
Sinapic Acid Suppresses SARS CoV-2 Replication by Targeting Its Envelope Protein

 , , ,
, , ,  , ,
, ,  and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
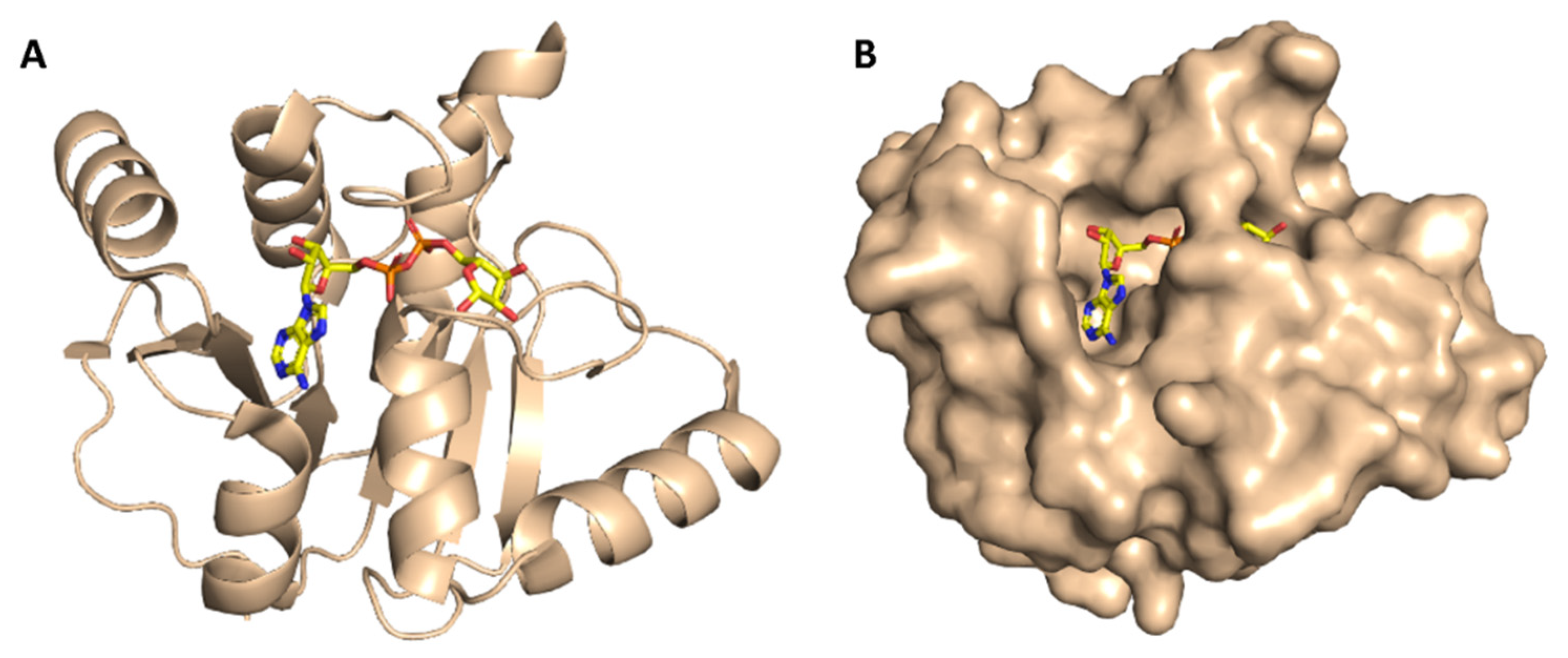
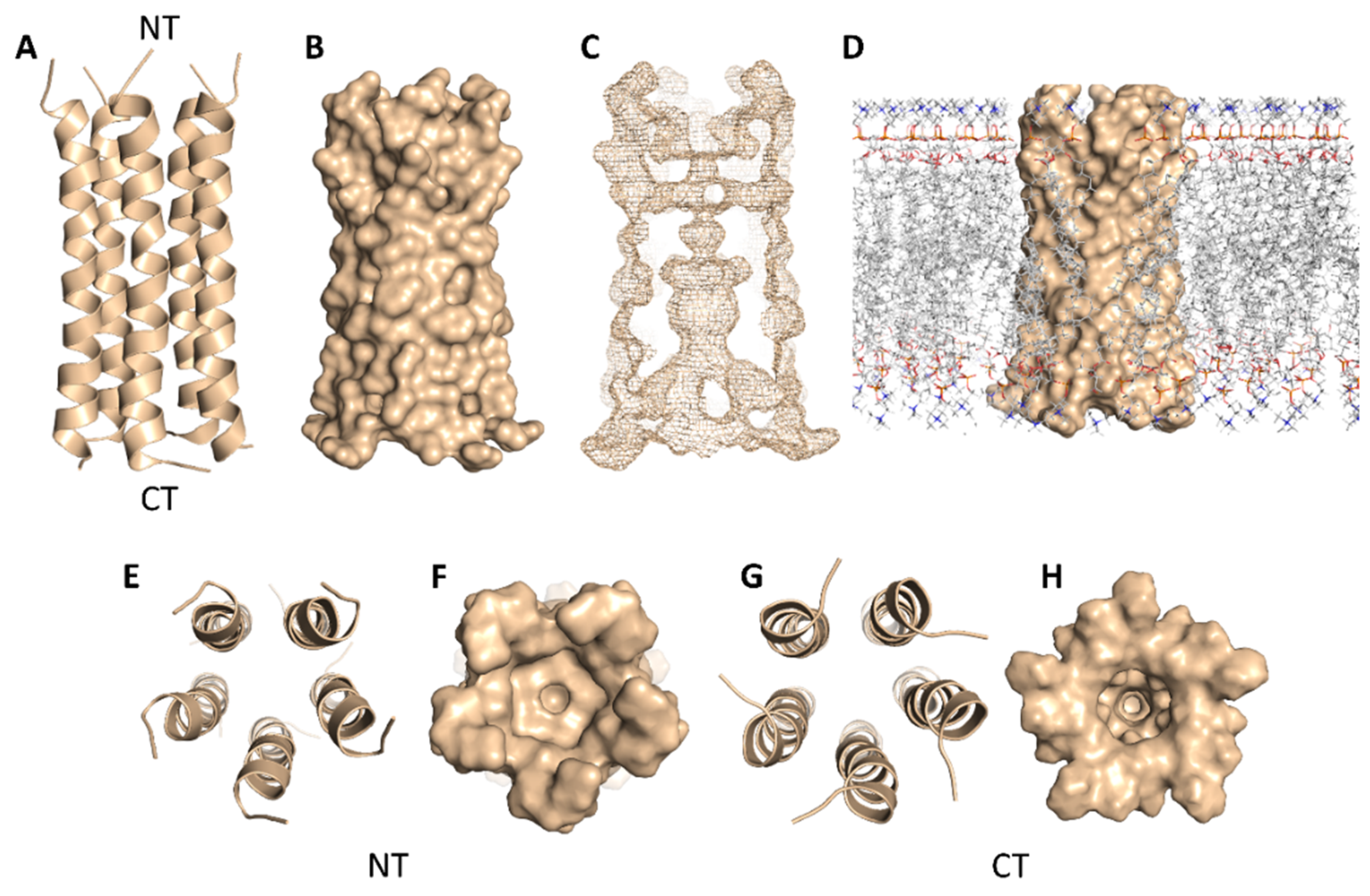
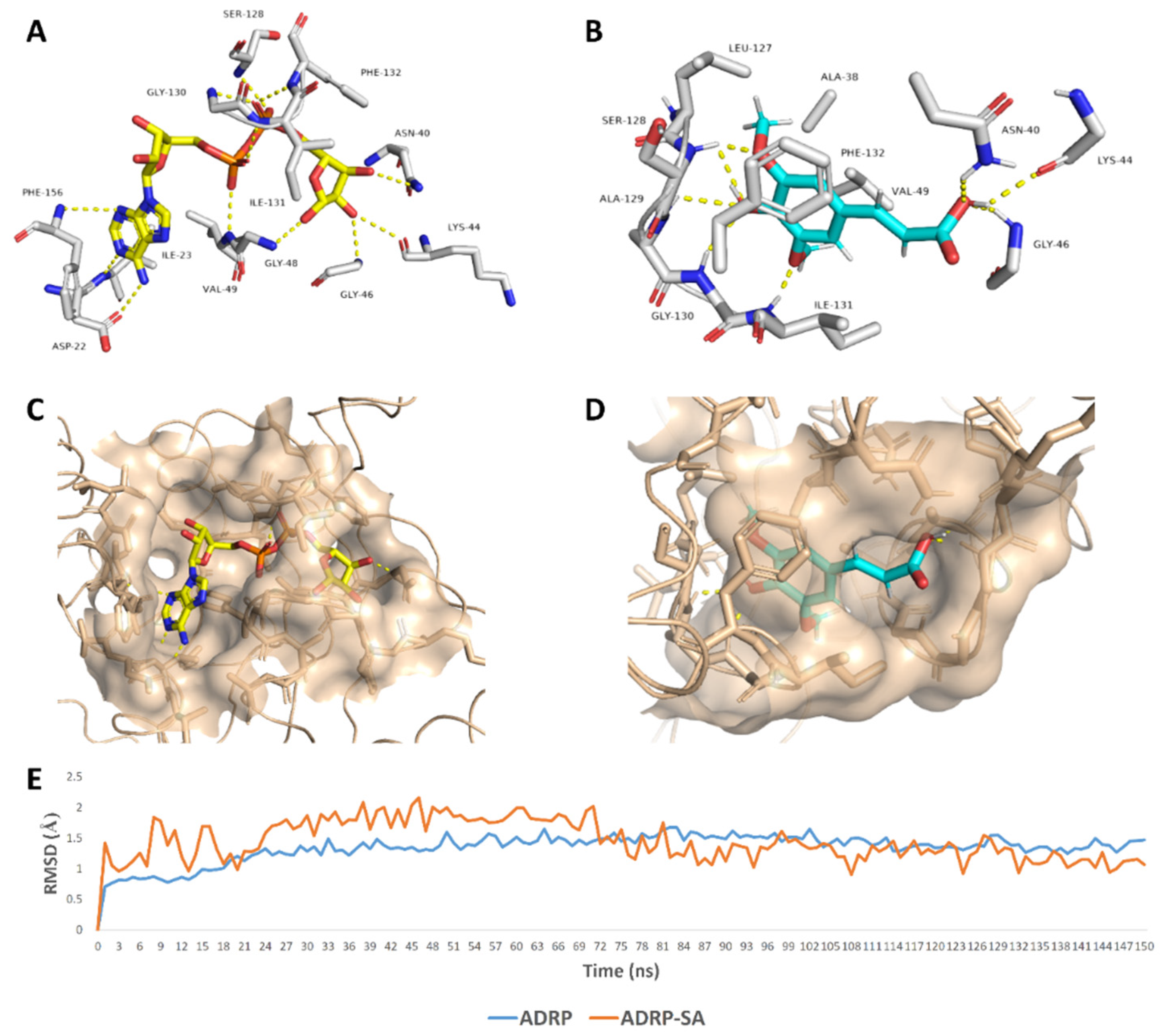
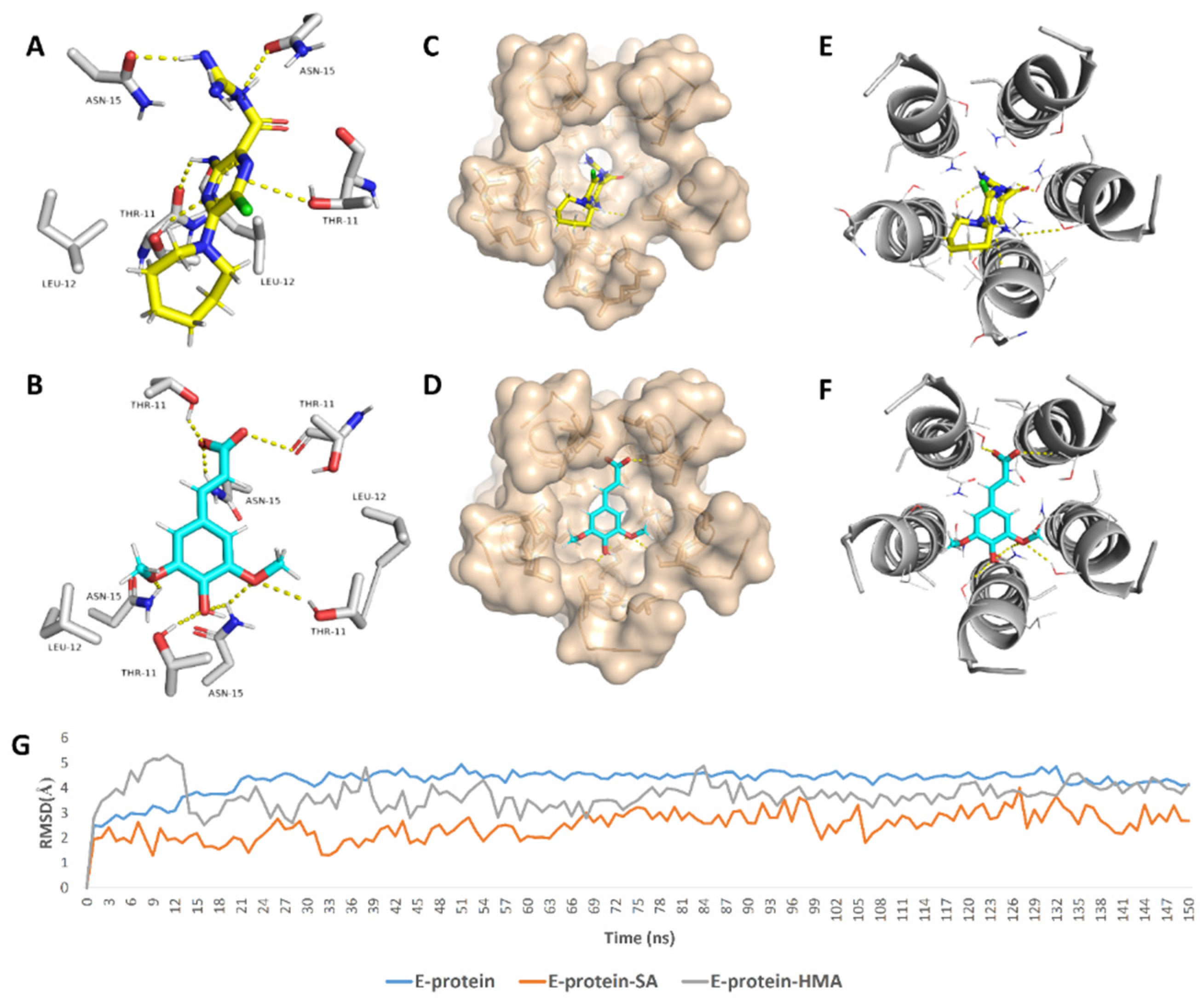
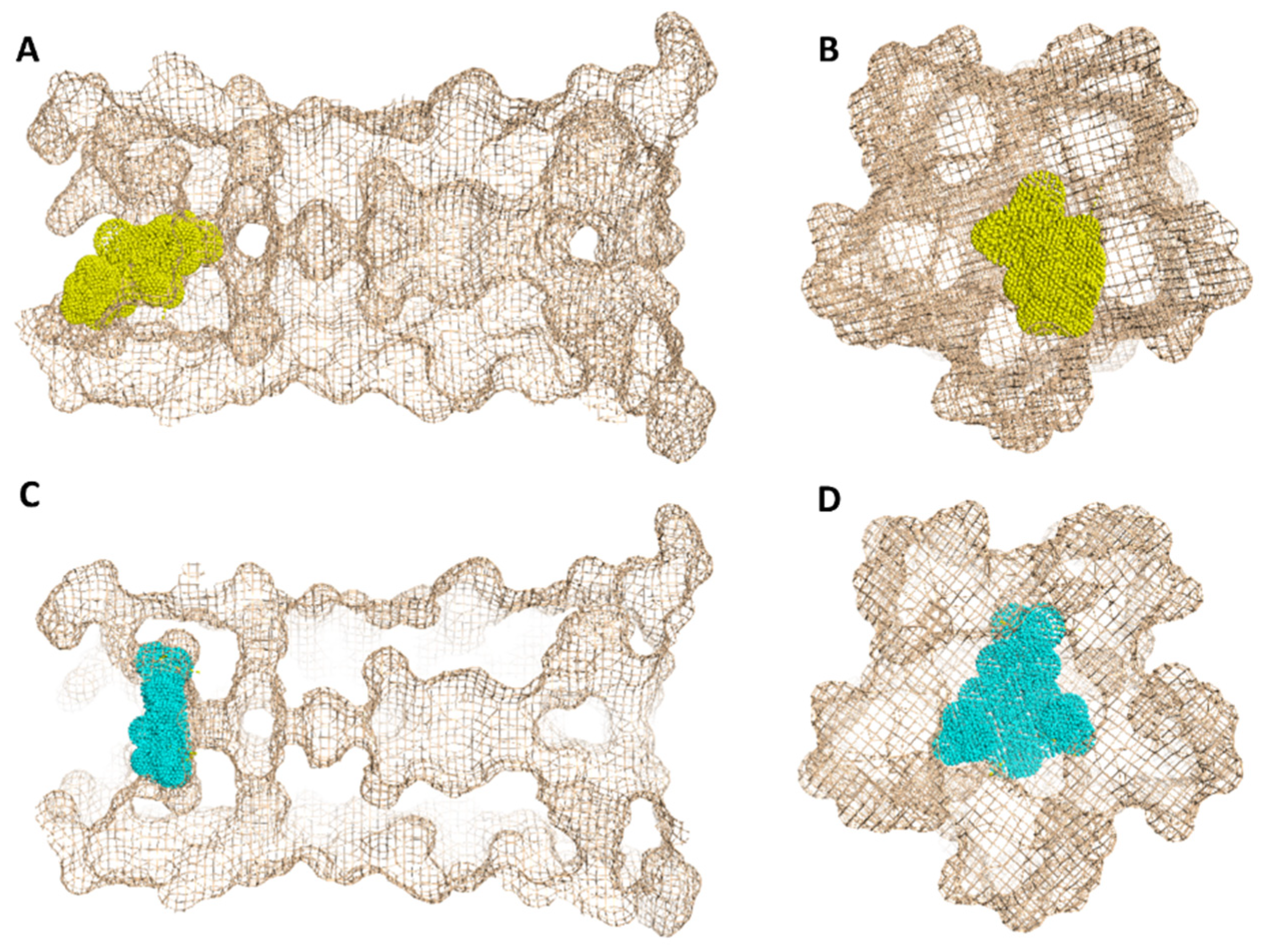
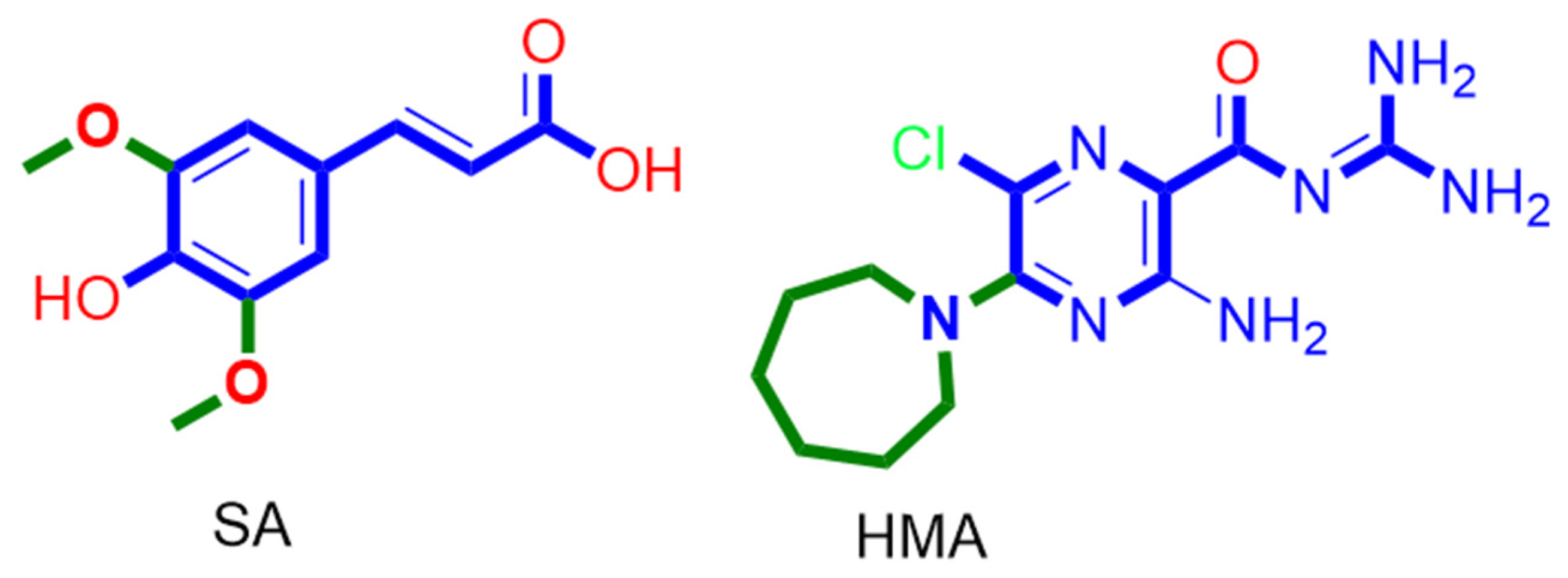
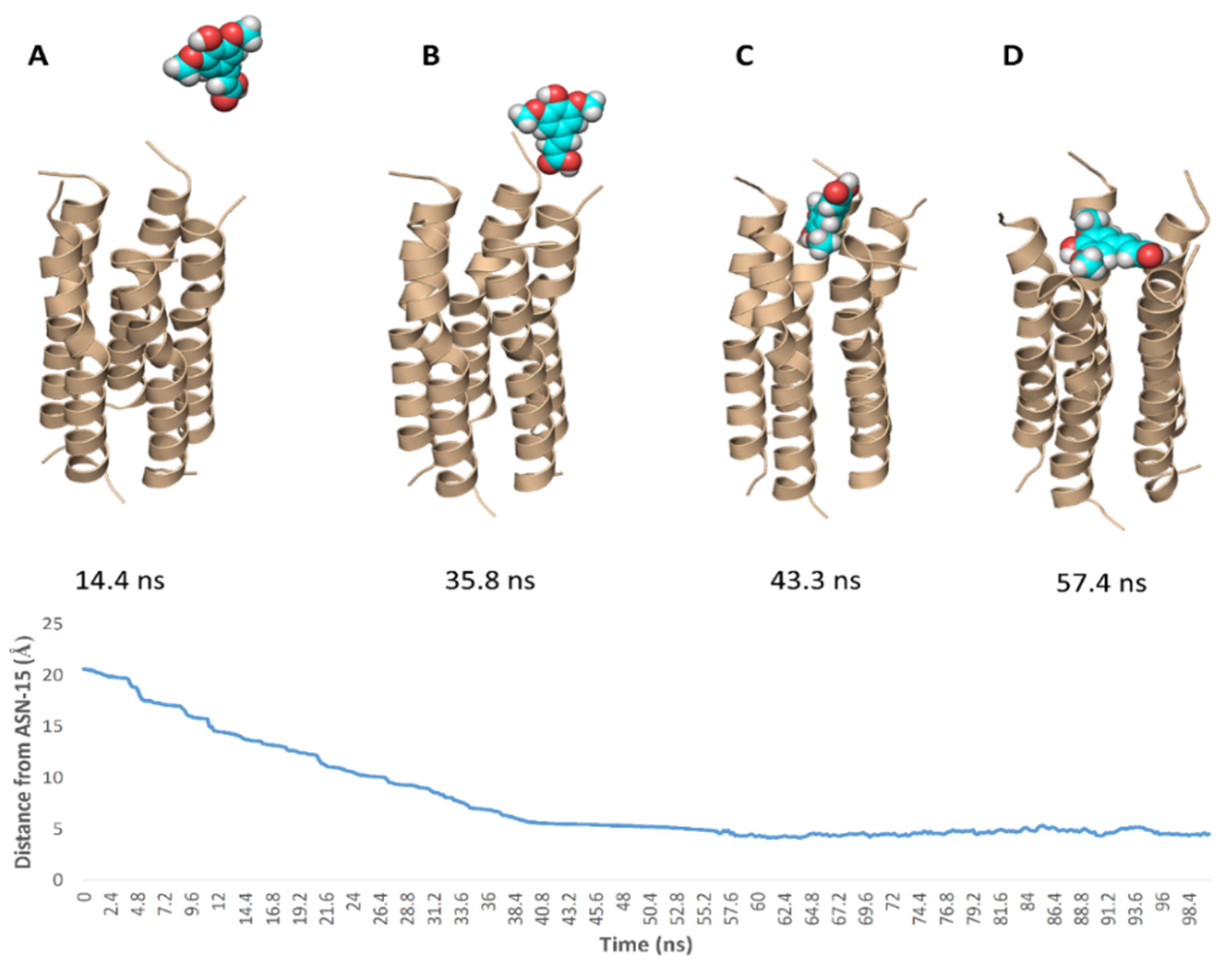
2. Results
3. Discussion
4. Materials and Methods
4.1. Isolation of Compounds
4.2. Data Preparation
4.3. Ensemble Docking
4.4. Molecular Dynamic Simulation
4.5. In Vitro Antiviral Assay
4.5.1. Virus and Cells
4.5.2. MTT Cytotoxicity Assay
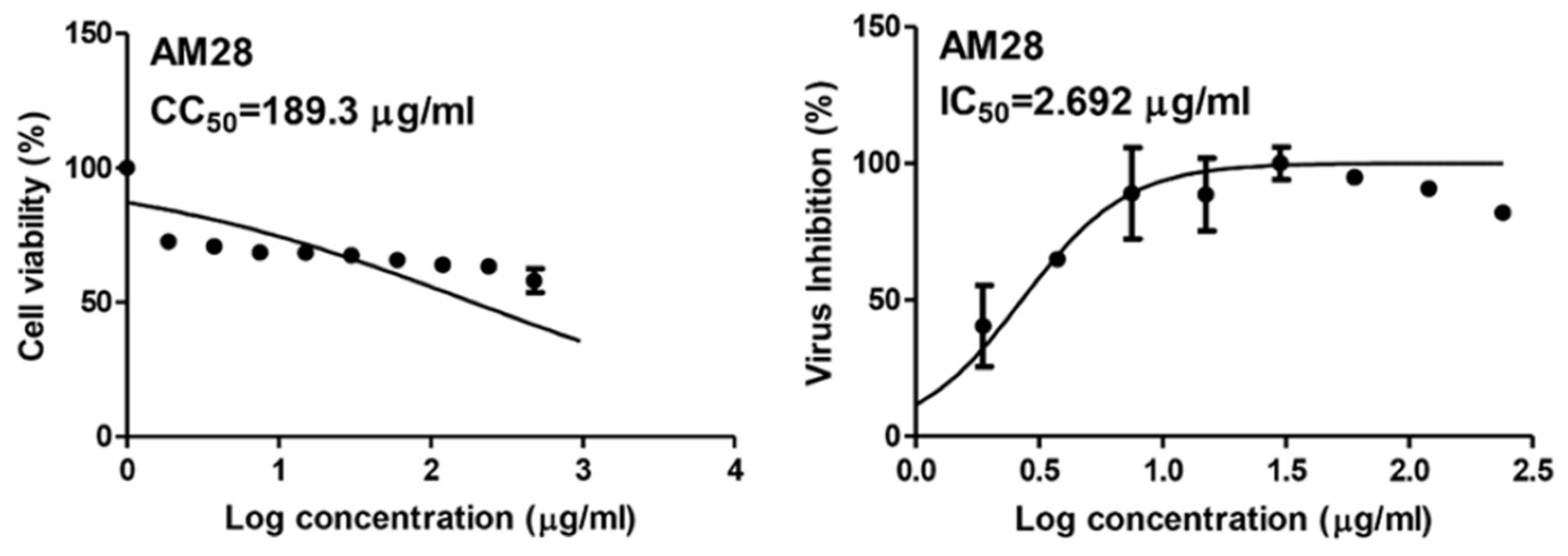
4.5.3. Viral Inhibitory Concentration 50 (IC50) Determination
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- González, J.M.; Gomez-Puertas, P.; Cavanagh, D.; Gorbalenya, A.E.; Enjuanes, L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch. Virol. 2003, 148, 2207–2235. [Google Scholar] [CrossRef]
- Sayed, A.M.; Khattab, A.R.; Aboul Magd, A.M.; Hassan, H.M.; Rateb, M.E.; Zaid, H.; Abdelmohsen, U.R. Nature as a treasure trove of potential anti-SARS-CoV drug leads: A structural/mechanistic rationale. RSC Adv. 2020, 10, 19790–19802. [Google Scholar] [CrossRef]
- Adamson, C.S.; Chibale, K.; Goss, R.J.; Jaspars, M.; Newman, D.J.; Dorrington, R.A. Antiviral drug discovery: Preparing for the next pandemic. Chem. Soc. Rev. 2021, 50, 3647–3655. [Google Scholar] [CrossRef]
- Sayed, A.M.; Alhadrami, H.A.; El-Gendy, A.O.; Shamikh, Y.I.; Belbahri, L.; Hassan, H.M.; Rateb, M.E. Microbial natural products as potential inhibitors of SARS-CoV-2 main protease (Mpro). Microorganisms 2020, 8, 970. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Mordy, F.M.; El-Hamouly, M.M.; Ibrahim, M.T.; Abd El-Rheem, G.; Aly, O.M.; Abd El-kader, A.M.; Abdelmohsen, U.R. Inhibition of SARS-CoV-2 main protease by phenolic compounds from Manilkara hexandra (Roxb.) Dubard assisted by metabolite profiling and in silico virtual screening. RSC Adv. 2020, 10, 32148–32155. [Google Scholar] [CrossRef]
- Hassan, H.A.; Abdelmohsen, U.R.; Aly, O.M.; Desoukey, S.Y.; Mohamed, K.M.; Kamel, M.S. Potential of Ficus microcarpa metabolites against SARS-CoV-2 main protease supported by docking studies. Nat. Prod. Res. 2020. [Google Scholar] [CrossRef]
- El Hawary, S.S.; Khattab, A.R.; Marzouk, H.S.; El Senousy, A.S.; Alex, M.G.; Aly, O.M.; Abdelmohsen, U.R. In silico identification of SARS-CoV-2 spike (S) protein-ACE2 complex inhibitors from eight Tecoma species and cultivars analyzed by LC-MS. RSC Adv. 2020, 10, 43103–43108. [Google Scholar] [CrossRef]
- Albohy, A.; Zahran, E.M.; Abdelmohsen, U.R.; Salem, M.A.; Al-Warhi, T.; Al-Sanea, M.M.; Kamel, M.S. Multitarget in silico studies of Ocimum menthiifolium, family Lamiaceae against SARS-CoV-2 supported by molecular dynamics simulation. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Sharun, K.; Dhama, K.; Patel, S.K.; Pathak, M.; Tiwari, R.; Singh, B.R.; Sah, R.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J.; Leblebicioglu, H. Ivermectin, a new candidate therapeutic against SARS-CoV-2/COVID-19. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 1–5. [Google Scholar] [CrossRef]
- Behera, P.; Patro, B.K.; Singh, A.K.; Chandanshive, P.D.; Pradhan, S.K.; Pentapati, S.S.K.; Mohanty, R.R. Role of ivermectin in the prevention of SARS-CoV-2 infection among healthcare workers in India: A matched case-control study. PLoS ONE 2021, 16, e0247163. [Google Scholar] [CrossRef]
- Cao, R.; Hu, H.; Li, Y.; Wang, X.; Xu, M.; Liu, J.; Zhong, W. Anti-SARS-CoV-2 potential of artemisinins in vitro. ACS Infect. Dis. 2020, 6, 2524–2531. [Google Scholar] [CrossRef]
- Uckun, F.M.; Saund, S.; Windlass, H.; Trieu, V. Repurposing Anti-Malaria Phytomedicine Artemisinin as a COVID-19 Drug. Front. Pharmacol. 2021, 12, 407. [Google Scholar] [CrossRef]
- Abdallah, H.; El-Halawany, A.; Sirwi, A.; El-Araby, A.; Mohamed, G.; Ibrahim, S.; Koshak, A.; Asfour, H.; Awan, Z.; Elfaky, M.A. Repurposing of Some Natural Product Isolates as SARS-COV-2 Main Protease Inhibitors via In Vitro Cell Free and Cell-Based Antiviral Assessments and Molecular Modeling Approaches. Pharmaceuticals 2021, 14, 213. [Google Scholar] [CrossRef]
- Su, H.X.; Yao, S.; Zhao, W.F.; Li, M.J.; Liu, J.; Shang, W.J.; Xu, Y.C. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.A.; Kato-Weinstein, J.; Li, Y.; Deng, Y.; Granet, R.; Garner, L.; Liu, C.Y.; Polshakov, D.; Gessner, C.; Watkins, S. Potential Therapeutic Agents and Associated Bioassay Data for COVID-19 and Related Human Coronavirus Infections. ACS Pharmacol. Transl. Sci. 2020, 3, 813–834. [Google Scholar] [CrossRef] [PubMed]
- Shady, N.H.; Youssif, K.A.; Sayed, A.M.; Belbahri, L.; Oszako, T.; Hassan, H.M.; Abdelmohsen, U.R. Sterols and Triterpenes: Antiviral Potential Supported by In-Silico Analysis. Plants 2020, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Michalska, K.; Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Stols, L.; Endres, M.; Joachimiak, A. Crystal structures of SARS-CoV-2 ADP-ribose phosphatase: From the apo form to ligand complexes. IUCrJ 2020, 7, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Alhammad, Y.M.O.; Fehr, A.R. The Viral Macrodomain Counters Host Antiviral ADP-Ribosylation. Viruses 2020, 12, 384. [Google Scholar] [CrossRef] [PubMed]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef]
- Pervushin, K.; Tan, E.; Parthasarathy, K.; Lin, X.; Jiang, F.L.; Yu, D.; Vararattanavech, A.; Soong, T.W.; Liu, D.X.; Torres, J. Structure and Inhibition of the SARS Coronavirus Envelope Protein Ion Channel. PLoS Pathog. 2009, 5, e1000511. [Google Scholar] [CrossRef]
- Wilson, L.; Gage, P.; Ewart, G. Hexamethylene amiloride blocks E protein ion channels and inhibits coronavirus replication. Virology 2006, 353, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Paśko, P.; Tyszka-Czochara, M.; Galanty, A.; Gdula-Argasińska, J.; Żmudzki, P.; Bartoń, H.; Gorinstein, S. Comparative study of predominant phytochemical compounds and proapoptotic potential of broccoli sprouts and florets. Plant Foods Hum. Nutr. 2018, 73, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Karakida, F.; Ikeya, Y.; Tsunakawa, M.; Yamaguchi, T.; Ikarashi, Y.; Takeda, S.; Aburada, M. Cerebral Protective and Cognition-Improving Effects of Sinapic Acid in Rodents. Biol. Pharm. Bull. 2007, 30, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Cherng, Y.-G.; Tsai, C.-C.; Chung, H.-H.; Lai, Y.-W.; Kuo, S.-C.; Cheng, J.-T. Antihyperglycemic Action of Sinapic Acid in Diabetic Rats. J. Agric. Food Chem. 2013, 61, 12053–12059. [Google Scholar] [CrossRef]
- Yun, K.-J.; Koh, D.-J.; Kim, S.-H.; Park, S.J.; Ryu, J.H.; Kim, D.-G.; Lee, J.-Y.; Lee, K.-T. Anti-Inflammatory Effects of Sinapic Acid through the Suppression of Inducible Nitric Oxide Synthase, Cyclooxygase-2 and Proinflammatory Cytokines Expressions via Nuclear Factor-κB Inactivation. J. Agric. Food Chem. 2008, 56, 10265–10272. [Google Scholar] [CrossRef]
- Martinović, N.; Ulrih, N.P.; Abramovič, H. Sinapic Acid and its Derivatives Increase Oxidative Stability in Different Model Lipid Systems. Eur. J. Lipid Sci. Technol. 2019, 121, 1800326. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; Tummino, T.A. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Seeliger, D.; De Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Release, S. 3: Desmond Molecular Dynamics System, DE Shaw Research, New York, NY, 2017; Maestro-Desmond Interoperability Tools, Schrödinger: New York, NY, USA, 2017. [Google Scholar]
- Schrodinger LLC. Maestro; Version 9.0; Schrodinger LLC: New York, NY, USA, 2009. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. Charmm-Gui: A web-based graphical user interface for Charmm. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Alhadrami, H.A.; Hamed, A.A.; Hassan, H.M.; Belbahri, L.; Rateb, M.E.; Sayed, A.M. Flavonoids as Potential anti-MRSA Agents through Modulation of PBP2a: A Computational and Experimental Study. Antibiotics 2020, 9, 562. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orfali, R.; Rateb, M.E.; Hassan, H.M.; Alonazi, M.; Gomaa, M.R.; Mahrous, N.; GabAllah, M.; Kandeil, A.; Perveen, S.; Abdelmohsen, U.R.; et al. Sinapic Acid Suppresses SARS CoV-2 Replication by Targeting Its Envelope Protein. Antibiotics 2021, 10, 420. https://doi.org/10.3390/antibiotics10040420
Orfali R, Rateb ME, Hassan HM, Alonazi M, Gomaa MR, Mahrous N, GabAllah M, Kandeil A, Perveen S, Abdelmohsen UR, et al. Sinapic Acid Suppresses SARS CoV-2 Replication by Targeting Its Envelope Protein. Antibiotics. 2021; 10(4):420. https://doi.org/10.3390/antibiotics10040420
Chicago/Turabian StyleOrfali, Raha, Mostafa E. Rateb, Hossam M. Hassan, Mona Alonazi, Mokhtar R. Gomaa, Noura Mahrous, Mohamed GabAllah, Ahmed Kandeil, Shagufta Perveen, Usama Ramadan Abdelmohsen, and et al. 2021. "Sinapic Acid Suppresses SARS CoV-2 Replication by Targeting Its Envelope Protein" Antibiotics 10, no. 4: 420. https://doi.org/10.3390/antibiotics10040420
APA StyleOrfali, R., Rateb, M. E., Hassan, H. M., Alonazi, M., Gomaa, M. R., Mahrous, N., GabAllah, M., Kandeil, A., Perveen, S., Abdelmohsen, U. R., & Sayed, A. M. (2021). Sinapic Acid Suppresses SARS CoV-2 Replication by Targeting Its Envelope Protein. Antibiotics, 10(4), 420. https://doi.org/10.3390/antibiotics10040420