
Combining Functional Genomics and Whole-Genome Sequencing to Detect Antibiotic Resistance Genes in Bacterial Strains Co-Occurring Simultaneously in a Brazilian Hospital

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
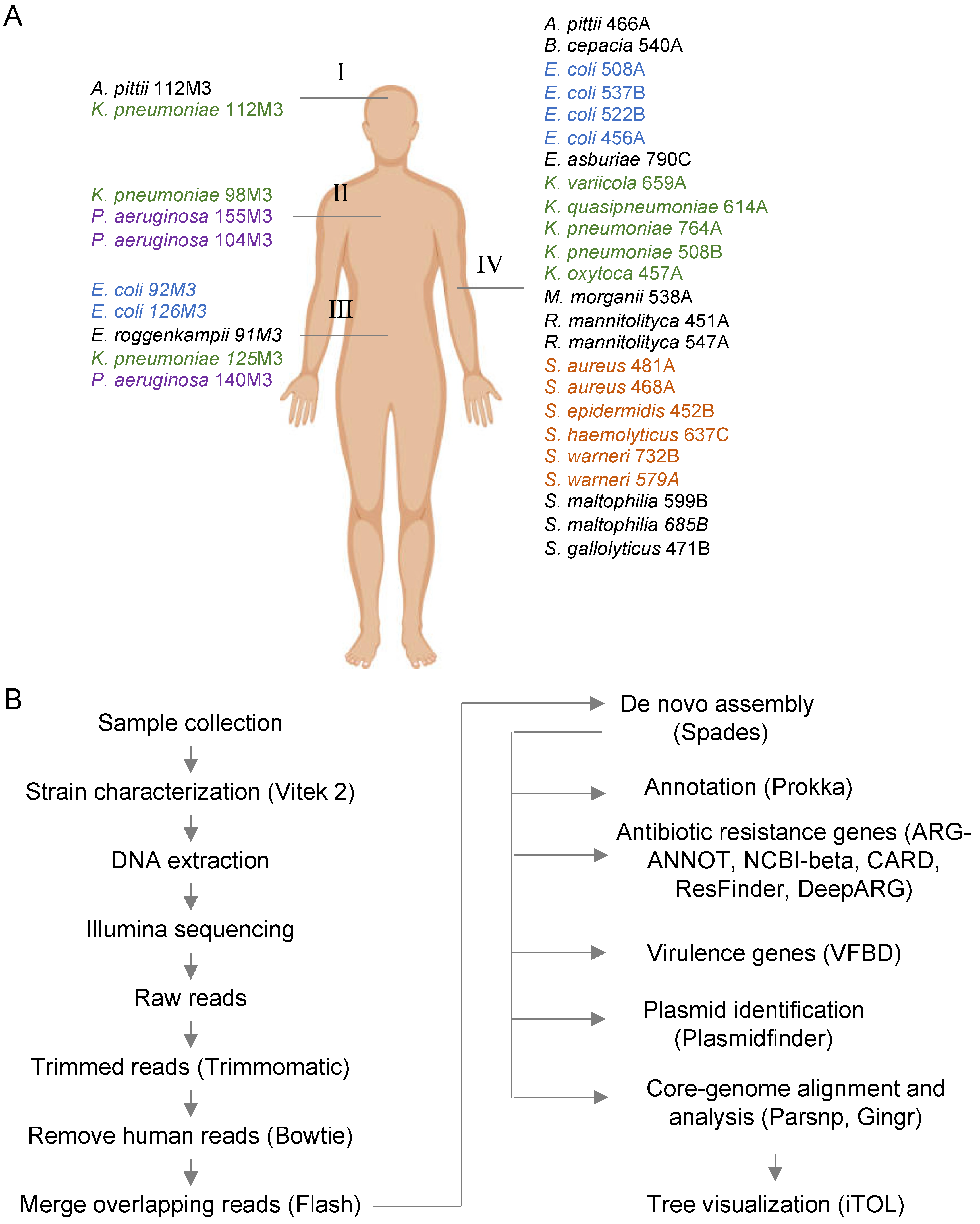
2.1. Sample Collection
2.2. DNA Extraction and Genome Sequencing
2.3. Identification of Antibiotic Resistance Genes, Plasmids, and Phylogenomic Analysis
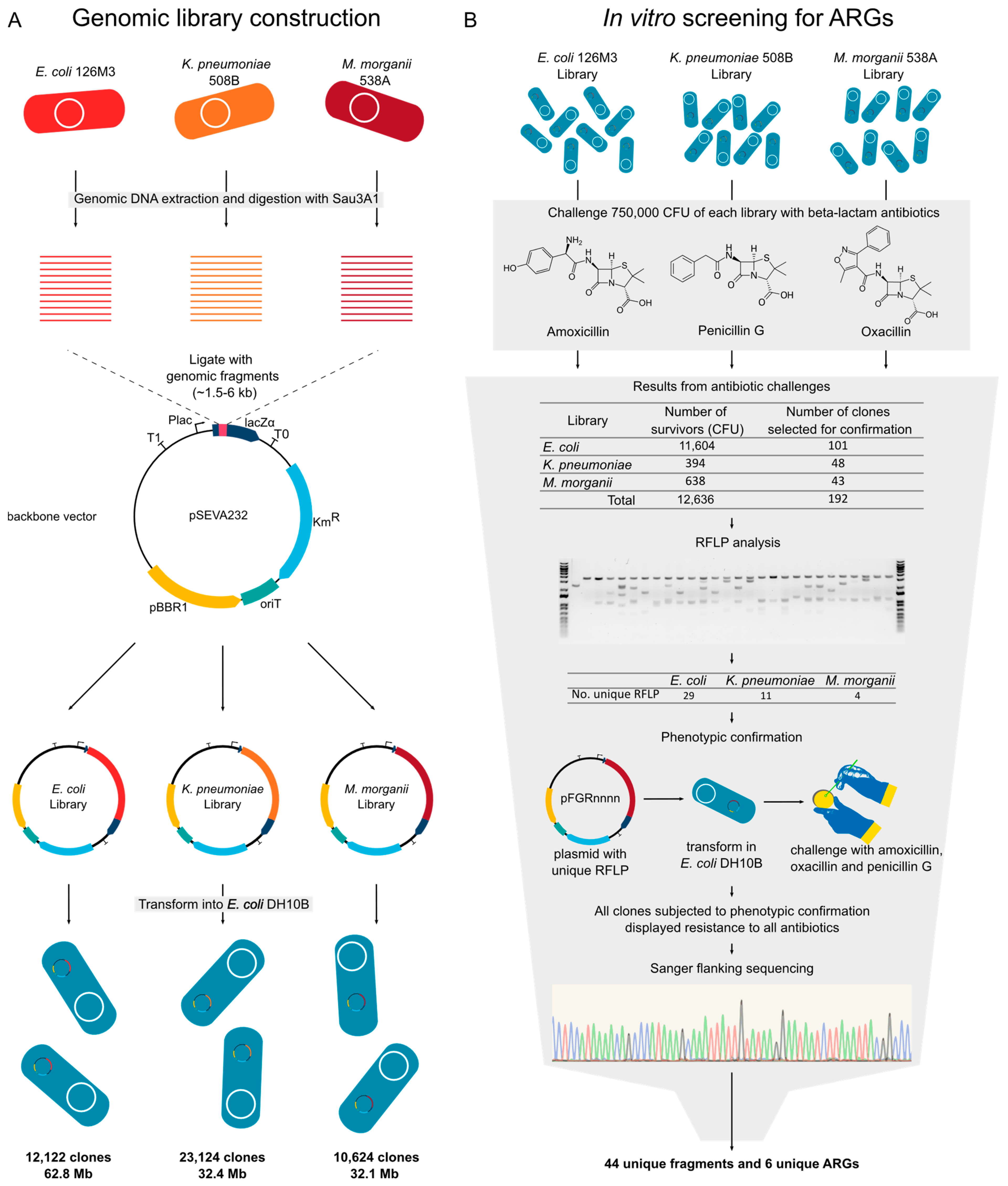
2.4. Genomic Library Construction
2.5. Determination of Minimum Inhibitory Concentrations (MICs)
2.6. Screening and Phenotype Confirmation
2.7. Extraction of Insert Sequence from Assembled Genomes
3. Results and Discussion
3.1. WGS Analysis of Clinical Strains Isolated Over the Same Two Weeks
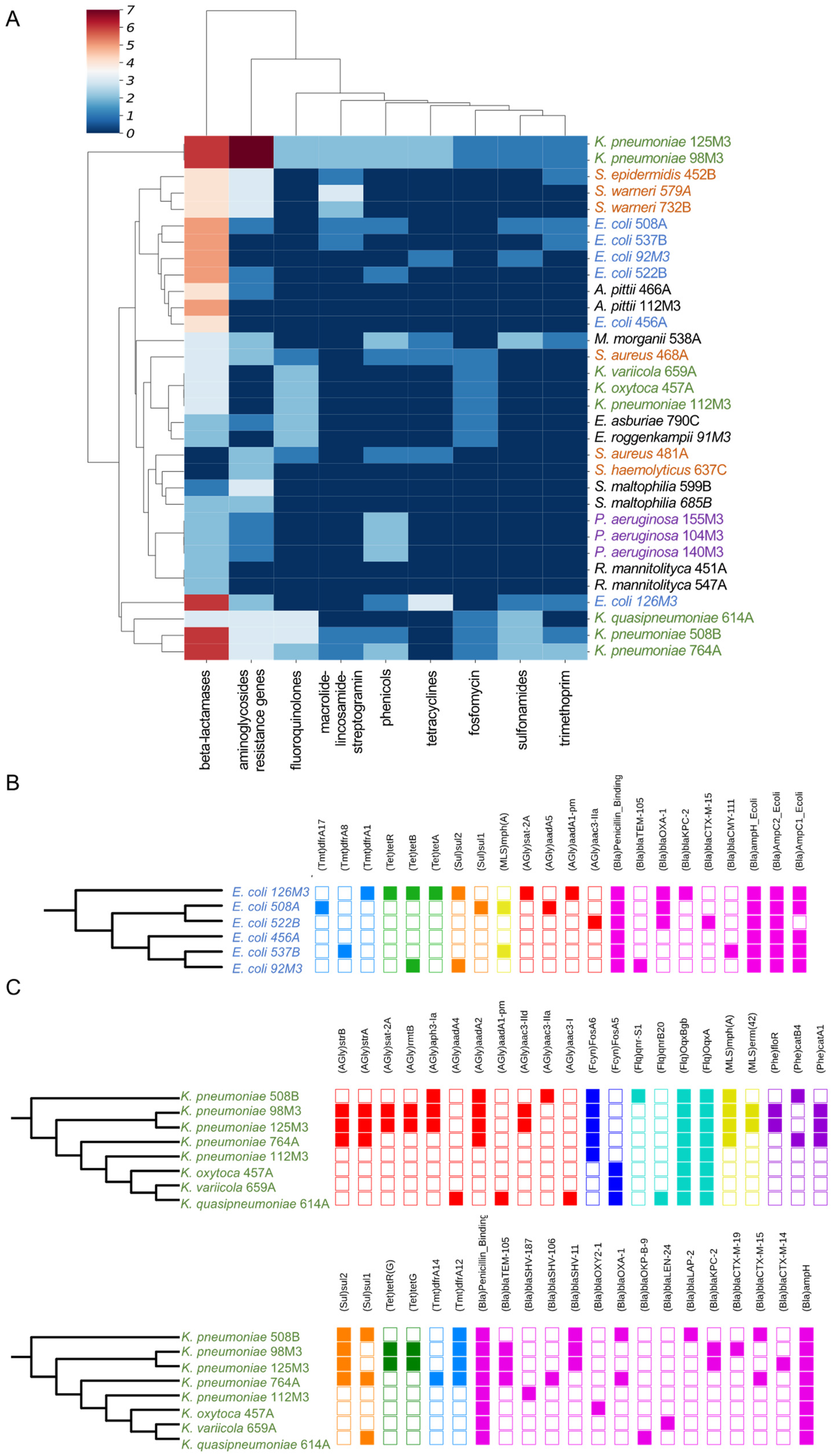
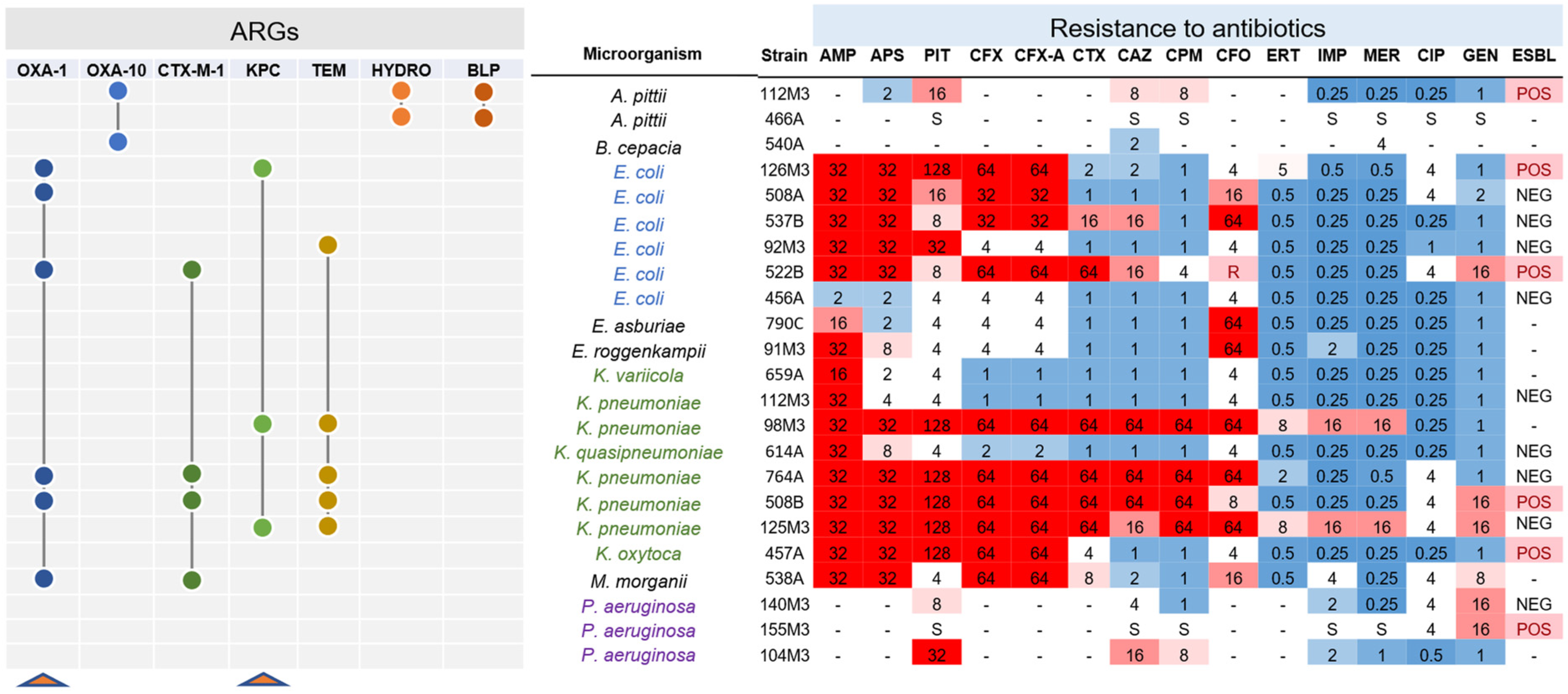
3.2. Identification of Resistance Genes in Clinical Strains
3.3. Intrinsic Resistome and Hospital-Associated Resistome
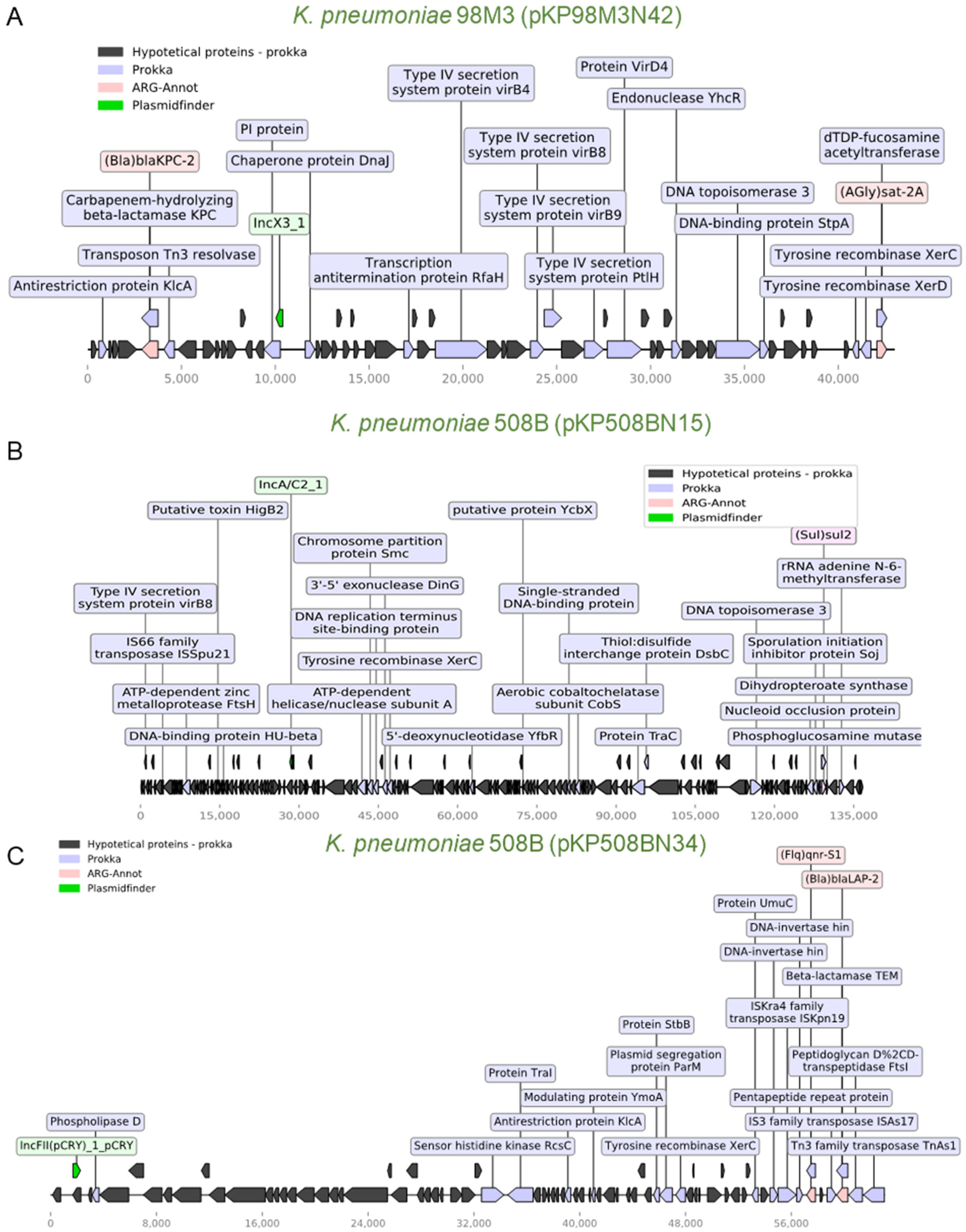
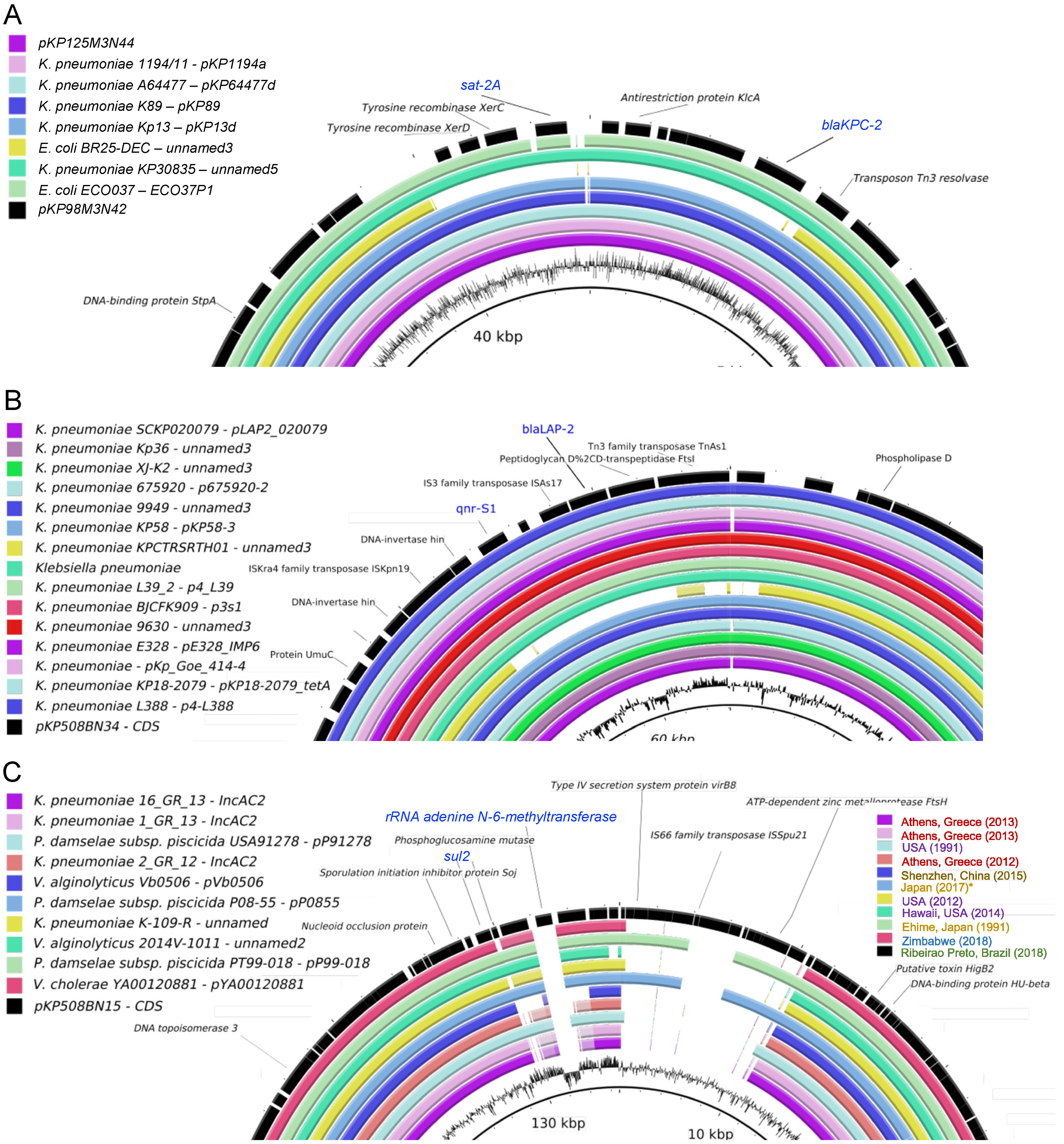
3.4. Identification of ARGs Located in Plasmids
3.5. Experimental Validation of Functional Beta-Lactamases from High-Resistance Bacteria
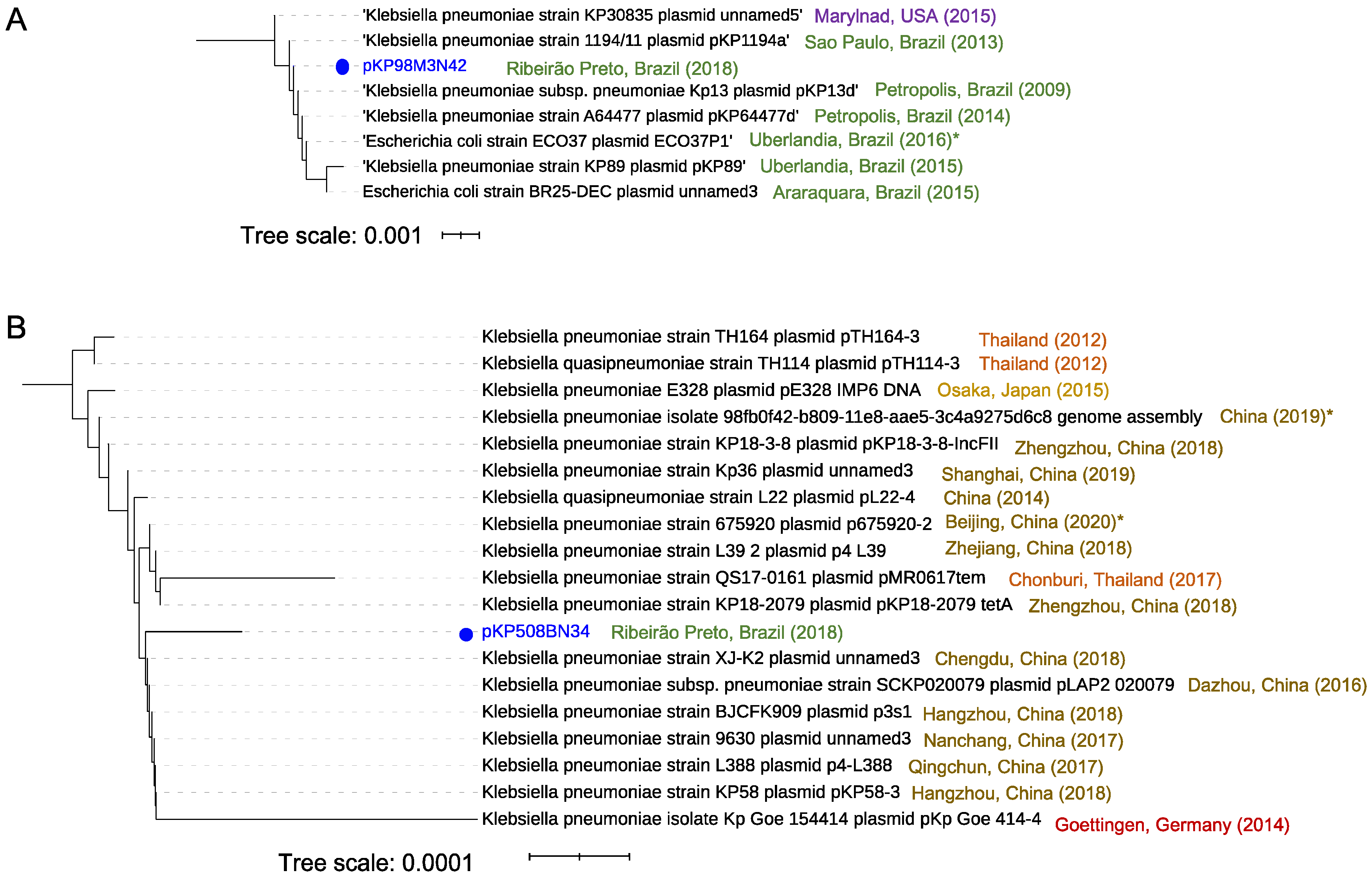
3.6. Mapping of Plasmids in the Brazilian Territory
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shetty, N.; Hill, G.; Ridgway, G.L. The Vitek analyser for routine bacterial identification and susceptibility testing: Protocols, problems, and pitfalls. J. Clin. Pathol. 1998, 51, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, P.; Carroll, K.C.; Buchan, B.W.; Chan, R.C.; Dhiman, N.; Ford, B.; Granato, P.A.; Harrington, A.T.; Hernandez, D.R.; Humphries, R.M.; et al. Multicenter Evaluation of the Accelerate PhenoTest BC Kit for Rapid Identification and Phenotypic Antimicrobial Susceptibility Testing Using Morphokinetic Cellular Analysis. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef]
- Köser, C.U.; Ellington, M.J.; Cartwright, E.J.P.; Gillespie, S.H.; Brown, N.M.; Farrington, M.; Holden, M.T.G.; Dougan, G.; Bentley, S.D.; Parkhill, J.; et al. Routine Use of Microbial Whole Genome Sequencing in Diagnostic and Public Health Microbiology. PLoS Pathog. 2012, 8, e1002824. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Goglin, K.; Molyneaux, N.; Hujer, K.M.; Lavender, H.; Jamison, J.J.; Macdonald, I.J.; Martin, K.M.; Russo, T.; Campagnari, A.A.; et al. Comparative Genome Sequence Analysis of Multidrug-Resistant Acinetobacter baumannii. J. Bacteriol. 2008, 190, 8053–8064. [Google Scholar] [CrossRef] [PubMed]
- Wyres, K.L.; Lam, M.M.C.; Holt, K.E. Population genomics of Klebsiella pneumoniae. Nat. Rev. Genet. 2020, 18, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Snitkin, E.S.; Won, S.; Pirani, A.; Lapp, Z.; Weinstein, R.A.; Lolans, K.; Hayden, M.K. Integrated genomic and interfacility patient-transfer data reveal the transmission pathways of multidrug-resistantKlebsiella pneumoniaein a regional outbreak. Sci. Transl. Med. 2017, 9, eaan0093. [Google Scholar] [CrossRef]
- Snitkin, E.S.; Zelazny, A.M.; Thomas, P.J.; Stock, F.; Henderson, D.K.; Palmore, T.N.; Segre, J.A.; Nisc Comparative Sequencing NISC Comparative Sequencing Program Group. Tracking a Hospital Outbreak of Carbapenem-Resistant Klebsiella pneumoniae with Whole-Genome Sequencing. Sci. Transl. Med. 2012, 4, 148ra116. [Google Scholar] [CrossRef]
- Quick, J.; Cumley, N.; Wearn, C.M.; Niebel, M.; Constantinidou, C.; Thomas, C.M.; Pallen, M.J.; Moiemen, N.S.; Bamford, A.; Oppenheim, B.; et al. Seeking the source ofPseudomonas aeruginosainfections in a recently opened hospital: An observational study using whole-genome sequencing. BMJ Open 2014, 4, e006278. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Bowden, R.; Wilson, D.J.; Peto, T.E.A.; Crook, D.W. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 2012, 13, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Pankhurst, L.J.; Elias, C.D.O.; Votintseva, A.A.; Walker, T.M.; Cole, K.; Davies, J.; Fermont, J.M.; Gascoyne-Binzi, D.M.; Kohl, T.A.; Kong, C.; et al. Rapid, comprehensive, and affordable mycobacterial diagnosis with whole-genome sequencing: A prospective study. Lancet Respir. Med. 2016, 4, 49–58. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Naccache, S.N.; Federman, S.; Veeraraghavan, N.; Zaharia, M.; Lee, D.; Samayoa, E.; Bouquet, J.; Greninger, A.L.; Luk, K.-C.; Enge, B.; et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 2014, 24, 1180–1192. [Google Scholar] [CrossRef]
- Greninger, A.L.; Naccache, S.N.; Federman, S.; Yu, G.; Mbala, P.; Bres, V.; Stryke, D.; Bouquet, J.; Somasekar, S.; Linnen, J.M.; et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015, 7, 1–13. [Google Scholar] [CrossRef]
- Aytan-Aktug, D.; Clausen, P.T.L.C.; Bortolaia, V.; Aarestrup, F.M.; Lund, O. Prediction of Acquired Antimicrobial Resistance for Multiple Bacterial Species Using Neural Networks. mSystems 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.F.; Olm, M.R.; Morowitz, M.J.; Banfield, J.F. Machine Learning Leveraging Genomes from Metagenomes Identifies Influential Antibiotic Resistance Genes in the Infant Gut Microbiome. mSystems 2018, 3, e00123-17. [Google Scholar] [CrossRef]
- Macesic, N.; Walk, O.J.B.D.; Pe’Er, I.; Tatonetti, N.P.; Peleg, A.Y.; Uhlemann, A.-C. Predicting Phenotypic Polymyxin Resistance in Klebsiella pneumoniae through Machine Learning Analysis of Genomic Data. mSystems 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.A.; Reyes, J.; Carvajal, L.P.; Rincon, S.; Diaz, L.; Panesso, D.; Ibarra, G.; Rios, R.; Munita, J.M.; Salles, M.J.; et al. A Prospective Cohort Multicenter Study of Molecular Epidemiology and Phylogenomics of Staphylococcus aureus Bacteremia in Nine Latin American Countries. Antimicrob. Agents Chemother. 2017, 61, e00816-17. [Google Scholar] [CrossRef] [PubMed]
- David, S.; The EuSCAPE Working Group; Reuter, S.; Harris, S.R.; Glasner, C.; Feltwell, T.; Argimon, S.; Abudahab, K.; Goater, R.; Giani, T.; et al. Epidemic of carbapenem-resistant Klebsiella pneumoniae in Europe is driven by nosocomial spread. Nat. Microbiol. 2019, 4, 1919–1929. [Google Scholar] [CrossRef]
- Arimizu, Y.; Kirino, Y.; Sato, M.P.; Uno, K.; Sato, T.; Gotoh, Y.; Auvray, F.; Brugere, H.; Oswald, E.; Mainil, J.G.; et al. Large-scale genome analysis of bovine commensal Escherichia coli reveals that bovine-adapted E. coli lineages are serving as evolutionary sources of the emergence of human intestinal pathogenic strains. Genome Res. 2019, 29, 1495–1505. [Google Scholar] [CrossRef]
- Bennett, P.M. Plasmid encoded antibiotic resistance: Acquisition and transfer of antibiotic resistance genes in bacteria. Br. J. Pharmacol. 2008, 153, S347–S357. [Google Scholar] [CrossRef]
- Millan, A.S. Evolution of Plasmid-Mediated Antibiotic Resistance in the Clinical Context. Trends Microbiol. 2018, 26, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Lerminiaux, N.A.; Cameron, A.D. Horizontal transfer of antibiotic resistance genes in clinical environments. Can. J. Microbiol. 2019, 65, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Porse, A.; Schønning, K.; Munck, C.; Sommer, M.O. Survival and Evolution of a Large Multidrug Resistance Plasmid in New Clinical Bacterial Hosts. Mol. Biol. Evol. 2016, 33, 2860–2873. [Google Scholar] [CrossRef]
- Buckner, M.M.C.; Ciusa, M.L.; Meek, R.W.; Moorey, A.R.; McCallum, G.E.; Prentice, E.L.; Reid, J.P.; Alderwick, L.J.; Di Maio, A.; Piddock, L.J.V. HIV Drugs Inhibit Transfer of Plasmids Carrying Extended-Spectrum β-Lactamase and Carbapenemase Genes. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Arango-Argoty, G.; Garner, E.; Pruden, A.; Heath, L.S.; Vikesland, P.; Zhang, L. DeepARG: A deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 2018, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Silva-Rocha, R.; Martínez-García, E.; Calles, B.; Chavarría, M.; Arce-Rodríguez, A.; Heras, A.D.L.; Páez-Espino, A.D.; Durante-Rodríguez, G.; Kim, J.; Nikel, P.I.; et al. The Standard European Vector Architecture (SEVA): A coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 2012, 41, D666–D675. [Google Scholar] [CrossRef]
- Martínez-García, E.; Aparicio, T.; Goñi-Moreno, A.; Fraile, S.; De Lorenzo, V. SEVA 2.0: An update of the Standard European Vector Architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res. 2015, 43, D1183–D1189. [Google Scholar] [CrossRef]
- Martínez-García, E.; Goñi-Moreno, A.; Bartley, B.; McLaughlin, J.; Sánchez-Sampedro, L.; Del Pozo, H.P.; Hernández, C.P.; Marletta, A.S.; De Lucrezia, D.; Sánchez-Fernández, G.; et al. SEVA 3.0: An update of the Standard European Vector Architecture for enabling portability of genetic constructs among diverse bacterial hosts. Nucleic Acids Res. 2019, 48, D1164–D1170. [Google Scholar] [CrossRef]
- Drury, L. Transformation of Bacteria by Electroporation. Basic DNA RNA Protoc. 2003, 58, 249–256. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 28th ed.; CLSI Supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.S.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Johnson, S.L.; Bishop-Lilly, K.A.; Ladner, J.T.; Daligault, H.E.; Davenport, K.W.; Jaissle, J.; Frey, K.G.; Koroleva, G.I.; Bruce, D.C.; Coyne, S.R.; et al. Complete Genome Sequences for 59BurkholderiaIsolates, Both Pathogenic and Near Neighbor: TABLE 1. Genome Announc. 2015, 3, e00159-15. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.A.; Melano, R.; Cárdenas, P.A.; Trueba, G. Mobile genetic elements associated with carbapenemase genes in South American Enterobacterales. Braz. J. Infect. Dis. 2020, 24, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Filho, R.A.C.P.; Andrade, L.N.; Junior, A.B.; Darini, A.L.C. Evaluation and characterization of plasmids carrying CTX-M genes in a non-clonal population of multidrug-resistant Enterobacteriaceae isolated from poultry in Brazil. Diagn. Microbiol. Infect. Dis. 2016, 85, 444–448. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, 1–61. [Google Scholar] [CrossRef]
- Cantón, R.; Novais, A.; Valverde, A.; Machado, E.; Peixe, L.; Baquero, F.; Coque, T. Prevalence and spread of extended-spectrum β-lactamase-producing Enterobacteriaceae in Europe. Clin. Microbiol. Infect. 2008, 14, 144–153. [Google Scholar] [CrossRef]
- Olivares, J.; Ebernardini, A.; Egarcia-Leon, G.; Ecorona, F.; Esanchez, M.B.; Martinez, J.L. The intrinsic resistome of bacterial pathogens. Front. Microbiol. 2013, 4, 103. [Google Scholar] [CrossRef]
- Cox, G.; Wright, G.D. Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol. 2013, 303, 287–292. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Naas, T.; Oueslati, S.; Bonnin, R.A.; Dabos, M.L.; Zavala, A.; Dortet, L.; Retailleau, P.; Iorga, B.I. Beta-lactamase database (BLDB)–structure and function. J. Enzym. Inhib. Med. Chem. 2017, 32, 917–919. [Google Scholar] [CrossRef]
- Peter, S.; Bosio, M.; Gross, C.; Bezdan, D.; Gutierrez, J.; Oberhettinger, P.; Liese, J.; Vogel, W.; Dörfel, D.; Berger, L.; et al. Tracking of Antibiotic Resistance Transfer and Rapid Plasmid Evolution in a Hospital Setting by Nanopore Sequencing. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Prussing, C.; Snavely, E.A.; Singh, N.; Lapierre, P.; Lasek-Nesselquist, E.; Mitchell, K.; Haas, W.; Owsiak, R.; Nazarian, E.; Musser, K.A. Nanopore MinION Sequencing Reveals Possible Transfer of blaKPC–2 Plasmid Across Bacterial Species in Two Healthcare Facilities. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zou, S.; Chen, H.; Yu, Y.; Ruan, Z. BacWGSTdb 2.0: A one-stop repository for bacterial whole-genome sequence typing and source tracking. Nucleic Acids Res. 2021, 49, D644–D650. [Google Scholar] [CrossRef]
- Ruan, Z.; Feng, Y. BacWGSTdb, a database for genotyping and source tracking bacterial pathogens. Nucleic Acids Res. 2016, 44, D682–D687. [Google Scholar] [CrossRef] [PubMed]
- Zulkower, V.; Rosser, S. DNA Features Viewer: A sequence annotation formatting and plotting library for Python. Bioinformatics 2020, 36, 4350–4352. [Google Scholar] [CrossRef] [PubMed]
- Rozwandowicz, M.; Brouwer, M.S.M.; Fischer, J.; Wagenaar, J.A.; Gonzalez-Zorn, B.; Guerra, B.; Mevius, D.J.; Hordijk, J. Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 2018, 73, 1121–1137. [Google Scholar] [CrossRef] [PubMed]
- Wein, T.; Hülter, N.F.; Mizrahi, I.; Dagan, T. Emergence of plasmid stability under non-selective conditions maintains antibiotic resistance. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Martínez, J.L.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Genet. 2015, 13, 116–123. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Larsson, D.G.J. Antibiotic resistance genes in the environment: Prioritizing risks. Nat. Rev. Genet. 2015, 13, 396. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Naas, T.; Poirel, L. Global Spread of Carbapenemase-producingEnterobacteriaceae. Emerg. Infect. Dis. 2011, 17, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Liakopoulos, A.; Van Der Goot, J.; Bossers, A.; Betts, J.; Brouwer, M.S.M.; Kant, A.; Smith, H.; Ceccarelli, D.; Mevius, D. Genomic and functional characterisation of IncX3 plasmids encoding blaSHV-12 in Escherichia coli from human and animal origin. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Ho, P.-L.; Cheung, Y.-Y.; Lo, W.-U.; Li, Z.; Chow, K.-H.; Lin, C.-H.; Chan, J.F.-W.; Cheng, V.C.-C. Molecular Characterization of an Atypical IncX3 Plasmid pKPC-NY79 Carrying bla KPC-2 in a Klebsiella pneumoniae. Curr. Microbiol. 2013, 67, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Cerdeira, L.T.; Cunha, M.P.; Francisco, G.R.; Bueno, M.F.C.; Araujo, B.F.; Ribas, R.M.; Gontijo-Filho, P.P.; Knöbl, T.; Garcia, D.D.O.; Lincopan, N. IncX3 plasmid harboring a non-Tn 4401 genetic element (NTE KPC) in a hospital-associated clone of KPC-2-producing Klebsiella pneumoniae ST340/CG258. Diagn. Microbiol. Infect. Dis. 2017, 89, 164–167. [Google Scholar] [CrossRef]
- Fuga, B.; Ferreira, M.L.; Cerdeira, L.T.; De Campos, P.A.; Dias, V.L.; Rossi, I.; Machado, L.G.; Lincopan, N.; Gontijo-Filho, P.P.; Ribas, R.M. Novel small IncX3 plasmid carrying the blaKPC-2 gene in high-risk Klebsiella pneumoniae ST11/CG258. Diagn. Microbiol. Infect. Dis. 2020, 96, 114900. [Google Scholar] [CrossRef] [PubMed]
- Partridge, S.R.; Ginn, A.N.; Wiklendt, A.M.; Ellem, J.; Wong, J.S.; Ingram, P.; Guy, S.; Garner, S.; Iredell, J.R. Emergence of blaKPC carbapenemase genes in Australia. Int. J. Antimicrob. Agents 2015, 45, 130–136. [Google Scholar] [CrossRef]
- Fortini, D.; Villa, L.; Feudi, C.; Pires, J.P.D.C.; Bonura, C.; Mammina, C.; Endimiani, A.; Carattoli, A. Double Copies of blaKPC-3::Tn4401a on an IncX3 Plasmid in Klebsiella Pneumoniae Successful Clone ST512 from Italy. Antimicrob. Agents Chemother. 2016, 60, 646–649. [Google Scholar] [CrossRef]
- Kassis-Chikhani, N.; Frangeul, L.; Drieux, L.; Sengelin, C.; Jarlier, V.; Brisse, S.; Arlet, G.; Decré, D. Complete Nucleotide Sequence of the First KPC-2- and SHV-12-Encoding IncX Plasmid, pKpS90, from Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2013, 57, 618–620. [Google Scholar] [CrossRef]
- Kim, J.O.; Song, S.A.; Yoon, E.-J.; Shin, J.H.; Lee, H.; Jeong, S.H.; Lee, K. Outbreak of KPC-2-producing Enterobacteriaceae caused by clonal dissemination of Klebsiella pneumoniae ST307 carrying an IncX3-type plasmid harboring a truncated Tn4401a. Diagn. Microbiol. Infect. Dis. 2017, 87, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Kim, J.O.; Yoon, E.-J.; Bae, I.K.; Lee, W.; Lee, H.; Park, Y.; Lee, K.; Jeong, S.H. Extensively Drug-Resistant Escherichia coli Sequence Type 1642 Carrying an IncX3 Plasmid Containing the blaKPC-2 Gene Associated with Transposon Tn4401a. Ann. Lab. Med. 2018, 38, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Fang, L.; Fu, Y.; Du, X.; Shen, Y.; Yu, Y. Dissemination of NDM-1-Producing Enterobacteriaceae Mediated by the IncX3-Type Plasmid. PLoS ONE 2015, 10, e0129454. [Google Scholar] [CrossRef] [PubMed]
- Acton, Q.A. Klebsiella Pneumoniae: New Insights for the Healthcare Professional, 2011 ed.; Scholarly Brief; Scholarly Editions: Atlanta, GA, USA, 2012. [Google Scholar]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Library | E. coli 126M3 | K. pneumoniae 508B | M. morganii 538A |
|---|---|---|---|
| Total number of clones | 13,442 | 25,693 | 11,804 |
| Percentage of clones with insert (%) | 90 | 90 | 90 |
| Number of clones with insert | 12,122 | 23124 | 10,624 |
| Insert size variation (kb) | 1.0–10.5 | 0.2–2.2 | 0.2–6.5 |
| Average insert size (kb) | 5.2 | 1.4 | 3.0 |
| Total genomic library size (mb) | 62.8 | 32.4 | 32.1 |
| Estimated genome coverage | 11.9× | 5.8× | 7.8× |
| Strain | In Silico (Argannot) | Number of Sequenced Resistant Clones |
|---|---|---|
| E. coli 126M3 | Penicillin_Binding_Protein_Ecoli | - |
| AmpC1_Ecoli | - | |
| blaKPC-2 | 28 | |
| AmpC2_Ecoli | 1 | |
| blaOXA-1 | - | |
| ampH_Ecoli | - | |
| K. pneumoniae 508B | blaSHV-11 | - |
| Penicillin_Binding_Protein_Ecoli | - | |
| blaLAP-2 | 2 | |
| ampH | - | |
| blaCTX-M-15 | 8 | |
| blaOXA-1 | 1 | |
| M. morganii 538A | blaCTX-M-15 | - |
| blaOXA-1 | - | |
| blaMOR-2 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borelli, T.C.; Lovate, G.L.; Scaranello, A.F.T.; Ribeiro, L.F.; Zaramela, L.; Pereira-dos-Santos, F.M.; Silva-Rocha, R.; Guazzaroni, M.-E. Combining Functional Genomics and Whole-Genome Sequencing to Detect Antibiotic Resistance Genes in Bacterial Strains Co-Occurring Simultaneously in a Brazilian Hospital. Antibiotics 2021, 10, 419. https://doi.org/10.3390/antibiotics10040419
Borelli TC, Lovate GL, Scaranello AFT, Ribeiro LF, Zaramela L, Pereira-dos-Santos FM, Silva-Rocha R, Guazzaroni M-E. Combining Functional Genomics and Whole-Genome Sequencing to Detect Antibiotic Resistance Genes in Bacterial Strains Co-Occurring Simultaneously in a Brazilian Hospital. Antibiotics. 2021; 10(4):419. https://doi.org/10.3390/antibiotics10040419
Chicago/Turabian StyleBorelli, Tiago Cabral, Gabriel Lencioni Lovate, Ana Flavia Tonelli Scaranello, Lucas Ferreira Ribeiro, Livia Zaramela, Felipe Marcelo Pereira-dos-Santos, Rafael Silva-Rocha, and María-Eugenia Guazzaroni. 2021. "Combining Functional Genomics and Whole-Genome Sequencing to Detect Antibiotic Resistance Genes in Bacterial Strains Co-Occurring Simultaneously in a Brazilian Hospital" Antibiotics 10, no. 4: 419. https://doi.org/10.3390/antibiotics10040419
APA StyleBorelli, T. C., Lovate, G. L., Scaranello, A. F. T., Ribeiro, L. F., Zaramela, L., Pereira-dos-Santos, F. M., Silva-Rocha, R., & Guazzaroni, M.-E. (2021). Combining Functional Genomics and Whole-Genome Sequencing to Detect Antibiotic Resistance Genes in Bacterial Strains Co-Occurring Simultaneously in a Brazilian Hospital. Antibiotics, 10(4), 419. https://doi.org/10.3390/antibiotics10040419