
Genomic Background and Phylogeny of cfiA-Positive Bacteroides fragilis Strains Resistant to Meropenem-EDTA

 ,
,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
2.1. Species Identification and Susceptibility Phenotype
2.2. Susceptibility Genotype
2.3. IS Elements
2.4. Plasmids
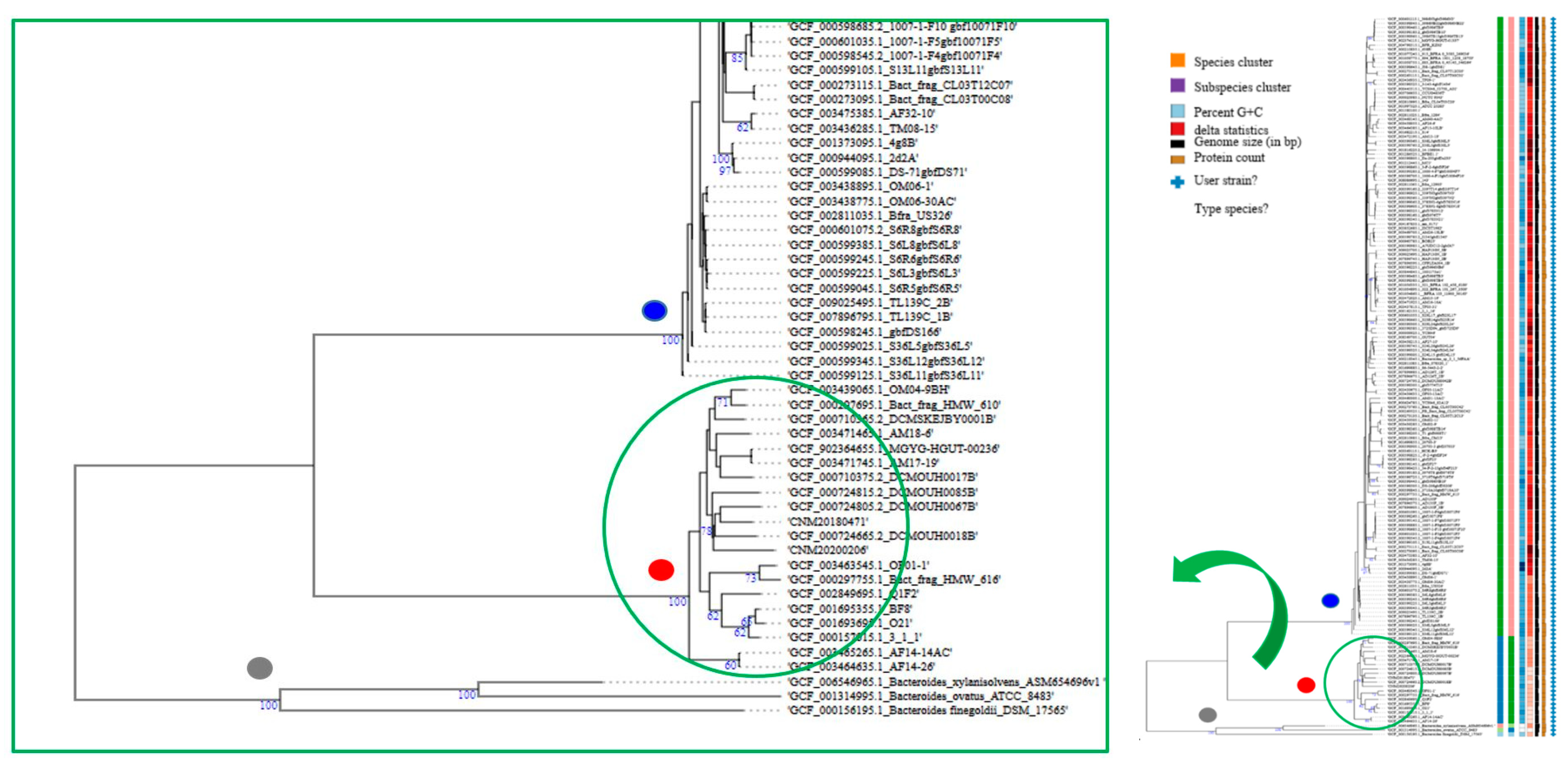
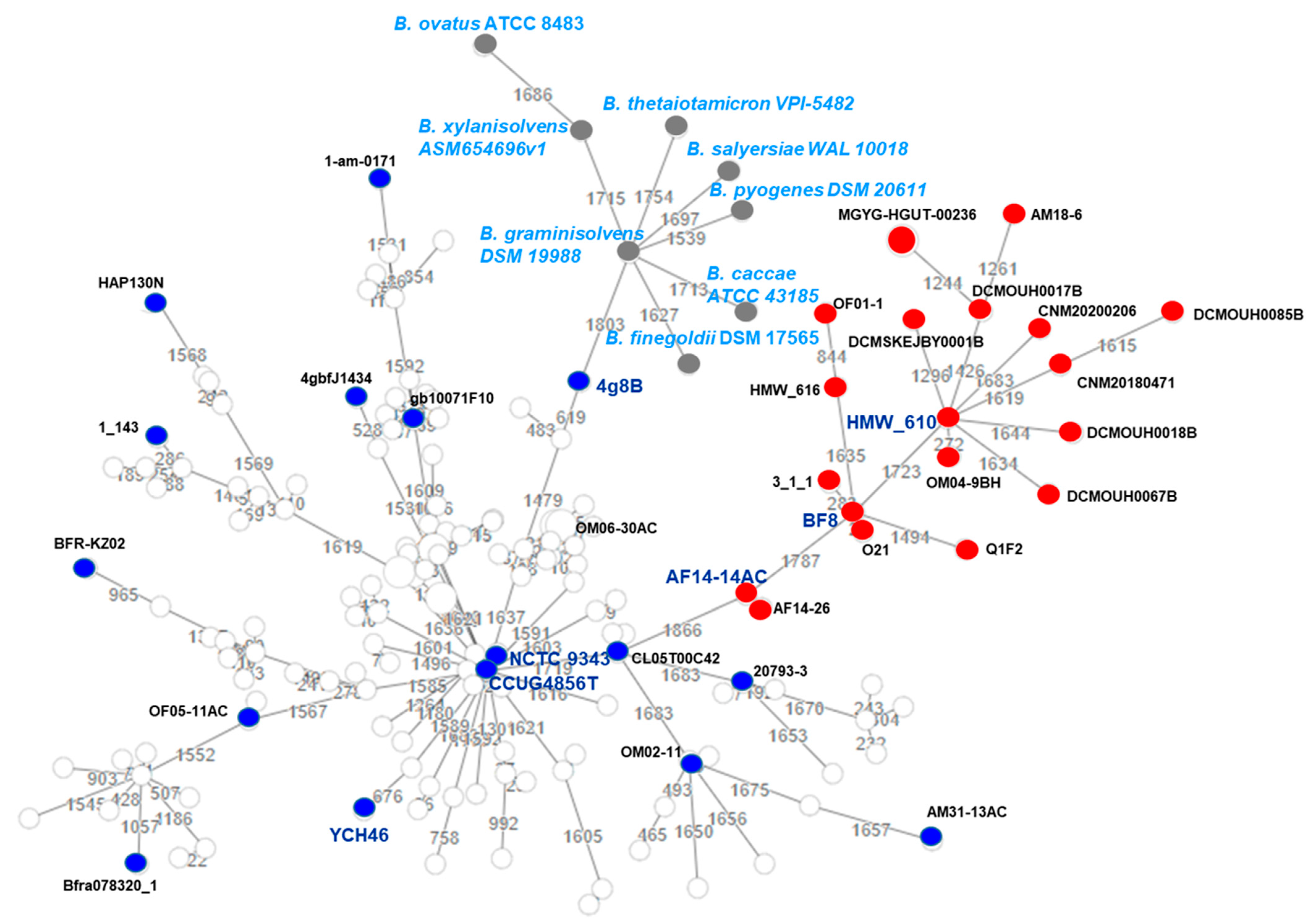
2.5. Genome-Based Taxonomy and B. fragilis Phylogeny
3. Materials and Methods
3.1. Case Reports
3.2. Identification and Antimicrobial Susceptibility Testing
3.3. Whole Genome Sequencing and de novo Assembly
3.4. Antimicrobial Resistance Genes, IS Elements, and Plasmids
3.5. Genome-Based Taxonomy and Phylogeny
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wexler, H.M. Bacteroides: The good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef]
- Husain, F.; Tang, K.; Veeranagouda, Y.; Boente, R.; Patrick, S.; Blakely, G.; Wexler, H.M. Novel large-scale chromosomal transfer in Bacteroides fragilis contributes to its pan-genome and rapid environmental adaptation. Microb. Genom. 2017, 3, e000136. [Google Scholar] [CrossRef]
- Nguyen, M.; Vedantam, G. Mobile genetic elements in the genus Bacteroides, and their mechanism(s) of dissemination. Mob. Genet Elem. 2011, 3, 187–196. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sóki, J.; Hedberg, M.; Patrick, S.; Bálint, B.; Herczeg, R.; Nagy, I.; Hecht, D.W.; Nagy, E.; Urbán, E. Emergence and evolution of an international cluster of MDR Bacteroides fragilis isolates. J. Antimicrob Chemother. 2016, 71, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Veloo, A.C.M.; Tokman, H.B.; Jean-Pierre, H.; Dumont, Y.; Jeverica, S.; Lienhardf, R.; Novak, A.; Rodloff, A.; Rotimi, V.; Wybo, I.; et al. Antimicrobial susceptibility profiles of anaerobic bacteria, isolated from human clinical specimens, within different European and surrounding countries. A joint ESCAI study. Anaerobe 2020, 61, 102111. [Google Scholar] [CrossRef] [PubMed]
- Treviño, M.; Areses, P.; Peñalver, M.D.; Cortizo, S.; Pardo, F.; Perez del Molino, M.L.; García-Riestra, C.; Hernández, M.; Llovo, J.; Regueiro, B.J. Susceptibility trends of Bacteroides fragilis group and characterisation of carbapenemase-producing strains by automated REP-PCR and MALDI-TOF. Anaerobe 2012, 18, 37–43. [Google Scholar] [CrossRef]
- Cobo, F.; Rodríguez-Granger, J.; Pérez-Zapata, I.; Sampedro, A.; Aliaga, L.; Navarro-Marí, J.M. Antimicrobial susceptibility and clinical findings of significant anaerobic bacteria in southern Spain. Anaerobe 2019, 59, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Alauzet, C.; Aujoulat, F.; Lozniewski, A.; Marchandin, H. A sequence database analysis of 5-nitroimidazole reductase and related proteins to expand knowledge on enzymes responsible for metronidazole inactivation. Anaerobe 2019, 55, 29–34. [Google Scholar] [CrossRef]
- Eitel, Z.; Sóki, J.; Urbán, E.; Nagy, E.; ESCMID Study Group on Anaerobic Infection. The prevalence of antibiotic resistance genes in Bacteroides fragilis group strains isolated in different European countries. Anaerobe 2013, 1, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Sydenham, T.V.; Overballe-Petersen, S.; Hasman, H.; Wexler, H.; Kemp, M.; Justesen, U.S. Complete hybrid genome assembly of clinical multidrug-resistant Bacteroides fragilis isolates enables comprehensive identification of antimicrobial-resistance genes and plasmids. Microb Genom. 2019, 5, e000312. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, G.R.; Shelby, A.; Shoemaker, N.B.; Salyers, A.A. A newly discovered Bacteroides conjugative transposon, CTnGERM1, contains genes also found in gram-positive bacteria. Appl. Environ. Microbiol. 2003, 69, 4595–4603. [Google Scholar] [CrossRef] [PubMed]
- 1Goto, T.; Tanaka, K.; Minh Tran, C.; Watanabe, K. Complete sequence of pBFUK1, a carbapenemase-harboring mobilizable plasmid from Bacteroides fragilis, and distribution of pBFUK1-like plasmids among carbapenem-resistant B. fragilis clinical isolates. J. Antibiot. 2013, 66, 239–242. [Google Scholar] [CrossRef]
- Gutacker, M.; Valsangiacomo, C.; Bernasconi, M.V.; Piffaretti, J.C. recA and glnA sequences separate the Bacteroides fragilis population into two genetic divisions associated with the antibiotic resistance genotypes cepA and cfiA. J. Med. Microbiol. 2002, 51, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Jeverica, S.; Sóki, J.; Premru, M.M.; Nagy, E.; Papst, L. High prevalence of division II (cfiA positive) isolates among blood stream Bacteroides fragilis in Slovenia as determined by MALDI-TOF MS. Anaerobe 2019, 58, 30–34. [Google Scholar] [CrossRef]
- Schwensen, S.A.; Acar, Z.; Sydenham, T.V.; Johansson, A.C.; Justesen, U.S. Phenotypic Detection of the cfiA Metallo-β-Lactamase in Bacteroides fragilis with the meropenem-EDTA Double-Ended Etest and the ROSCO KPC/MBL Confirm Kit. J. Antimicrob. Chemother. 2017, 72, 437–440. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Interpretive Criteria for Identification of Bacteria and Fungi by DNA Target Sequencing: Approved Guideline MM18-A; CLSI: Wayne, PA, USA, 2008. [Google Scholar]
- Litterio, M.R.; Cejas, G.; Gutkind, D.; Radice, M. Identification of CfiA coding genes in Bacteroides fragilis isolates recovered in Argentina. Inconsistencies in CfiA organization and nomenclature. Anaerobe 2017, 48, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Sydenham, T.V.; Sóki, J.; Hasman, H.; Wang, M.; Justesen, U.S.; ESGAI (ESCMID Study Group on Anaerobic Infections). Identification of antimicrobial resistance genes in multidrug-resistant clinical Bacteroides fragilis isolates by whole genome shotgun sequencing. Anaerobe 2015. [Google Scholar] [CrossRef]
- Miyamae, S.; Ueda, O.; Yoshimura, F.; Hwang, J.; Tanaka, Y.; Nikaido, H. A MATE family multidrug efflux transporter pumps out fluoroquinolones in Bacteroides Thetaiotaomicron. Antimicrob. Agents Chemother. 2001, 45, 3341–3346. [Google Scholar] [CrossRef]
- Markley, J.L.; Wencewicz, T.A. Tetracycline-Inactivating Enzymes. Front. Microbiol. 2018, 30, 1058. [Google Scholar] [CrossRef]
- Outten, F.W.; Huffman, D.L.; Hale, J.A.; O’Halloran, T.V. The independent cue and cus systems confer copper tolerance during aerobic and anaerobic growth in Escherichia coli. J. Biol. Chem. 2001, 276, 30670–30677. [Google Scholar] [CrossRef]
- Ghotaslou, R.; Yekani, M.; Memar, M.Y. The role of efflux pumps in Bacteroides fragilis resistance to antibiotics. Microbiol. Res. 2018, 210, 1–5. [Google Scholar] [CrossRef]
- Pumbwe, L.; Glass, D.; Wexler, H.M. Efflux pump overexpression in multiple antibiotic-resistant mutants of Bacteroides fragilis. Antimicrob. Agents Chemother. 2006, 50, 3150–3153. [Google Scholar] [CrossRef]
- Silva, C.M.G.; Silva, D.N.D.S.; Costa, S.B.; Bezerra, S.; Soares, J.; Ferreira, R.; Teixeira, F.L.; Domingues, R.M.C.P.; Lobo, L.A. Inactivation of MarR gene homologs increases susceptibility to antimicrobials in Bacteroides fragilis. Braz J. Microbiol. 2018, 49, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, T.; Yamashita, A.; Hirakawa, H.; Nakayama, H.; Toh, H.; Okada, N.; Kuhara, S.; Hattori, M.; Hayashi, T.; Ohnishi, Y. Genomic analysis of Bacteroides fragilis reveals extensive DNA inversions regulating cell surface adaptation. Proc. Natl. Acad. Sci. USA 2004, 101, 14919–14924. [Google Scholar] [CrossRef] [PubMed]
- Robillard, N.J.; Tally, F.P.; Malamy, M.H. Tn4400, a compound transposon isolated from Bacteroides fragilis, functions in Escherichia coli. J. Bacteriol. 1985, 164, 1248–1255. [Google Scholar] [CrossRef]
- Gao, Q.; Wu, S.; Xu, T.; Zhao, X.; Huang, H.; Hu, F. Emergence of carbapenem resistance in Bacteroides fragilis in China. Int. J. Antimicrob. Agents. 2019, 53, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Sóki, J.; Wareham, D.W.; Rátkai, C.; Aduse-Opoku, J.; Urbán, E.; Nagy, E. Prevalence, nucleotide sequence and expression studies of two proteins of a 5.6kb, class III, Bacteroides plasmid frequently found in clinical isolates from European countries. Plasmid 2010, 63, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Varghese, N.J.; Mukherjee, S.; Ivanova, N.; Konstantinidis, K.T.; Mavrommatis, K.; Kyrpides, N.C.; Pati, A. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 2015, 43, 6761–6771. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.-W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Silva, M.; Machado, M.; Silva, D.; Rossi, M.; Moran-Gilad, J.; Santos, S.; Ramirez, M.; Carriço, J.A. chewBBACA: A complete suite for gene-by-gene schema creation and strain identification. Microb. Genom. 2018, 4, e000166. [Google Scholar] [CrossRef]
- Baker, G.C.; Smith, J.J.; Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 2003, 55, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Galata, V.; Fehlmann, T.; Backes, C.; Keller, A. PLSDB: A resource of complete bacterial plasmids. Nucleic Acids Res. 2019, 47, D195–D202. [Google Scholar] [CrossRef] [PubMed]
- Salipante, S.J.; Kalapila, A.; Pottinger, P.S.; Hoogestraat, D.R.; Cummings, L.; Duchin, J.S.; Sengupta, D.J.; Pergam, S.A.; Cookson, B.T.; Butler-Wu, S.M. Characterization of a multidrug-resistant, novel Bacteroides genomospecies. Emerg. Infect. Dis. 2015, 21, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Urbán, E.; Nord, C.; EESCMID Study Group on Antimicrobial Resistance in Anaerobic Bacteria. Antimicrobial susceptibility of Bacteroides fragilis group isolates in Europe: 20 years of experience. Clin. Microbiol. Infect. 2011, 17, 371–379. [Google Scholar] [CrossRef]
- Snydmana, D.R.; Jacobus, N.V.; McDermott, L.A.; Golan, Y.; Goldstein, E.J.C.; Harrell, L.; Jenkins, S.; Newton, D.; Pierson, C.; Rosenblatt, J.; et al. Update on resistance of Bacteroides fragilis group and related species with special attention to carbapenems 2006–2009. Anaerobe 2011, 17, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Woerther, P.L.; Lepeule, R.; Burdet, C.; Decousser, J.W.; Ruppé, E.; Barbier, F. Carbapenems and alternative beta-lactams for the treatment of infections due to extended-spectrum beta-lactamase-producing Enterobacteriaceae: What impact on intestinal colonisation resistance? Int. J. Antimicrob. Agents 2018, 52, 762–770. [Google Scholar] [CrossRef]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef]
- European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 10.0. 2020. Available online: http://www.eucast.org/clinicalbreakpoints/ (accessed on 12 May 2020).
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, 29th ed.; CLSI supplement M100; CLSI: Wayne, PA, USA, 2019. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Lukjancenko, O.; Saputra, D.; Rasmussen, S.; Hasman, H.; Sicheritz-Pontén, T.; Aarestrup, F.M.; Ussery, D.W.; Lund, O. Benchmarking of methods for genomic taxonomy. J. Clin. Microbiol. 2014, 52, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Inouye, M.; Dashnow, H.; Raven, L.A.; Schultz, M.B.; Pope, B.J.; Tomita, T.; Zobel, J.; Holt, K.E. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014, 6, 90. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.F.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Mungan, M.D.; Alanjary, M.; Blin, K.; Weber, T.; Medema, M.H.; Ziemert, N. ARTS 2.0: Feature updates and expansion of the Antibiotic Resistant Target Seeker for comparative genome mining. Nucleic Acids Res. 2020, 48, W546–W552. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 8, D517–D525. [Google Scholar] [CrossRef]
- Kanehis, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 1, D32–D36. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods Mol. Biol. 2020, 2075, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Cerdeno-Tarraga, A.M.; Patrick, S.; Crossman, L.C.; Blakely, G.; Abratt, V.; Lennard, N.; Poxton, I.; Duerden, B.; Harris, B.; Quail, M.A.; et al. Extensive DNA inversions in the B. fragilis genome control variable gene expression. Science 2005, 307, 1463–1465. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Alikhan, N.F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Strain | Antimicrobial Agent | MIC (mg/L) 1 | Interpretation 2 EUCAST/CLSI | Antimicrobial Resistance (AMR) Gene (Resistance Mechanism) | % Identity 3 (% Length) | IS 4 Element |
|---|---|---|---|---|---|---|
| CNM20180471 | ||||||
| Penicillin Amoxicillin-clavunalate Piperacillin/tazobactam | >32 >256 >256 | R/R R/R R/R | ||||
| Imipenem Imipenem+EDTA 5 Meropenem Meropenem+EDTA | >32 1 >32 >2 | R/R MBL-positive 6 R/R MBL-negative | cfiA14b 7, metallo-β-lactamase (antibiotic inactivation) | 100 (100) | ||
| Amikacin Gentamicin Tobramycin | >32 >32 >32 | NA 8 NA NA | aadS, aminoglycoside 6-adenylyltransferase aac(3’), N-acetyltransferase (antibiotic inactivation) | 100 (100) 99.7 (100) | ||
| Erythromycin Clindamycin | >256 >256 | NA/NA R/R | ermF, 23S ribosomal RNA methyltransferase lnu(AN2), lincosamide nucleotidyltransferase vatA, virginiamycin A acetyltransferase (antibiotic target alteration) mef(En2),, major facilitator superfamily (antibiotic efflux pump) | 97.7 (100) 100 (100) 99.2(100) | ISBaov1 | |
| Tetracycline Tigecycline | 8 0.5 | NA NA | tetX, tetracycline-inactivating monooxygenase (antibiotic inactivation) tetQ, tetracycline-resistant ribosomal protection protein (antibiotic target protection) | 100 (100) 96.4 (97.5) | ||
| Metronidazole | 0.38 | S/S | ||||
| Chloramphenicol | ND 8 | NA | cat, chloramphenicol acetyltransferase (antibiotic target alteration) | 95.77 (100) | ||
| Linezolid | 1 | NA | ||||
| Ciprofloxacin Moxifloxacin | >32 >32 | NA/NA NA/R | bexA 9 bexB, multidrug efflux MATE transporter (antibiotic efflux) | 98.2 (100) 83.2 (100) | ||
| Quaternary ammonium compounds | - | qacE, quaternary ammonium compound resistance (antibiotic efflux) | 95.4 (100) | |||
| CNM20200206 | ||||||
| Penicillin Amoxicillin-clavunalate Piperacillin/tazobactam | >32 >256 >256 | R/R R/R R/R | ||||
| Imipenem Imipenem+EDTA Meropenem Meropenem+EDTA | >32 <1 >32 >2 | R/R MBL-positive R/R MBL-negative | New cfiA28, metallo-β-lactamase (antibiotic inactivation) | |||
| Amikacin Gentamicin Tobramycin | >32 >32 >32 | NA NA NA | aac(3´), N-acetyltransferase (antibiotic inactivation) | 100 (100) | ||
| Erythromycin Clindamycin | >256 >256 | NA/NA R/R | vatA, virginiamycin A acetyltransferase (antibiotic target alteration) | 100 (100) | ||
| Tetracycline Tigecycline | 16 6 | NA NA | tetQ, tetracycline-resistant ribosomal protection protein (antibiotic target protection) | 96.41 (97.5) | ||
| Metronidazole | 0.5 | S /S | ||||
| Chloramphenicol | ND | NA | cat, chloramphenicol acetyltransferase (antibiotic target alteration) | 95.77 (100) | ||
| Linezolid | 1.5 | NA | ||||
| Ciprofloxacin Moxifloxacin | >32 4 | R/R NA/I | bexA bexB, multidrug efflux MATE transporter (antibiotic efflux) | 83.8 (100) 98.0 (100) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdezate, S.; Cobo, F.; Monzón, S.; Medina-Pascual, M.J.; Zaballos, Á.; Cuesta, I.; Pino-Rosa, S.; Villalón, P. Genomic Background and Phylogeny of cfiA-Positive Bacteroides fragilis Strains Resistant to Meropenem-EDTA. Antibiotics 2021, 10, 304. https://doi.org/10.3390/antibiotics10030304
Valdezate S, Cobo F, Monzón S, Medina-Pascual MJ, Zaballos Á, Cuesta I, Pino-Rosa S, Villalón P. Genomic Background and Phylogeny of cfiA-Positive Bacteroides fragilis Strains Resistant to Meropenem-EDTA. Antibiotics. 2021; 10(3):304. https://doi.org/10.3390/antibiotics10030304
Chicago/Turabian StyleValdezate, Sylvia, Fernando Cobo, Sara Monzón, María J. Medina-Pascual, Ángel Zaballos, Isabel Cuesta, Silvia Pino-Rosa, and Pilar Villalón. 2021. "Genomic Background and Phylogeny of cfiA-Positive Bacteroides fragilis Strains Resistant to Meropenem-EDTA" Antibiotics 10, no. 3: 304. https://doi.org/10.3390/antibiotics10030304
APA StyleValdezate, S., Cobo, F., Monzón, S., Medina-Pascual, M. J., Zaballos, Á., Cuesta, I., Pino-Rosa, S., & Villalón, P. (2021). Genomic Background and Phylogeny of cfiA-Positive Bacteroides fragilis Strains Resistant to Meropenem-EDTA. Antibiotics, 10(3), 304. https://doi.org/10.3390/antibiotics10030304