
Investigating the OXA Variants of ESKAPE Pathogens


Abstract
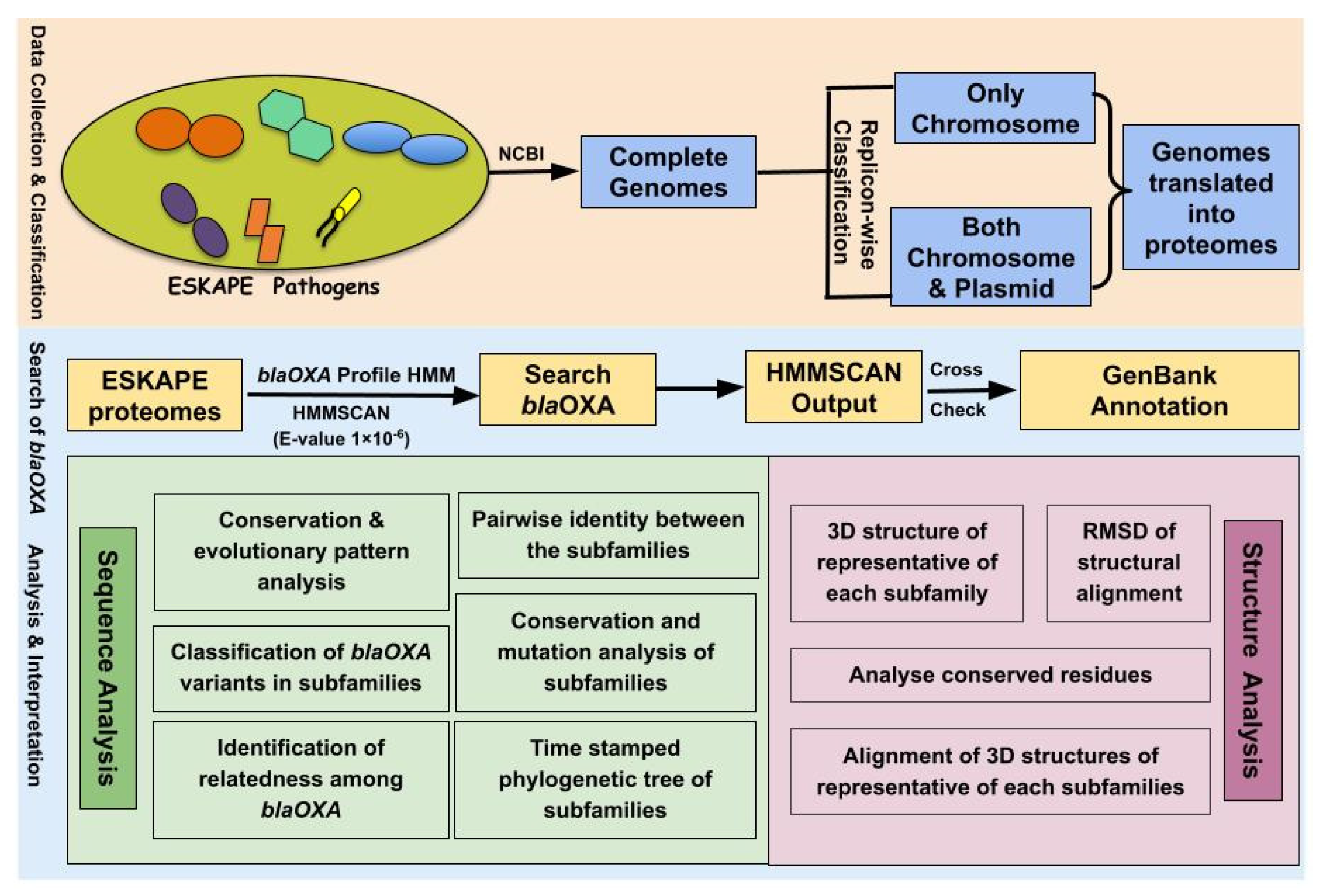
:1. Introduction
2. Results and Discussion
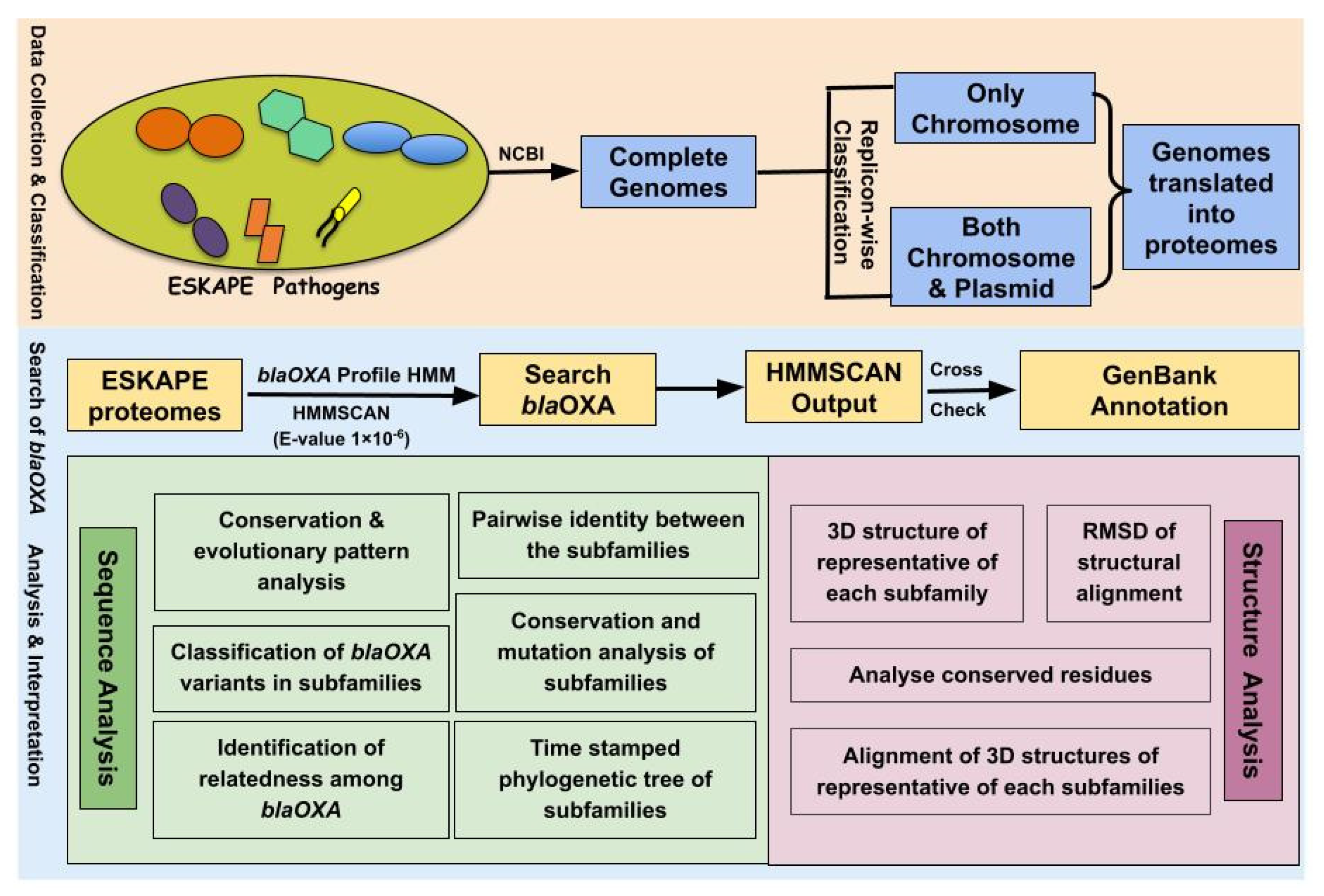
2.1. Data Statistics and Classification of ESKAPE Pathogens
2.2. Construction of Profile HMM of BlaOXA
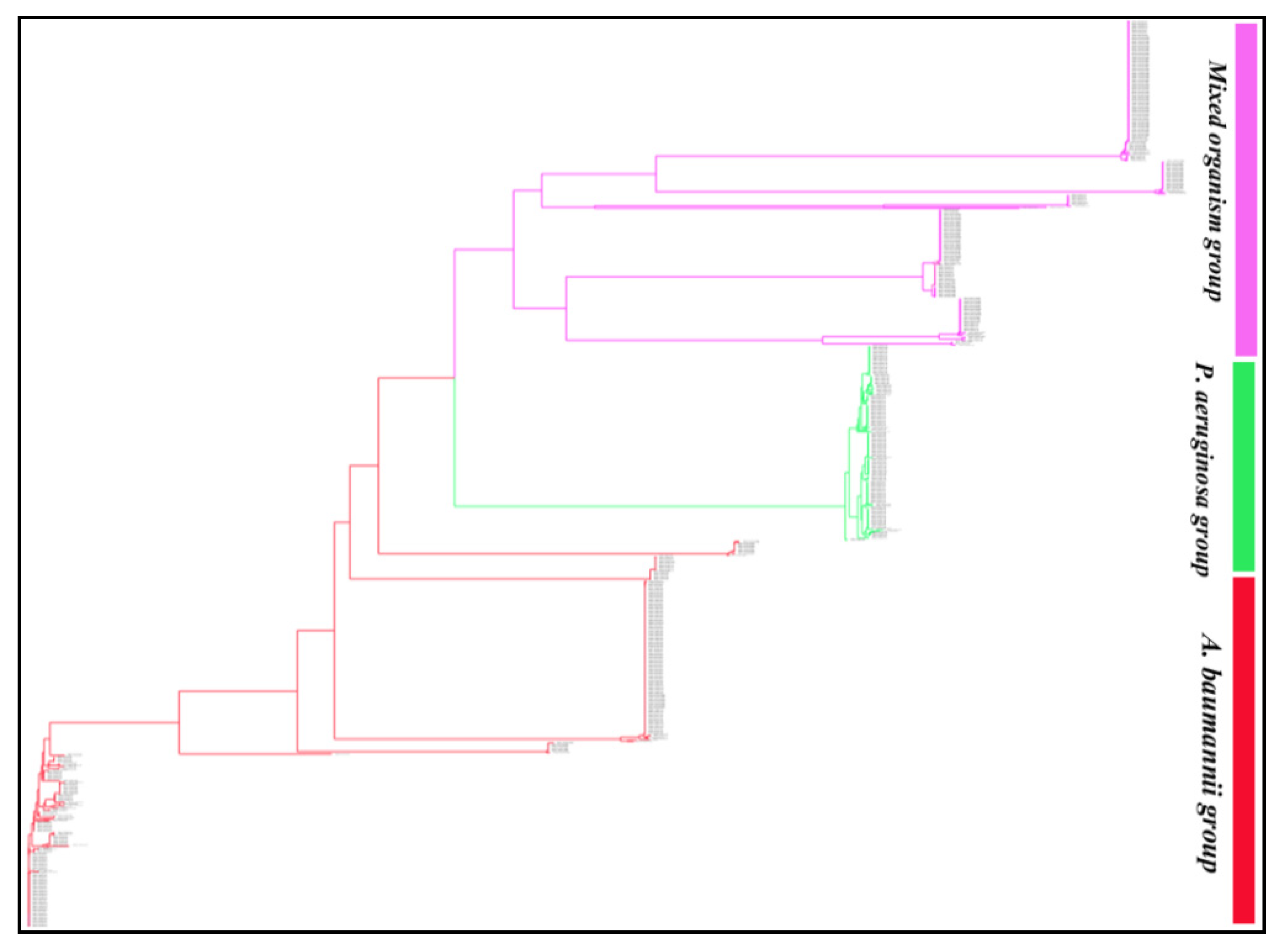
2.3. Distribution and Localization of BlaOXA in ESKAPE Pathogens
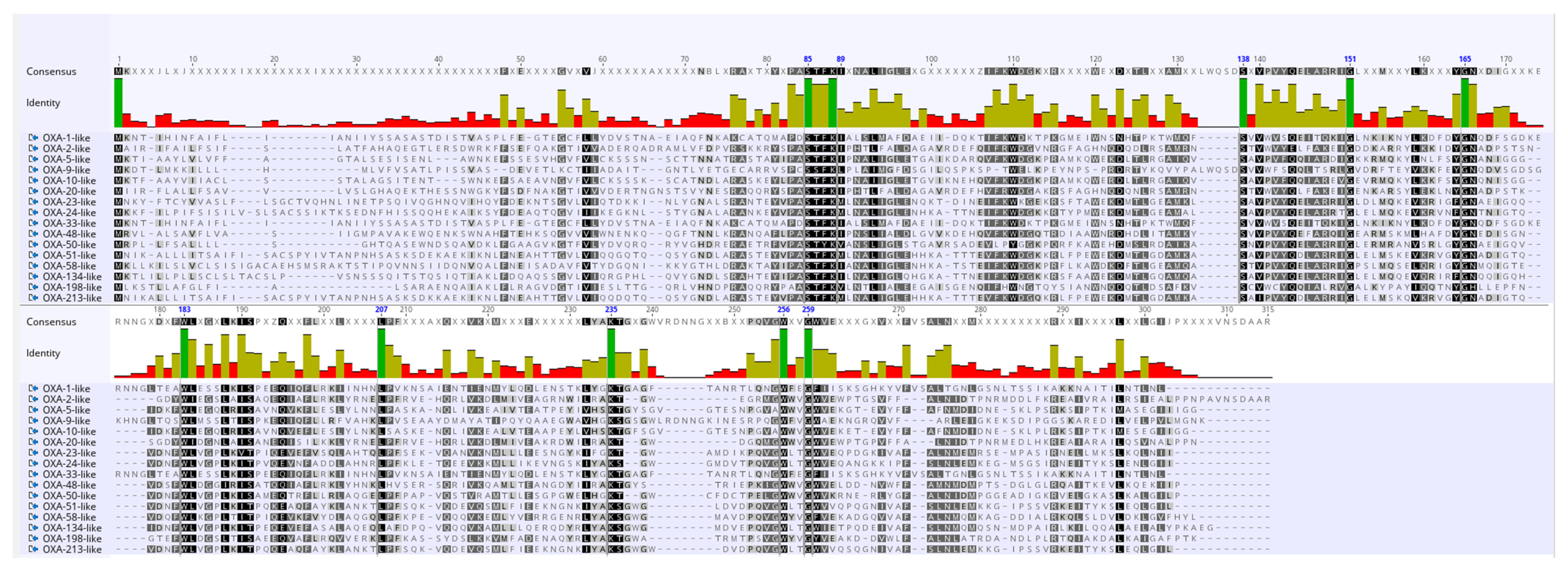
2.4. Classification of BlaOXA Variants in Subfamilies
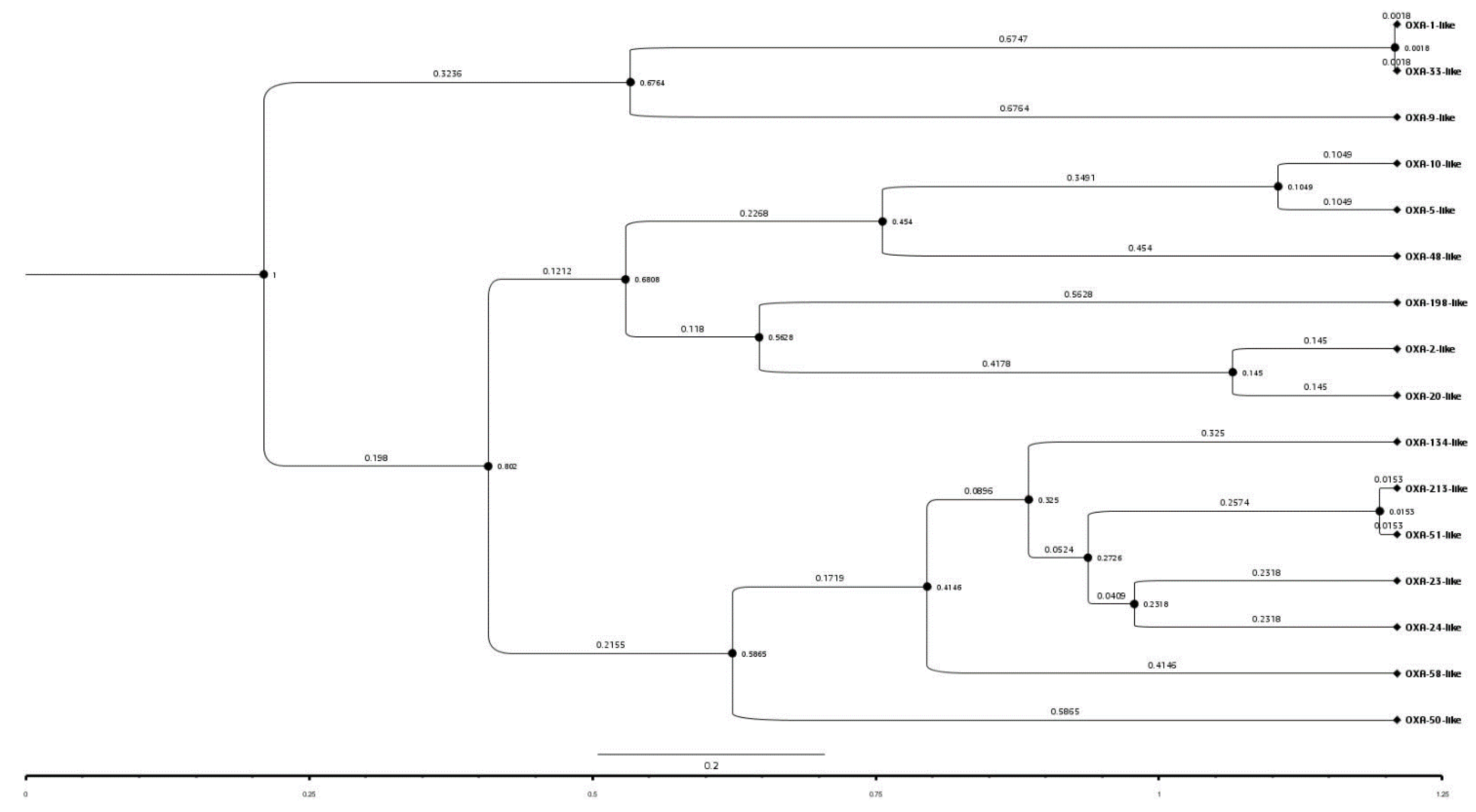
2.5. Identification of Relatedness among BlaOXA
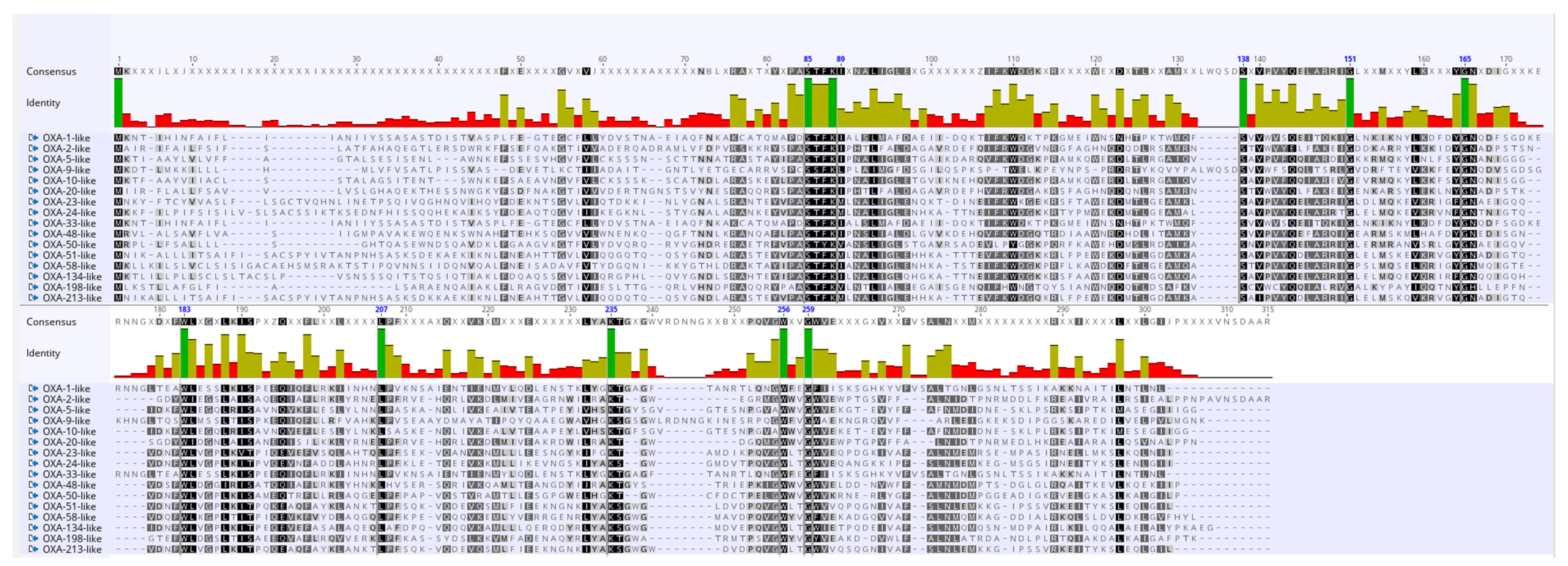
2.6. Analysis of Amino Acid Variations in OXA Subfamilies
2.7. Pairwise Identity between the Subfamilies
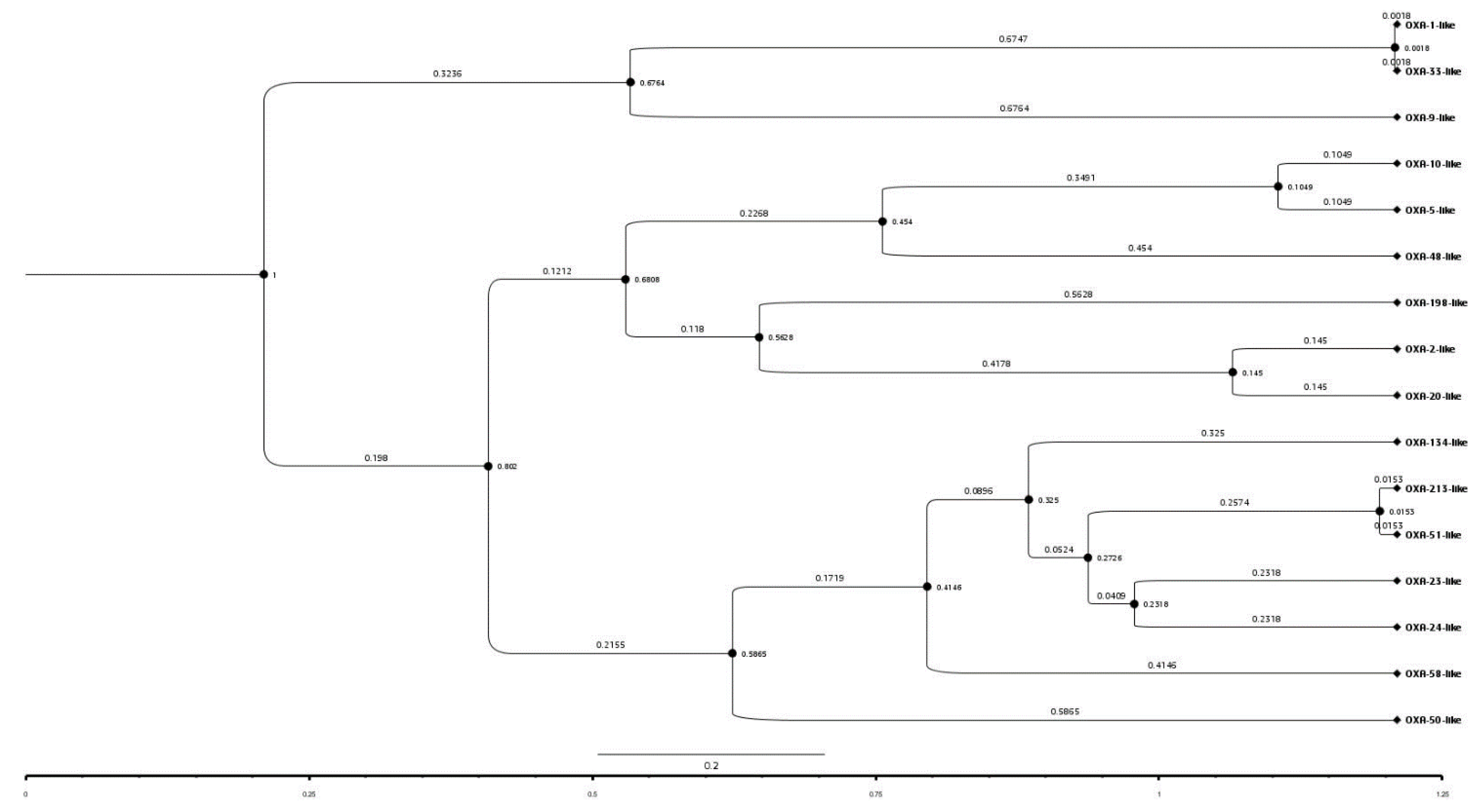
2.8. Amino Acid Analysis of Representatives of OXA Subfamilies and Their Phylogenetic Study
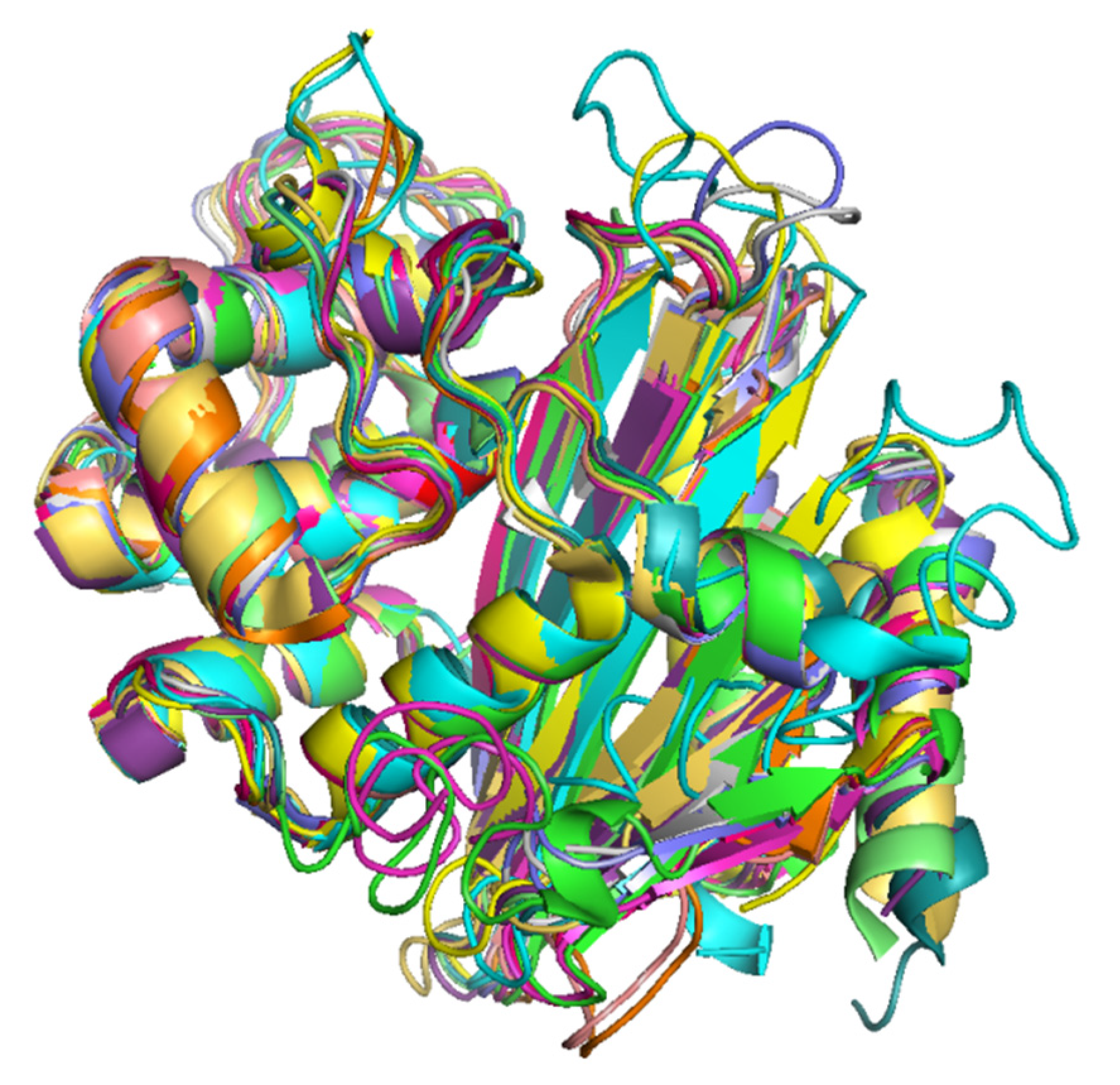
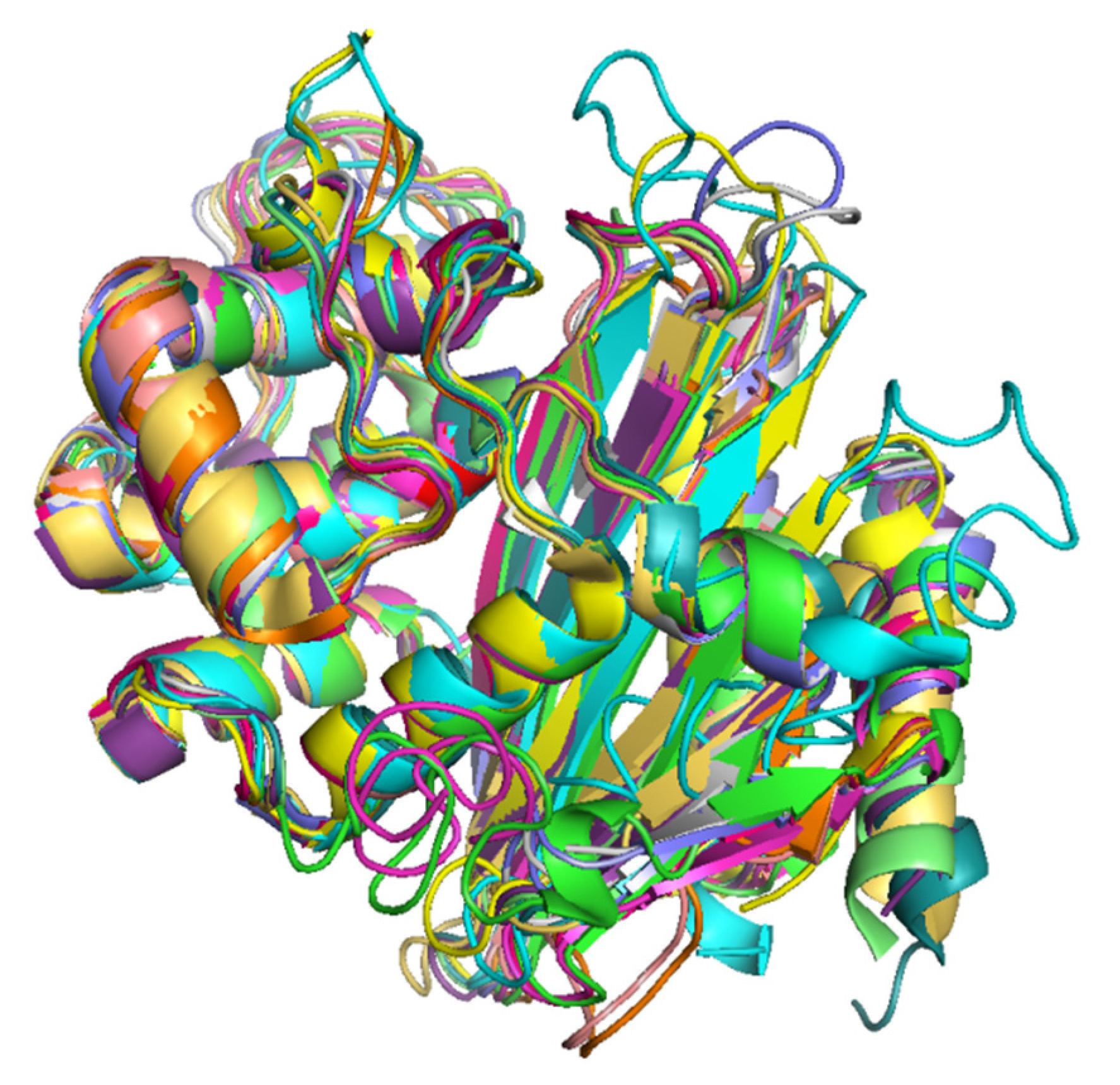
2.9. Structural Variations in the 3D Protein Models of the OXA Subfamilies
3. Materials and Methods
3.1. Data Collection
3.2. Building Profile HMM and OXA Search in the Genomes of ESKAPE Pathogens
3.3. Multiple Sequence Alignment and Phylogenetic Tree
3.4. Determination of Pairwise Sequence Identity in Representatives of OXA Subfamilies and Phylogenetic Analysis
3.5. 3D Modeling and Assessment of Structural Variations in the OXA Subfamilies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OXA | Oxacillinases |
| ESKAPE | Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species |
| WHO | World Health Organization |
| AMR | Antimicrobial Resistance |
| MDR | Multidrug Resistance |
| blaOXA | OXA β-lactamase |
| ARG | Antibiotic Resistance Gene |
| MSA | Multiple Sequence Alignment |
| HMM | Hidden Markov Model |
| RMSD | Root Mean Square Deviation |
| CBMAR | Comprehensive Beta-lactamase Molecular Annotation Resource |
| NCBI | National Center for Biotechnology Information |
| NR | Non Redundant |
| BLDB | Beta-Lactamase Database |
| NJ | Neighbor joining |
| BLAST | Basic Local Alignment Search Tool |
| BEAST | Bayesian evolutionary analysis through a simple tutorial |
| BEAUti | Bayesian Evolutionary Analysis Utility |
| MCMC | Markov chain Monte Carlo |
References
- Llor, C.; Bjerrum, L. Antimicrobial resistance: Risk associated with antibiotic overuse and initiatives to reduce the problem. Ther. Adv. Drug Saf. 2014, 5, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santajit, S.; Indrawattana, N. Mechanisms of antimicrobial resistance in ESKAPE pathogens. BioMed Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria. Drugs 2004, 64, 159–204. [Google Scholar] [CrossRef]
- Tacconelli, E. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Available online: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed on 25 February 2017).
- Wright, G.D. Bacterial resistance to antibiotics: Enzymatic degradation and modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef]
- Egorov, A.M.; Ulyashova, M.M.; Rubtsova, M.Y. Bacterial Enzymes and antibiotic resistance. Acta Nat. 2018, 10, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Bonomo, R.A. β-Lactamases: A focus on current challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a025239. [Google Scholar] [CrossRef] [PubMed]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-lactamase inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef]
- Bush, K. Past and present perspectives on β-Lactamases. Antimicrob. Agents Chemother. 2018, 62, e01076-18. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.A.; Amyes, S.G.B. OXA β-lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar] [CrossRef] [Green Version]
- Ambler, R.P. The structure of β-lactamases. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1980, 289, 321–331. [Google Scholar]
- Bush, K. Metallo-beta-lactamases: A class apart. Clin. Infect. Dis. 1998, 27 (Suppl. 1), S48–S53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behzadi, P.; García-Perdomo, A.H.; Karpiński, T.M.; Issakhanian, L. Metallo-ß-lactamases: A review. Mol. Biol. Rep. 2020, 47, 6281–6294. [Google Scholar] [CrossRef]
- Baurin, S.; Vercheval, L.; Bouillenne, F.; Falzone, C.; Brans, A.; Jacquamet, L.; Ferrer, J.L.; Sauvage, E.; Dehareng, D.; Frere, J.M.; et al. Critical role of tryptophan 154 for the activity and stability of class D β-lactamases. Biochemistry 2009, 48, 11252–11263. [Google Scholar] [CrossRef]
- Gniadkowski, M. Evolution of extended-spectrum beta-lactamases by mutation. Clin. Microbiol. Infect. 2008, 14 (Suppl. 1), 11–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, K.; Jacoby, G.A. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, present, and future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, D.A.; Bonomo, R.A.; Powers, R.A. Class D β-lactamases: A reappraisal after five decades. Acc. Chem. Res. 2013, 46, 2407–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlynarcik, P.; Chalachanova, A.; Vagnerovă, I.; Holy, O.; Zatloukalova, S.; Kolar, M. PCR detection of oxacillinases in bacteria. Microb. Drug Resist. 2020, 26, 1023–1037. [Google Scholar] [CrossRef] [Green Version]
- Bush, K.; Fisher, J.F. Epidemiological expansion, structural studies, and clinical challenges of new β-lactamases from gram-negative bacteria. Annu. Rev. Microbiol. 2011, 65, 455–478. [Google Scholar] [CrossRef] [PubMed]
- Sanschagrin, F.; Couture, F.; Levesque, R.C. Primary structure of OXA-3 and phylogeny of oxacillin-hydrolyzing class D beta-lactamases. Antimicrob. Agents Chemother. 1995, 39, 887–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirel, L.; Héritier, C.; Tolün, V.; Nordmann, P. Emergence of oxacillinase-mediated resistance to imipenem in klebsiella pneumoniae. Antimicrob. Agents Chemother. 2004, 48, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, K. Proliferation and significance of clinically relevant β-lactamases. Ann. N.Y. Acad. Sci. 2013, 1277, 84–90. [Google Scholar] [CrossRef]
- Bush, K. The ABCD’s of β-Lactamase nomenclature. J. Infect. Chemother. 2013, 19, 549–559. [Google Scholar] [CrossRef]
- Pitout, J.D.D.; Peirano, G.; Kock, M.M.; Strydom, K.-A.; Matsumura, Y. The global ascendency of OXA-48-type carbapenemases. Clin. Microbiol. Rev. 2019, 33, e00102-19. [Google Scholar] [CrossRef]
- Birck, C.; Cha, J.Y.; Cross, J.; Schulze-Briese, C.; Meroueh, S.O.; Schlegel, H.B.; Mobashery, S.; Samama, J.-P. X-ray crystal structure of the acylated beta-lactam sensor domain of blar1 from staphylococcus aureus and the mechanism of receptor activation for signal transduction. J. Am. Chem. Soc. 2004, 126, 13945–13947. [Google Scholar] [CrossRef]
- Kerff, F.; Charlier, P.; Colombo, M.-L.; Sauvage, E.; Brans, A.; Frère, J.-M.; Joris, B.; Fonzé, E. Crystal structure of the sensor domain of the BLaR penicillin receptor from bacillus licheniformis. Biochemistry 2003, 42, 12835–12843. [Google Scholar] [CrossRef]
- Marrero, A.; Mallorquí-Fernández, G.; Guevara, T.; García-Castellanos, R.; Gomis-Rüth, F.X. Unbound and acylated structures of the mecr1 extracellular antibiotic-sensor domain provide insights into the signal-transduction system that triggers methicillin resistance. J. Mol. Biol. 2006, 361, 506–521. [Google Scholar] [CrossRef]
- Wilke, M.S.; Hills, T.L.; Zhang, H.-Z.; Chambers, H.F.; Strynadka, N.C.J. Crystal structures of the apo and penicillin-acylated forms of the BlaR1 beta-lactam sensor of staphylococcus aureufs. J. Biol. Chem. 2004, 279, 47278–47287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngoi, S.T.; Chong, C.W.; Ponnampalavanar, S.S.L.S.; Tang, S.N.; Idris, N.; Abdul Jabar, K.; Gregory, M.J.; Husain, T.; Teh, C.S.J. genetic mechanisms and correlated risk factors of antimicrobial-resistant ESKAPEE pathogens isolated in a tertiary hospital in Malaysia. Antimicrob. Resist. Infect. Control 2021, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Naas, T.; Oueslati, S.; Bonnin, R.A.; Dabos, M.L.; Zavala, A.; Dortet, L.; Retailleau, P.; Iorga, B.I. Beta-lactamase database (BLDB)-structure and function. J. Enzym. Inhib. Med. Chem. 2017, 32, 917–919. [Google Scholar] [CrossRef]
- Antunes, N.T.; Fisher, J.F. Acquired class D β-lactamases. Antibiotics 2014, 3, 398–434. [Google Scholar] [CrossRef]
- Poirel, L.; Potron, A.; Nordmann, P. OXA-48-like carbapenemases: The Phantom Menace. J. Antimicrob. Chemother. 2012, 67, 1597–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignoli, R.; Cordeiro, N.; Seija, V.; Schelotto, F.; Radice, M.; Ayala, J.; Power, P.; Gutkind, G. Genetic environment of CTX-M-2 in Klebsiella pneumoniae isolates from hospitalized patients in Uruguay. Rev. Argent. Microbiol. 2006, 38, 84–88. [Google Scholar]
- Mossakowska, D.; Ali, N.A.; Dale, J.W. Oxacillin-hydrolysing beta-lactamases. A comparative analysis at nucleotide and amino acid sequence levels. Eur. J. Biochem. 1989, 180, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Singhal, N.; Goel, M.; Virdi, J.S.; Kumar, M. CBMAR: A comprehensive β-lactamase molecular annotation resource. Database J. Biol. Databases Curation 2014. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web Server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X Version 2.0. Bioinfromatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, N.; Mikheyev, A.S. Interactive web-based visualization and sharing of phylogenetic trees using phylogeny.IO. Nucleic Acids Res. 2019, 47, W266–W269. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Total No. of Genomes (NCBI) | No. of Complete Genomes/No. of blaOXA | Replicons | |||
|---|---|---|---|---|---|---|
| No. of Chromosomal Genomes/No. of blaOXA | Both Chromosome and Plasmid | |||||
| No. of Genomes Containing Both Chromosomes and Plasmids/No. of blaOXA (Total) | No. of Chromosomes/No. of blaOXA | No. of Plasmids/No. of blaOXA | ||||
| E. faecium | 2256 | 187/0 | 12/0 | 175/0 | 175/0 | 851/0 |
| S. aureus | 12,397 | 590/0 | 285/0 | 305/0 | 305/0 | 467/0 |
| K. pneumoniae | 10,383 | 732/263 | 63/6 | 669/257 | 669/39 | 2362/218 |
| A. baumannii | 5057 | 233/408 | 50/82 | 183/326 | 183/283 | 384/43 |
| P. aeruginosa | 5711 | 291/235 | 254/199 | 37/36 | 37/25 | 50/11 |
| Enterobacter spp. | 2680 | 225/23 | 53/0 | 172/23 | 172/4 | 593/19 |
| Total | 2258/929 | 717/287 | 1541/642 | 1541/351 | 4707/291 | |
| S. No. | Enzyme Group | Organism and Gene Location(s) | Number of Sequences | Variations |
|---|---|---|---|---|
| 1 | OXA-48-like | K. pneumoniae (B: chromosome, B: plasmid, O: chromosome) Enterobacter spp. (B: plasmid) | 86 | T104A, N110D, E168Q, S171A, R214S |
| 2 | OXA-10-like | K. pneumoniae (B: plasmid) P. aeruginosa (B: chromosome, B: plasmid, O: chromosome) Enterobacter spp. (B: plasmid) A. baumannii (O: chromosome) | 42 | I10T, G20S, S27F, D55N, T107S, Y174F, E229G, S245N, E259A |
| 3 | OXA-9-like | K. pneumoniae (B: plasmid) P. aeruginosa (O: chromosome) Enterobacter spp. (B: plasmid) | 32 | Not found |
| 4 | OXA-2-like | Enterobacter spp. (B: chromosome) K. pneumoniae (B: plasmid) P. aeruginosa (B: chromosome, O: chromosome) | 12 | D321V |
| 5 | OXA-51-like | A. baumannii (B: chromosome, B: plasmid, O: chromosome) | 200 | A5T, T24S, E36V, E36D, E36K, A38G, A48V, Q57H, A96T, D105N, K107Q, K107E, D117, V129I, P130Q, K146N, L167V, K177Q, Q194P, K195E, D198H, L222W, D225N |
| 6 | OXA-20-like | A. baumannii (B: chromosome) | 1 | NA |
| 7 | OXA-23-like | A. baumannii (B: chromosome, B: plasmid, O: chromosome) P. aeruginosa (B: chromosome) | 156 | Not found |
| 8 | OXA-213-like | A. baumannii (B: chromosome) | 1 | NA |
| 9 | OXA-134-like | A. baumannii (B: chromosome, B: plasmid, O: chromosome) | 24 | D208G |
| 10 | OXA-58-like | A. baumannii (B: plasmid) | 14 | F114L, I161M, A256D |
| 11 | OXA-24-like | 11 | D224G | |
| 12 | OXA-1-like | K. pneumoniae (B: chromosome, B: plasmid, O: chromosome) P. aeruginosa (B: chromosome, B: plasmid, O: chromosome) Enterobacter spp. (B: chromosome, B: plasmid) | 154 | L28F, V64A, I102M, D103N, N2111E E223D, N268S |
| 13 | OXA-50-like | P. aeruginosa (B: chromosome, O: chromosome) | 189 | F6L, A8T, T17A, Q26R, R50C, R84K, D110E, R168H, K113E |
| 14 | OXA-33-like | P. aeruginosa (B: chromosome) | 2 | Not found |
| 15 | OXA-198-like | P. aeruginosa (O: chromosome) | 1 | NA |
| 16 | OXA-5-like | Enterobacter spp. (B: plasmid) P. aeruginosa (O: chromosome) | 4 | V117L |
| OXA- 1-like | OXA- 2-like | OXA- 5-like | OXA- 9-like | OXA- 10-like | OXA- 20-like | OXA- 23-like | OXA- 24-like | OXA- 33-like | OXA- 48-like | OXA- 50-like | OXA- 51-like | OXA- 58-like | OXA- 134-like | OXA- 198-like | OXA- 213-like | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OXA- 1-like | 100.00 | |||||||||||||||
| OXA- 2-like | 26.00 | 100.00 | ||||||||||||||
| OXA- 5-like | 27.50 | 32.30 | 100.00 | |||||||||||||
| OXA- 9-like | 31.00 | 21.20 | 26.40 | 100.00 | ||||||||||||
| OXA- 10-like | 27.10 | 34.00 | 80.90 | 26.30 | 100.00 | |||||||||||
| OXA- 20-like | 26.90 | 72.70 | 35.60 | 21.70 | 33.90 | 100.00 | ||||||||||
| OXA- 23-like | 24.10 | 27.00 | 33.90 | 19.90 | 33.20 | 28.80 | 100.00 | |||||||||
| OXA- 24-like | 26.90 | 25.20 | 33.10 | 22.10 | 36.30 | 28.90 | 59.90 | 100.00 | ||||||||
| OXA- 33-like | 100.00 | 26.00 | 27.60 | 31.00 | 27.10 | 26.90 | 24.10 | 26.90 | 100.00 | |||||||
| OXA- 48-like | 27.20 | 39.20 | 44.10 | 22.50 | 45.40 | 40.90 | 35.60 | 31.80 | 27.20 | 100.00 | ||||||
| OXA- 50-like | 25.00 | 32.90 | 35.80 | 25.60 | 33.30 | 35.80 | 40.90 | 36.20 | 25.00 | 36.10 | 100.00 | |||||
| OXA- 51-like | 26.40 | 26.40 | 34.50 | 25.60 | 32.60 | 31.00 | 56.90 | 63.40 | 26.40 | 36.30 | 40.40 | 100.00 | ||||
| OXA- 58-like | 21.80 | 28.40 | 35.40 | 25.30 | 36.00 | 29.60 | 47.70 | 48.60 | 21.80 | 34.00 | 39.40 | 48.30 | 100.00 | |||
| OXA- 134-like | 25.00 | 28.70 | 34.50 | 23.70 | 35.20 | 28.50 | 55.90 | 55.90 | 25.00 | 32.10 | 38.10 | 54.50 | 52.70 | 100.00 | ||
| OXA- 198-like | 26.40 | 34.80 | 30.70 | 22.50 | 31.30 | 36.30 | 27.20 | 31.00 | 26.40 | 35.30 | 33.00 | 28.40 | 32.50 | 32.60 | 100.00 | |
| OXA- 213-like | 24.70 | 26.70 | 33.80 | 24.70 | 33.00 | 30.00 | 56.50 | 62.00 | 24.70 | 36.60 | 39.60 | 97.10 | 47.20 | 53.00 | 28.10 | 100.00 |
| Super-Imposition | OXA-1-like | OXA-2-like | OXA-5-like | OXA-9-like | OXA-10-like | OXA-20-like | OXA-23-like | OXA-24-like | OXA-33-like | OXA-48-like | OXA-50-like | OXA-51-like | OXA-58-like | OXA-134-like | OXA-198-like | OXA-213-like |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OXA-1-like | 0 | |||||||||||||||
| OXA-2-like | 0.972 | 0 | ||||||||||||||
| OXA-5-like | 1.112 | 0.704 | 0 | |||||||||||||
| OXA-9-like | 0.784 | 1.099 | 1.756 | 0 | ||||||||||||
| OXA-10-like | 1.058 | 0.762 | 0.268 | 1.738 | 0 | |||||||||||
| OXA-20-like | 0.96 | 0.226 | 0.715 | 1.174 | 0.759 | 0 | ||||||||||
| OXA-23-like | 1.143 | 0.902 | 1.108 | 1.768 | 1.118 | 0.898 | 0 | |||||||||
| OXA-24-like | 1.023 | 1.007 | 1.027 | 1.052 | 1.07 | 0.946 | 0.426 | 0 | ||||||||
| OXA-33-like | 0 | 0.972 | 1.112 | 0.784 | 1.058 | 0.96 | 1.143 | 1.023 | 0 | |||||||
| OXA-48-like | 0.817 | 0.613 | 0.659 | 0.946 | 0.805 | 0.645 | 0.823 | 0.848 | 0.817 | 0 | ||||||
| OXA-50-like | 0.811 | 0.623 | 0.51 | 0.699 | 0.637 | 0.636 | 0.798 | 0.798 | 0.811 | 0.375 | 0 | |||||
| OXA-51-like | 0.963 | 0.893 | 1.081 | 1.364 | 1.131 | 0.912 | 0.41 | 0.478 | 0.963 | 0.817 | 0.759 | 0 | ||||
| OXA-58-like | 0.987 | 1.007 | 0.848 | 2.324 | 0.881 | 1.054 | 0.623 | 0.639 | 0.987 | 0.926 | 0.882 | 0.616 | 0 | |||
| OXA-134-like | 1.143 | 0.902 | 1.108 | 1.768 | 1.118 | 0.898 | 0 | 0.426 | 1.143 | 0.823 | 0.798 | 0.41 | 0.623 | 0 | ||
| OXA-198-like | 0.78 | 0.581 | 0.548 | 2.466 | 0.705 | 0.556 | 0.727 | 0.784 | 0.78 | 0.381 | 0.277 | 0.701 | 0.915 | 0.727 | 0 | |
| OXA-213-like | 0.963 | 0.893 | 1.081 | 1.364 | 1.131 | 0.912 | 0.41 | 0.478 | 0.963 | 0.817 | 0.759 | 0 | 0.616 | 0.41 | 0.701 | 0 |
| S. No. | Protein Name | PDB ID | Query Coverage (%) | Identity (%) |
|---|---|---|---|---|
| 1 | OXA-10-like | 1K6R | 92 | 100 |
| 2 | OXA-2-like | 1K38 | 92 | 99.60 |
| 3 | OXA-48-like | 5OE0 | 100 | 99.60 |
| 4 | OXA-24-like | 4WM9 | 89 | 99.50 |
| 5 | OXA-58-like | 4OH0 | 100 | 99.29 |
| 6 | OXA-1-like | 1M6K | 90 | 98.8 |
| 7 | OXA-213-like | 4ZDX | 100 | 97 |
| 8 | OXA-33-like | 1M6K | 90 | 98.80 |
| 9 | OXA-51-like | 4ZDX | 100 | 97 |
| 10 | OXA-23-like | 4JF4 | 89 | 100 |
| 11 | OXA-5-like | 1FOF | 91 | 84.01 |
| 12 | OXA-20-like | 6XJ3 | 92 | 77.30 |
| 13 | OXA-134-like | 4JF4 | 85 | 63.90 |
| 14 | OXA-9-like | 6NHU | 89 | 46.06 |
| 15 | OXA-198-like | 6NLW | 88 | 40.60 |
| 16 | OXA-50-like | 4JF4 | 90 | 44.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, D.; Singhal, N.; Kumar, M. Investigating the OXA Variants of ESKAPE Pathogens. Antibiotics 2021, 10, 1539. https://doi.org/10.3390/antibiotics10121539
Pandey D, Singhal N, Kumar M. Investigating the OXA Variants of ESKAPE Pathogens. Antibiotics. 2021; 10(12):1539. https://doi.org/10.3390/antibiotics10121539
Chicago/Turabian StylePandey, Deeksha, Neelja Singhal, and Manish Kumar. 2021. "Investigating the OXA Variants of ESKAPE Pathogens" Antibiotics 10, no. 12: 1539. https://doi.org/10.3390/antibiotics10121539
APA StylePandey, D., Singhal, N., & Kumar, M. (2021). Investigating the OXA Variants of ESKAPE Pathogens. Antibiotics, 10(12), 1539. https://doi.org/10.3390/antibiotics10121539