancing killing of methicillin-susceptible Staphylococcus aureus causing bacteremia by human cathelicidin LL-37. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1441–1447. [Google Scholar] [CrossRef]
- Smith, J.R.; Barber, K.E.; Raut, A.; Rybak, M.J. β-Lactams enhance daptomycin activity against vancomycin-resistant Enterococcus faecalis and Enterococcus faecium in in vitro pharmacokinetic/pharmacodynamic models. Antimicrob. Agents Chemother. 2015, 59, 4748–4754. [Google Scholar] [CrossRef]
- Sakoulas, G.; Bayer, A.S.; Pogliano, J.; Tsuji, B.T.; Yang, S.J.; Mishra, N.N.; Nizet, V.; Yeaman, M.R.; Moise, P.A. Ampicillin enhances daptomycin- and cationic host defense peptide-mediated killing of ampicillin- and vancomycin-resistant Enterococcus faecium. Antimicrob. Agents Chemother. 2012, 56, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Dahesh, S.; Wong, B.; Nizet, V.; Sakoulas, G.; Tran, T.T.; Aitken, S.L. Treatment of multidrug-resistant vancomycin-resistant enterococcus faecium hardware-associated vertebral osteomyelitis with oritavancin plus ampicillin. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Nuding, S.; Frasch, T.; Schaller, M.; Stange, E.F.; Zabel, L.T. Synergistic effects of antimicrobial peptides and antibiotics against clostridium difficile. Antimicrob. Agents Chemother. 2014, 58, 5719–5725. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, E.R.; Dillon, N.; Tsunemoto, H.; Pogliano, J.; Sakoulas, G.; Nizet, V. Avibactam Sensitizes Carbapenem-Resistant NDM-1-Producing Klebsiella pneumoniae to Innate Immune Clearance. J. Infect. Dis. 2019, 220, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Kumaraswamy, M.; Lin, L.; Olson, J.; Sun, C.F.; Nonejuie, P.; Corriden, R.; Döhrmann, S.; Ali, S.R.; Amaro, D.; Rohde, M.; et al. Standard susceptibility testing overlooks potent azithromycin activity and cationic peptide synergy against MDR Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 2016, 71, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Alaiwa, M.H.A.; Reznikov, L.R.; Gansemer, N.D.; Sheets, K.A.; Horswill, A.R.; Stoltz, D.A.; Zabner, J.; Welsh, M.J. pH modulates the activity and synergism of the airway surface liquid antimicrobials β-defensin-3 and LL-37. Proc. Natl. Acad. Sci. USA 2014, 111, 18703–18709. [Google Scholar] [CrossRef] [PubMed]
- Sakoulas, G.; Okumura, C.Y.; Thienphrapa, W.; Olson, J.; Nonejuie, P.; Dam, Q.; Dhand, A.; Pogliano, J.; Yeaman, M.R.; Hensler, M.E.; et al. Nafcillin enhances innate immune-mediated killing of methicillin-resistant Staphylococcus aureus. J. Mol. Med. 2014, 92, 139–149. [Google Scholar] [CrossRef]
- Huang, Y.; Alumasa, J.N.; Callaghan, L.T.; Samuel Baugh, R.; Rae, C.D.; Keiler, K.C.; McGillivray, S.M. A small-molecule inhibitor of trans-translation synergistically interacts with cathelicidin antimicrobial peptides to impair survival of staphylococcus aureus. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Niemirowicz, K.; Piktel, E.; Wilczewska, A.Z.; Markiewicz, K.H.; Durnaś, B.; Wątek, M.; Puszkarz, I.; Wróblewska, M.; Niklińska, W.; Savage, P.B.; et al. Core–shell magnetic nanoparticles display synergistic antibacterial effects against Pseudomonas aeruginosa and Staphylococcus aureus when combined with cathelicidin LL-37 or selected ceragenins. Int. J. Nanomed. 2016, 11, 5443. [Google Scholar] [CrossRef]
- Wang, S.; Yan, C.; Zhang, X.; Shi, D.; Chi, L.; Luo, G.; Deng, J. Antimicrobial peptide modification enhances the gene delivery and bactericidal efficiency of gold nanoparticles for accelerating diabetic wound healing. Biomater. Sci. 2018, 6, 5757–5772. [Google Scholar] [CrossRef]
- Malekkhaiat Häffner, S.; Nyström, L.; Nordström, R.; Xu, Z.P.; Davoudi, M.; Schmidtchen, A.; Malmsten, M. Membrane interactions and antimicrobial effects of layered double hydroxide nanoparticles. Phys. Chem. Chem. Phys. 2017, 19, 23832–23842. [Google Scholar] [CrossRef] [PubMed]
- Bitschar, K.; Sauer, B.; Focken, J.; Dehmer, H.; Moos, S.; Konnerth, M.; Schilling, N.A.; Grond, S.; Kalbacher, H.; Kurschus, F.C.; et al. Lugdunin amplifies innate immune responses in the skin in synergy with host- and microbiota-derived factors. Nat. Commun. 2019, 10, 2730. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 2017, 9, eaah4680. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.; Ahumada, M.; Franco, W.; Mah, T.F.; Seymour, R.; Suuronen, E.J.; Alarcon, E.I. Sprayable peptide-modified silver nanoparticles as a barrier against bacterial colonization. Nanoscale 2016, 8, 19200–19203. [Google Scholar] [CrossRef] [PubMed]
- Werth, B.J.; Sakoulas, G.; Rose, W.E.; Pogliano, J.; Tewhey, R.; Rybak, M.J. Ceftaroline increases membrane binding and enhances the activity of daptomycin against daptomycin-nonsusceptible vancomycin-intermediate Staphylococcus aureus in a pharmacokinetic/pharmacodynamic model. Antimicrob. Agents Chemother. 2013, 57, 66–73. [Google Scholar] [CrossRef]
- Miller, W.R.; Bayer, A.S.; Arias, C.A. Mechanism of action and resistance to daptomycin in Staphylococcus aureus and enterococci. Cold Spring Harb. Perspect. Med. 2016, 6, a026997. [Google Scholar] [CrossRef]
- Sakoulas, G.; Rose, W.; Nonejuie, P.; Olson, J.; Pogliano, J.; Humphries, R.; Nizet, V. Ceftaroline restores daptomycin activity against daptomycin-nonsusceptible vancomycin-resistant Enterococcus faecium. Antimicrob. Agents Chemother. 2014, 58, 1494–1500. [Google Scholar] [CrossRef]
- Sakoulas, G.; Nonejuie, P.; Nizet, V.; Pogliano, J.; Crum-Cianflone, N.; Haddad, F. Treatment of high-level gentamicin-resistant enterococcus faecalis endocarditis with daptomycin plus ceftaroline. Antimicrob. Agents Chemother. 2013, 57, 4042–4045. [Google Scholar] [CrossRef]
- Sakoulas, G.; Moise, P.A.; Casapao, A.M.; Nonejuie, P.; Olson, J.; Okumura, C.Y.M.; Rybak, M.J.; Kullar, R.; Dhand, A.; Rose, W.E.; et al. Antimicrobial salvage therapy for persistent staphylococcal bacteremia using daptomycin plus ceftaroline. Clin. Ther. 2014, 36, 1317–1333. [Google Scholar] [CrossRef]


{kind=link}
| Bacteria | Gene | Effect of Mutation/Deletion of Gene | Reference |
|---|---|---|---|
| Pseudomonas aeruginosa | vacJ | Increased membrane permeability | [108] |
| algD | Disruption to virulence factors | [100,101] | |
| Neisseria gonorrhoeae | mpg | Disruption to virulence factors | [106] |
| pilE | Disruption to virulence factors | [106] | |
| Streptococcus mutans | dltC, ciaRh | Disruption in biofilm formation | [37] |
| Streptococcus agalactiae | dltA | Decreased cell surface charge | [36] |
| bceR | Disruption to virulence factors | [49] | |
| Staphylococcus aureus | pheS, mprF, graR, vraF, trkA | Increased membrane permeability | [33] |
| lspA, vraR | Disruption in biofilm formation | [33] | |
| argC, lipA, pheS, yrF, thrB, acoB | Disruption to biosynthesis and metabolism | [33] | |
| secDF; blaI | Disruption to virulence factors | [56,102] | |
| dltA, mprF | Decreased cell surface charge | [38] | |
| Streptococcus pneumoniae | dltD, licD2; dlt | Decreased cell surface charge | [12,35] |
| Acinetobacter baumannii | ompA | Disruption in biofilm formation | [63] |
| Helicobacter pylori | lpxE, lpxF | Disruption to virulence factors, and decreased cell surface charge | [40] |
| Bacillus anthracis | clpX, lrgAB | Structural changes to cell envelope | [74] |
| Clostridioides difficile | clnR, clnAB | Disruption to biosynthesis and metabolism, and disruption to virulence factors | [105] |
| Mycobacterium tuberculosis | rv1258c | Increased membrane permeability | [109] |
| Salmonella enterica | phoPQ | Disruption to virulence factors | [26] |
| envC, zapB | Decreased cell surface charge, increased hydrophobicity | [107] | |
| Escherichia coli | yrfF, rcpA; ompT, pch, ler, lrp; ompT | Disruption to virulence factors | [61,65,104] |
| envC, zapB | Decreased cell surface charge, increased hydrophobicity | [107] | |
| waaP | Disruption to virulence factors | [44] | |
| Enterococcus faecalis | liaR | Structural changes to cell envelope | [30] |
| Porphyromonas gingivalis | pgm6/pgm7 | Structural changes to cell envelope | [64] |
| Streptococcus pyogenes | gacI | Increased hydrophobicity | [39] |
| hasA | Disruption to virulence factors | [80] | |
| Haemophilus ducreyi | cpxA, mtrC; sapBC, sapA | Disruption to virulence factors | [51,52,53] |
| Haemophilus influenzae | sapBC, htrB | Disruption to virulence factors | [54] |
| Vibrio cholerae | msbB | Disruption to virulence factors | [103] |
| Type of Derivative | LL-37 Derivative | Sequence | Reference |
|---|---|---|---|
| WT sequence | LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | [155] |
| Truncations (without amino acid substitutions) | LL-32 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLV | [156] |
| LL-31 * | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNL | [112,143,157] | |
| IG-19 | LLGDFFRKSKEKIGKEFKR | [157] | |
| RK-31 | RKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | [143] | |
| RK-25 | RKSKEKIGKEFKRIVQRIKDFLRNL | [143] | |
| LL7-27 | RKSKEKIGKEFKRIVQRIKDF | [147] | |
| KS-30 * | KSKEKIGKEFKRIVQRIKDFLRNLVPRTES | [158] | |
| LL-13 * | IGKEFKRIVQRIKDFLRNLVPRTES | [150] | |
| KE-18 * | KEFKRIVQRIKDFLRNLV | [153] | |
| LL-17 * | FKRIVQRIKDFLR | [150] | |
| FK-12 | FKRIVQRIKDFL | [146] | |
| KR-20 * | KRIVQRIKDFLRNLVPRTES | [158] | |
| KR-12 * | KRIVQRIKDFLR | ||
| Truncations (with amino acid substitutions) | IG-13-1 | IGKLFKRIVQLIK | [159] |
| IG-13-2 | IGKLFKRIVQLIL | [159] | |
| FK13-a1 * | WKRIVRRIKRWLR | [145] | |
| FK13-a7 * | WKRWVRRWKRWLR | [145] | |
| KR-12-a2 | KRIVQRIKKWLR | [154] | |
| KR-12-a3 | KRIVKRIKKWLR | [154] | |
| KR-12-a4 | KRIVKLIKKWLR | [154] | |
| KR-12-a5 * | KRIVKLILKWLR | [154,160,161] | |
| KR-12-a6 | LRIVKLILKWLR | [154] | |
| VQ-12V26 | VQRIKVFLRNLV | [146] | |
| Hybrids (with truncated LL-37) | AL32-P113 | ALGDFFRKSKEKIGKEFKRIVQRIKDFLRNLAKRHHGYKRK FHLEY | [152] |
| L31-P113 | LGDFFRKSKEKIGKEFKRIVQRIKDFLRNLAK | [152] | |
| M-L | GIGAVLKVLTTGLFKRIVQRIKDFLRN | [162] | |
| B1 | KFKKLFKKLSPVFKRIVQRIKDFLR | [148] | |
| C-L | KWKLFKKIFKRIVQRIKDFLRN | [163] | |
| FV-LL * | FRIRVRVFKRIVQRIKDFLR | [164] | |
| LI | GKEFKRIVKWPWWPWRR | [144] | |
| Peptides based on LL-37 | LLAP | GRKSAKKIGKRAKRI | [165] |
| P38 | TSVRQRWRWRQRVRTS | [166] | |
| RP557 * | RFCWKVCYKGICFKKCK | [167] | |
| GF-17 * | GFKRIVQRIKDFLRNLV | [149,168] | |
| OP-145 * | IGKEFKRIVERIKRFLRELVRPLR | [116,169] | |
| P60.4Ac * | IGKEFKRIVERIKRFLRELVRPLR | [151] | |
| P10 * | LAREYKKIVEKLKRWLRQVLRTLR | [151] | |
| SAAP-145 | LKRLYKRLAKLIKRLYRYLKKPVR | [135] | |
| SAAP-148 * | LKRVWKRVFKLLKRYWRQLKKPVR | [135,170] | |
| SAAP-159 | LKRLYKRVFRLLKRYYRQLRRPVR | [135] | |
| SAAP-276 | LKRVWKAVFKLLKRYWRQLKKPVR | [135] | |
| D-enantiomers | D-LL-31 * | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNL | [171,172] |
| D-LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | [121,124] | |
| 17tF-W * | GX1KRlVQRlKDWlRKLV, where 1 is a D amino acid and X1 = 4-t-butylphenylalanine | [173] | |
| 17BIPHE2 * | GX1KR1VQR1KDX21RNLV, where 1 is a D amino acid and X1 = X2 = biphenylalanine | [33,134,168,173,174] |
| Bacteria | Antibiotic Paired with LL-37 | Isolate | Reference |
|---|---|---|---|
| Pseudomonas aeruginosa | Colistin | AS1, MDRPA1, MDRPA2; 2 CIs | [126,187] |
| Imipenem | MDRPA1 | [187] | |
| Azithromycin | PA01 | [188] | |
| Ciprofloxacin | 2 CIs | [126] | |
| Tobramycin | 1 CI | [189] | |
| Staphylococcus aureus | Tazobactam | Sanger 252, VISA D712, hVISA D592 | [190] |
| Tobramycin | 4 CIs | [189] | |
| Teicoplanin | JAR060131, ATCC 49230, AMC201, LUH15101 | [38] | |
| Vancomycin | ATCC 25923 | [150] | |
| Amoxicillin with clavulanic acid | ATCC 29213, 3 MSSA CIs, 2 MRSA CIs | [177] | |
| Amikacin | MSSA CI | [177] | |
| Nafcillin | MSSA CIs | [191] | |
| Enterococcus faecium | Ceftaroline | DAP-susceptible parent strain, R6370, 8019 | [192] |
| Ampicillin | DAP-susceptible parent strain, 8019; AMP- and VAN-resistant isolate | [192,193] | |
| Ertapenem | DAP-susceptible parent strain, R6370, 8019 | [192] | |
| Oritavancin | VAN-resistant CI | [194] | |
| Oritavancin + Ampicillin | |||
| Enterococcus faecalis | Ceftaroline | R6981 | [192] |
| Ertapenem | R6981 | [192] | |
| Clostridioides difficile | Moxifloxacin | 9 toxinogenic and 10 non-toxinogenic CIs, DSM 1296 | [195] |
| Tigecycline | |||
| Piperacillin-tazobactam | |||
| Meropenem | |||
| Klebsiella pneumonaie | Azithromycin | K700603 | [188] |
| Avibactam | CDC1100192, KP1088 and KP1004 | [196] | |
| Zidebactam | CDC1100192 | [196] | |
| Acinetobacter baumannii | Tazobactam | AB5075, AB1 AB2, AB3, AB4 | [190] |
| Azithromycin | Ab19606 | [188] | |
| Stenotrophomonas maltophilia | Colistin | K279a (ATCC BAA-2423) | [197] |
| Micrococcus luteus | Gentamicin | CIP A270 | [185] |
| Streptococcus spp. | Tobramycin | 2 CIs | [189] |
| Bacteria | Antibiotic | LL-37 Derivative | Strain | Reference |
|---|---|---|---|---|
| Pseudomonas aeruginosa | Ciprofloxacin | KR-12-a5, KR-12-a5(6-DL), KR-12-a5(7-DL) | MDRPA (CCARM 2095) | [160] |
| Rifampicin | B1 | ATCC BAA-2114 | [148] | |
| Oxacillin | KR-12-a5, KR-12-a5(5-DK), KR-12-a5(7-DL) | MDRPA (CCARM 2095) | [160] | |
| Chloramphenicol | FK13; KR-12-a5, KR-12-a5(5-DK), KR-12-a5(6-DL) | MDRPA (CCARM 2095) | [145,160] | |
| Staphylococcus aureus | Ampicillin | B1 | BAA-41 | [148] |
| Levofloxacin | B1 | ATCC 33591, ATCC BAA-41 | [148] | |
| Chloramphenicol | B1; FK13-a1, FK13-a7; C-L | ATCC 43300; MRSA (CCARM 3095); ATCC 25923 | [145,148,163] | |
| Erythromycin | B1 | ATCC 33591 | [148] | |
| Thiamphenicol | C-L | ATCC 25923 | [163] | |
| Neomycin sulfate | C-L | ATCC 25923 | [163] | |
| Rifampicin | B1 | ATCC 33591, BAA-41 | [148] | |
| Burkholderia pseudomallei | Ceftazidime | D-LL-31 | 1026b, H777, M10 | [171] |
| Escherichia coli | Neomycin sulfate | C-L | ATCC 25923 | [163] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ridyard, K.E.; Overhage, J. The Potential of Human Peptide LL-37 as an Antimicrobial and Anti-Biofilm Agent. Antibiotics 2021, 10, 650. https://doi.org/10.3390/antibiotics10060650
Ridyard KE, Overhage J. The Potential of Human Peptide LL-37 as an Antimicrobial and Anti-Biofilm Agent. Antibiotics. 2021; 10(6):650. https://doi.org/10.3390/antibiotics10060650
Chicago/Turabian StyleRidyard, Kylen E., and Joerg Overhage. 2021. "The Potential of Human Peptide LL-37 as an Antimicrobial and Anti-Biofilm Agent" Antibiotics 10, no. 6: 650. https://doi.org/10.3390/antibiotics10060650
APA StyleRidyard, K. E., & Overhage, J. (2021). The Potential of Human Peptide LL-37 as an Antimicrobial and Anti-Biofilm Agent. Antibiotics, 10(6), 650. https://doi.org/10.3390/antibiotics10060650