
Determination of Tetracycline, Oxytetracycline, Sulfadiazine, Norfloxacin, and Enrofloxacin in Swine Manure Using a Coupled Method of On-Line Solid-Phase Extraction with the UHPLC–DAD

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Optimization of the Chromatographic Conditions
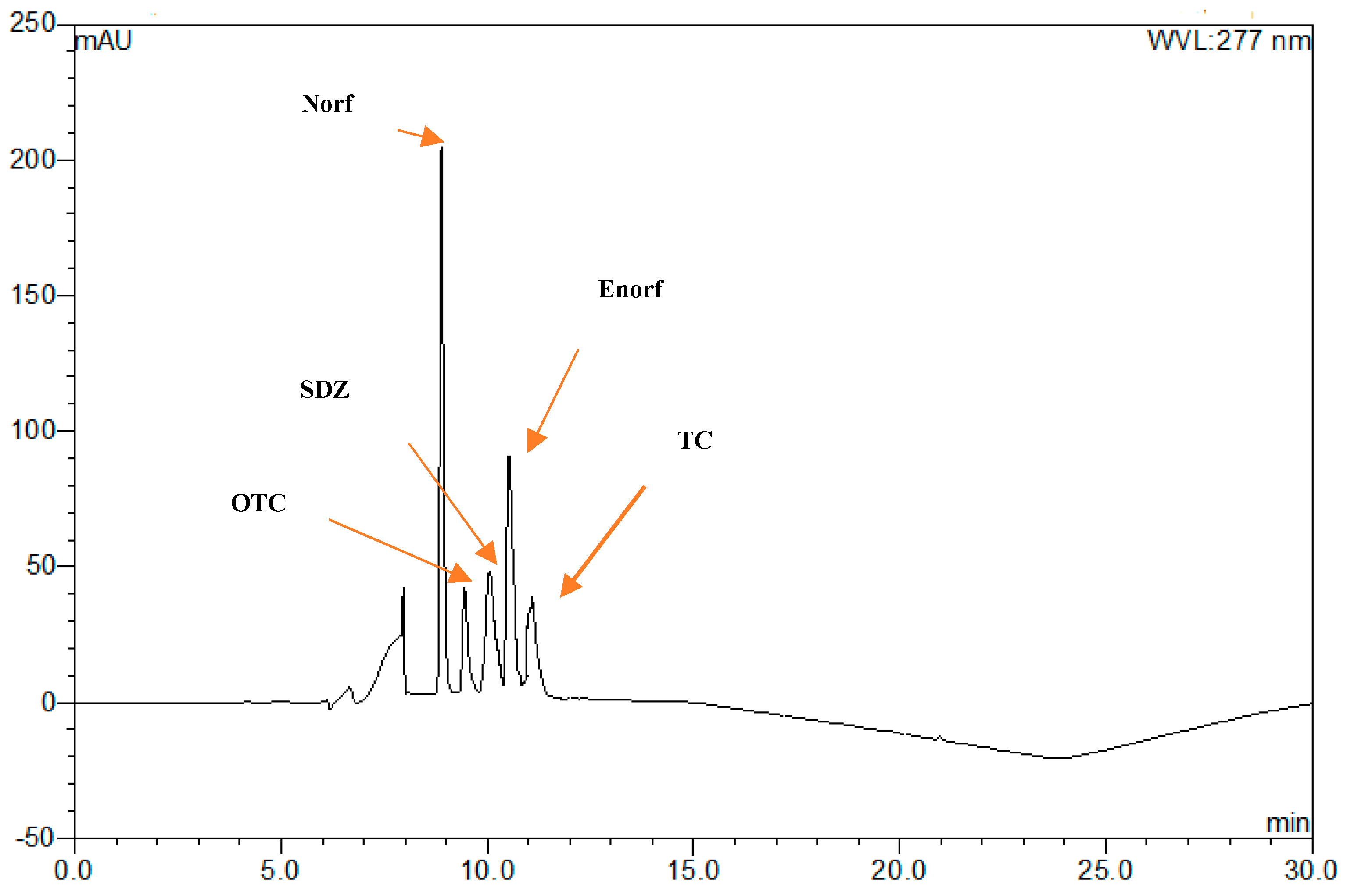
2.1.1. Selection of the Optimum Detection Wavelength
2.1.2. Selection of the Suitable Mobile Phase and Chromatographic Conditions
2.2. Optimization of SPE
2.2.1. The Choice of On-Line SPE Column Sorbent
2.2.2. Optimal Extraction Sorbent for VAs in Solid Fraction
2.3. The Suggested Method
2.4. Method Validation
2.4.1. Linearity of the Suggested Method
2.4.2. Limit of Detection (LOD) and Limit of Quantification (LOQ)
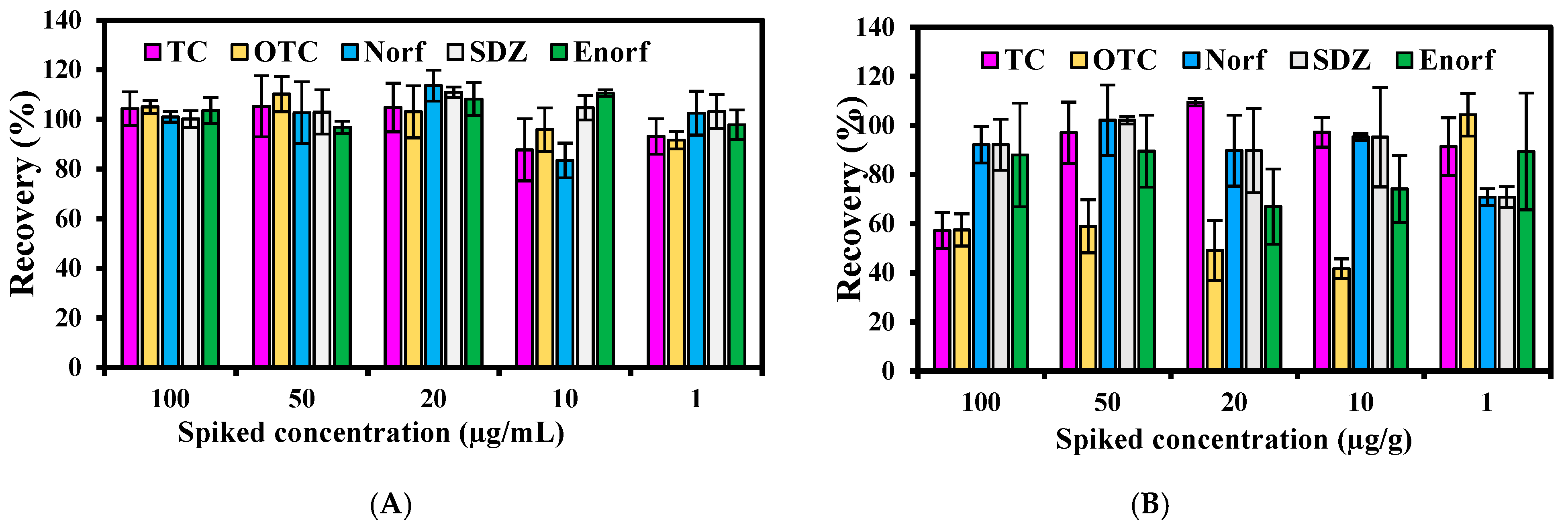
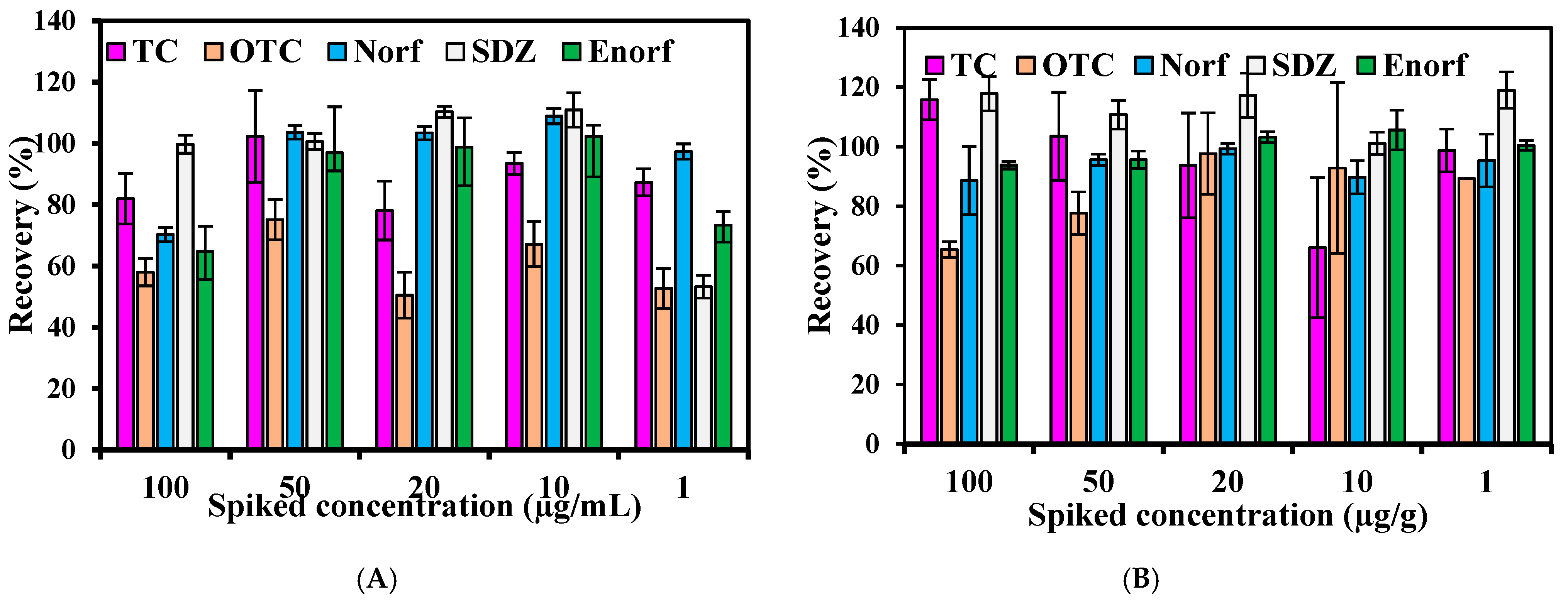
2.4.3. Precision and Accuracy
3. Materials and Methods
3.1. Reagents and Materials
3.2. Apparatus
3.3. Standard Solutions
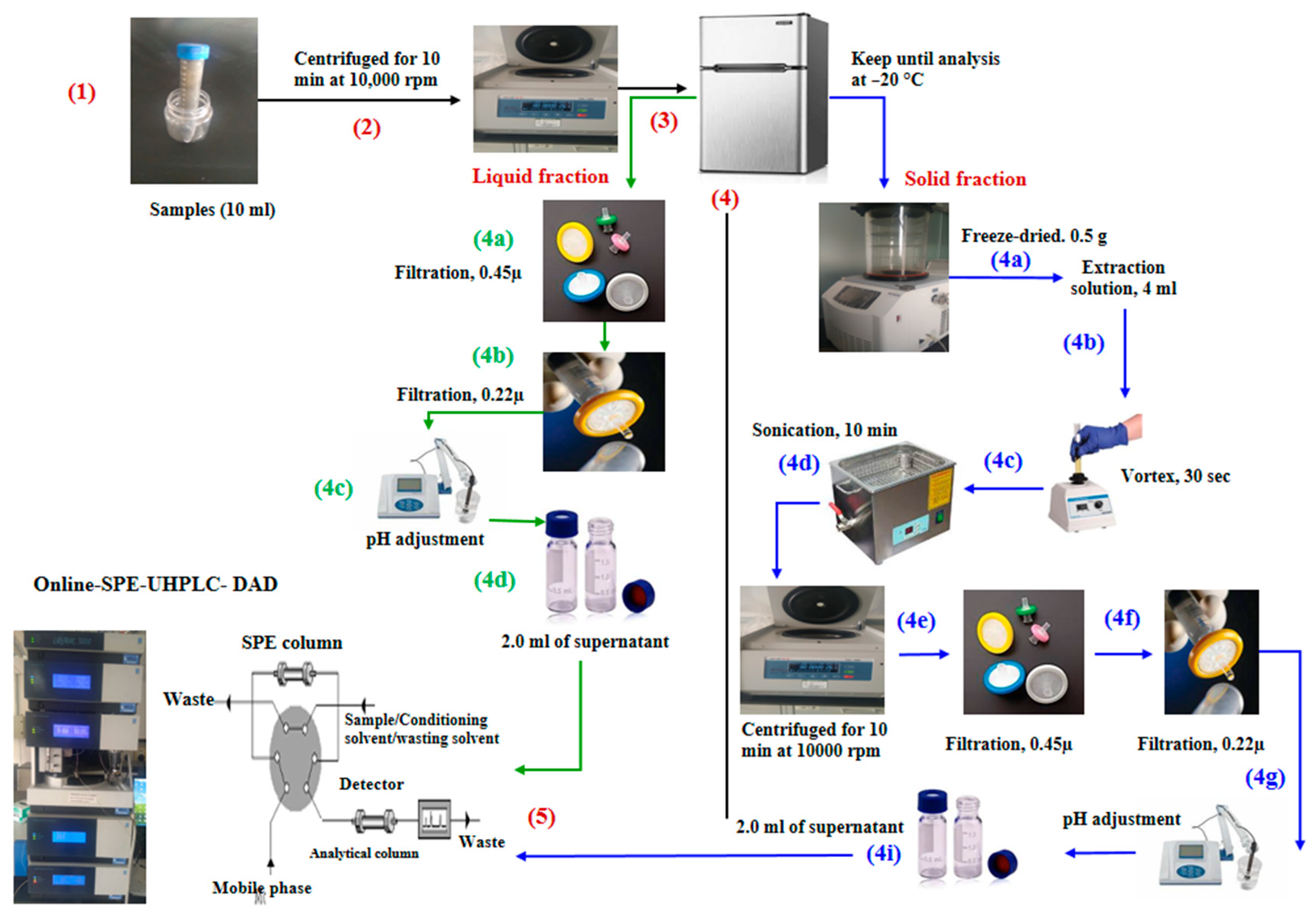
3.4. Sample Preparation
- i.
- (T1): a mixture (50:50 v/v) of MeOH/ACN and pH adjusted to 3.0;
- ii.
- (T2): saturated aqueous Na2EDTA, water, and Na2EDTA–McIlvaine buffer (pH 4.0) as developed by Li et al. [7];
- iii.
- (T3): a mixture between a McIlvaine buffer (0.1 M Na2EDTA) solution, and MeOH at a ratio of 25:25:50 (based on the volume v/v/v), and pH was adjusted to 7.2 by adding 6N NaOH solution. The McIlvaine buffer was prepared by mixing 0.2 M citric acid and 0.4 M Na2HPO4 solutions at a ratio of (90:60) (based on the volume v/v)) as reported by Karci and Balcioglu [16]. The samples’ pH was adjusted to 3.0 before HPLC injection.
3.5. Cleaning Up
3.6. Chromatographic Separation
3.7. Assay Validation
3.7.1. Specificity, Linearity, Limit of Detection (LOD), and Limit of Quantification (LOQ)
3.7.2. Precision
3.7.3. Accuracy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaballah, M.S.; Guo, J.; Sun, H.; Aboagye, D.; Sobhi, M.; Muhmood, A.; Dong, R. A review targeting veterinary antibiotics removal from livestock manure management systems and future outlook. Bioresour. Technol. 2021, 333, 125069. [Google Scholar] [CrossRef]
- Shalaby, A.R.; Salama, N.A.; Abou-Raya, S.H.; Emam, W.H.; Mehaya, F.M. Validation of HPLC method for determination of tetracycline residues in chicken meat and liver. Food Chem. 2011, 124, 1660–1666. [Google Scholar] [CrossRef]
- Spielmeyer, A. Occurrence and fate of antibiotics in manure during manure treatments: A short review. Sustain. Chem. Pharm. 2018, 9, 76–86. [Google Scholar] [CrossRef]
- Rufino, M.C.; Brandt, P.; Herrero, M. Reducing uncertainty in nitrogen budgets for African livestock systems. Environ. Res. Lett. Pap. 2014, 9, 105008. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Wei, R.; Chen, M.; Wang, T. A new, simple and rapid HPLC method for determination of chlortetracycline in pig solid manure. Ital. J. Anim. Sci. 2010, 9, 190–194. [Google Scholar] [CrossRef]
- Springer, V.; Jacksé, J.; Ek, P.; Lista, A.G.; Emmer, Å. Determination of fluoroquinolones in bovine milk samples using a pipette-tip SPE step based on multiwalled carbon nanotubes prior to CE separation. J. Sep. Sci. 2014, 37, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, L.; Wang, X.; Jin, H.; Ding, L.; Zhang, K.; Zhang, H. Determination of tetracyclines residues in honey by on-line solid-phase extraction high-performance liquid chromatography. Talanta 2008, 75, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Tao, Y.; Chen, D.; Wang, Y.; Yuan, Z. Development of an HPLC-UV method for the simultaneous determination of tetracyclines in muscle and liver of porcine, chicken and bovine with accelerated solvent extraction. Food Chem. 2011, 124, 1131–1138. [Google Scholar] [CrossRef]
- Sartini, I.; Beata, Ł.-W.; Tae, W.K.; Andrzej, L.; Amnart, P.; Mario, G. Pharmacokinetics and tissue analysis of levofloxacin in sheep (Ovis aries Linnaeus) after multiple-dose administration. Res. Vet. Med. 2020, 128, 124–128. [Google Scholar]
- Yu, H.; Mu, H.; Hu, Y.M. Determination of fluoroquinolones, sulfonamides, and tetracyclines multiresidues simultaneously in porcine tissue by MSPD and HPLC–DAD. J. Pharm. Anal. 2012, 2, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Panda, A.K.; Sharma, N. Determination of antibiotic residues in bovine milk by HPLC-DAD and assessment of human health risks in Northwestern Himalayan region, India. J. Food Sci. Technol. 2021, 1–10. [Google Scholar] [CrossRef]
- Sartini, I.; Łebkowska-Wieruszewska, B.; Lisowski, A.; Poapolathep, A.; Giorgi, M. Danofloxacin pharmacokinetics and tissue residues in Bilgorajska geese. Res. Vet. Sci. 2021, 136, 11–17. [Google Scholar] [PubMed]
- Kumar, A.; Gill, J.P.S.; Bedi, J.S.; Chhuneja, P.K.; Kumar, A. Determination of antibiotic residues in Indian honeys and assessment of potential risks to consumers. J. Apic. Res. 2020, 59, 25–34. [Google Scholar] [CrossRef]
- Sher, M.; Hussain, M.A.; Mehmood, M.H.; Hassan, M.N.; Bashir, S. Bioequivalence of norfloxacin by HPLC-UV method. J. Chil. Chem. Soc. 2010, 55, 203–205. [Google Scholar] [CrossRef] [Green Version]
- Vella, J.; Busuttil, F.; Bartolo, N.S.; Sammut, C.; Ferrito, V.; Serracino-Inglott, A.; Azzopardi, L.M.; LaFerla, G. A simple HPLC-UV method for the determination of ciprofloxacin in human plasma. J. Chromatogr. B. 2015, 989, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Karci, A.; Balcioglu, I.A. Investigation of the tetracycline, sulfonamide, and fluoroquinolone antimicrobial compounds in animal manure and agricultural soils in Turkey. Sci. Total Environ. 2009, 407, 4652–4664. [Google Scholar] [CrossRef]
- Barreca, S.; Busetto, M.; Colzani, L.; Clerici, L.; Daverio, D.; Dellavedova, P.; Balzamo, S.; Calabretta, E.; Ubaldi, V. Determination of estrogenic endocrine disruptors in water at sub-ng L-1 levels in compliance with Decision 2015/495/EU using offline-online solid phase extraction concentration coupled with high performance liquid chromatography-tandem mass spectrometry. Microchem. J. 2019, 147, 1186–1191. [Google Scholar] [CrossRef]
- Barreca, S.; Busetto, M.; Vitelli, M.; Colzani, L.; Clerici, L.; Dellavedova, P. Development of a rapid and accurate method for the determination of key. J. Chem. 2018, 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Orecchio, S.; Indelicato, R.; Barreca, S. Determination of selected phthalates by gas chromatography–mass spectrometry in personal perfumes. J. Toxicol. Environ. Health Part 2015, 114, 187–191. [Google Scholar] [CrossRef]
- Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results (2002/657/EC) (2002). Off. J. Eur. Commun. 2002, L221, 8–36.
- Marti, E.; Gros, M.; Boy-roura, M.; Ovejero, J.; Busquets, A.M.; Colón, J.; Petrovic, M.; Ponsá, S. Pharmaceuticals removal in an on-farm pig slurry treatment plant based on solid-liquid separation and nitrification-denitrification systems. Waste Manag. 2020, 102, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.M.; Nemati, M.; Babaeia, H.; Ansarin, M.; Nourdadgar, A.O.S. Solid-phase extraction and simultaneous determination of tetracycline residues in edible cattle tissues using an HPLC-FL method. Iran. J. Pharm. Res. 2012, 11, 781–787. [Google Scholar] [CrossRef]
- Zhao, L.; Dong, Y.H.; Wang, H. Residues of veterinary antibiotics in manures from feedlot livestock in eight provinces of China. Sci. Total Environ. 2010, 408, 1069–1075. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic | Wavelength λ (nm) | Typical Retention Time (min) |
|---|---|---|
| OTC | 270 | 9.14 |
| SDZ | 277 | 9.9 |
| Norf | 270 | 8.7 |
| Enorf | 277 | 10.2 |
| TC | 270 | 10.9 |
| Mobile Phase | Methanol (A) Acetonitrile (B) | ||||||||
| 0.05 and 0.01 M Oxalic Acid in Highly Purified Distilled Water (C) | |||||||||
| Chromatography Condition | |||||||||
| HPLC | On-Line SPE | ||||||||
| Time (min) | Flow Rate (mL∙min−1) | A (%) | B (%) | C (%) | Time (min) | Flow Rate (mL∙min−1) | A (%) | B (%) | C (%) |
| 0 | 0.8 | 0 | 40 | 60 | 0 | 0.8 | 0 | 5 | 95 |
| 10 | 0 | 40 | 60 | 15 | 0 | 5 | 95 | ||
| 20 | 0 | 60 | 40 | 20 | 0 | 80 | 20 | ||
| 25 | 0 | 40 | 60 | 25 | 0 | 5 | 95 | ||
| 30 | 0 | 40 | 60 | 30 | 0 | 5 | 95 | ||
| 35 | 100 | 0 | 0 | 35 | 100 | 0 | 0 | ||
| Solid Extraction Solution | Recovery (%) | ||||
|---|---|---|---|---|---|
| TC | OTC | SDZ | Enorf | Norf | |
| T1 | 84.5% | 2.85% | 11.37% | 1.43% | 21.87% |
| T2 | 77.2% | 67.15% | 80.63% | 62.11% | 51.28% |
| T3 | 95.58% | 84.58% | 113.2% | 99.73% | 93.71% |
| Antibiotics | Slope | Intercept | R2 | |||
|---|---|---|---|---|---|---|
| Liquid | Solid | Liquid | Solid | Liquid | Solid | |
| TC | 1.07 | 1.514 | −0.51 | −6.62 | 0.995 | 0.994 |
| OTC | 0.795 | 0.825 | 0.05 | 5.486 | 0.997 | 0.992 |
| Norf | 2.34 | 2.85 | 3.62 | 5.27 | 0.999 | 0.994 |
| SDZ | 1.43 | 1.62 | 0.19 | 5.68 | 0.999 | 0.991 |
| Enorf | 2.17 | 3.17 | 1.73 | 2.18 | 0.999 | 0.999 |
| Compound | Spiked Level (µg·g−1) | LOD | LOQ | Within-Day RSD (%) | Between-Day RSD (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Liquid (µg mL−1) | Solid (µg·g−1) | Liquid (µg mL−1) | Solid (µg·g−1) | Liquid | Solid | Liquid | Solid | ||
| TC | 1 | 0.15 | 0.29 | 0.47 | 0.89 | 11.27 | 1.63 | 9.20 | 10.58 |
| 10 | 6.97 | 4.59 | 9.02 | 19.48 | |||||
| 20 | 8.24 | 16.94 | 4.25 | 9.01 | |||||
| 50 | 16.9 | 15.17 | 3.40 | 2.26 | |||||
| 100 | 16.58 | 14.87 | 5.87 | 0.82 | |||||
| OTC | 1 | 0.1 | 0.58 | 0.32 | 1.77 | 15.52 | 7.14 | 6.57 | 3.65 |
| 10 | 11.00 | 17.3 | 5.24 | 10.51 | |||||
| 20 | 1.59 | 13.67 | 0.8 | 12.92 | |||||
| 50 | 10.87 | 12.69 | 11.34 | 17.45 | |||||
| 100 | 13.78 | 7.49 | 8.21 | 3.67 | |||||
| Norf | 1 | 0.42 | 0.032 | 1.27 | 0.096 | 17.78 | 9.94 | 8.39 | 5.56 |
| 10 | 9.28 | 13.17 | 2.81 | 9.17 | |||||
| 20 | 4.68 | 10.32 | 2.30 | 9.48 | |||||
| 50 | 5.5 | 17.22 | 4.80 | 18.06 | |||||
| 100 | 17.85 | 10.69 | 12.65 | 5.15 | |||||
| SDZ | 1 | 0.34 | 0.13 | 1.04 | 0.39 | 1.36 | 8.68 | 4.47 | 10.65 |
| 10 | 5.61 | 13.73 | 8.21 | 0.8 | |||||
| 20 | 2.15 | 10.79 | 1.09 | 17.28 | |||||
| 50 | 15.52 | 1.59 | 2.86 | 7.22 | |||||
| 100 | 9.16 | 19.8 | 0.68 | 4.48 | |||||
| Enorf | 1 | 0.26 | 0.05 | 0.79 | 0.16 | 7.03 | 16.93 | 2.13 | 12.02 |
| 10 | 5.37 | 19.07 | 6.08 | 0.44 | |||||
| 20 | 0.29 | 6.02 | 0.15 | 3.08 | |||||
| 50 | 12.69 | 0.87 | 2.74 | 10.23 | |||||
| 100 | 4.32 | 12.15 | 3.43 | 16.24 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaballah, M.S.; Li, X.; Zhang, Z.; Al-Anazi, A.; Sun, H.; Sobhi, M.; Philbert, M.; Ghorab, M.A.; Guo, J.; Dong, R. Determination of Tetracycline, Oxytetracycline, Sulfadiazine, Norfloxacin, and Enrofloxacin in Swine Manure Using a Coupled Method of On-Line Solid-Phase Extraction with the UHPLC–DAD. Antibiotics 2021, 10, 1397. https://doi.org/10.3390/antibiotics10111397
Gaballah MS, Li X, Zhang Z, Al-Anazi A, Sun H, Sobhi M, Philbert M, Ghorab MA, Guo J, Dong R. Determination of Tetracycline, Oxytetracycline, Sulfadiazine, Norfloxacin, and Enrofloxacin in Swine Manure Using a Coupled Method of On-Line Solid-Phase Extraction with the UHPLC–DAD. Antibiotics. 2021; 10(11):1397. https://doi.org/10.3390/antibiotics10111397
Chicago/Turabian StyleGaballah, Mohamed S., Xin Li, Zijia Zhang, Abdulaziz Al-Anazi, Hui Sun, Mostafa Sobhi, Mperejekumana Philbert, Mohamed A. Ghorab, Jianbin Guo, and Renjie Dong. 2021. "Determination of Tetracycline, Oxytetracycline, Sulfadiazine, Norfloxacin, and Enrofloxacin in Swine Manure Using a Coupled Method of On-Line Solid-Phase Extraction with the UHPLC–DAD" Antibiotics 10, no. 11: 1397. https://doi.org/10.3390/antibiotics10111397
APA StyleGaballah, M. S., Li, X., Zhang, Z., Al-Anazi, A., Sun, H., Sobhi, M., Philbert, M., Ghorab, M. A., Guo, J., & Dong, R. (2021). Determination of Tetracycline, Oxytetracycline, Sulfadiazine, Norfloxacin, and Enrofloxacin in Swine Manure Using a Coupled Method of On-Line Solid-Phase Extraction with the UHPLC–DAD. Antibiotics, 10(11), 1397. https://doi.org/10.3390/antibiotics10111397