Phosphate Modified Screen Printed Electrodes by LIFT Treatment for Glucose Detection

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
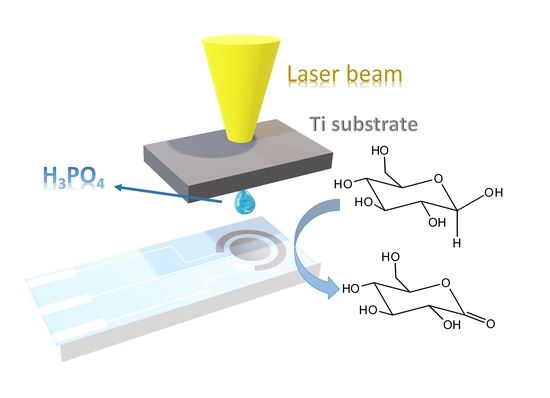
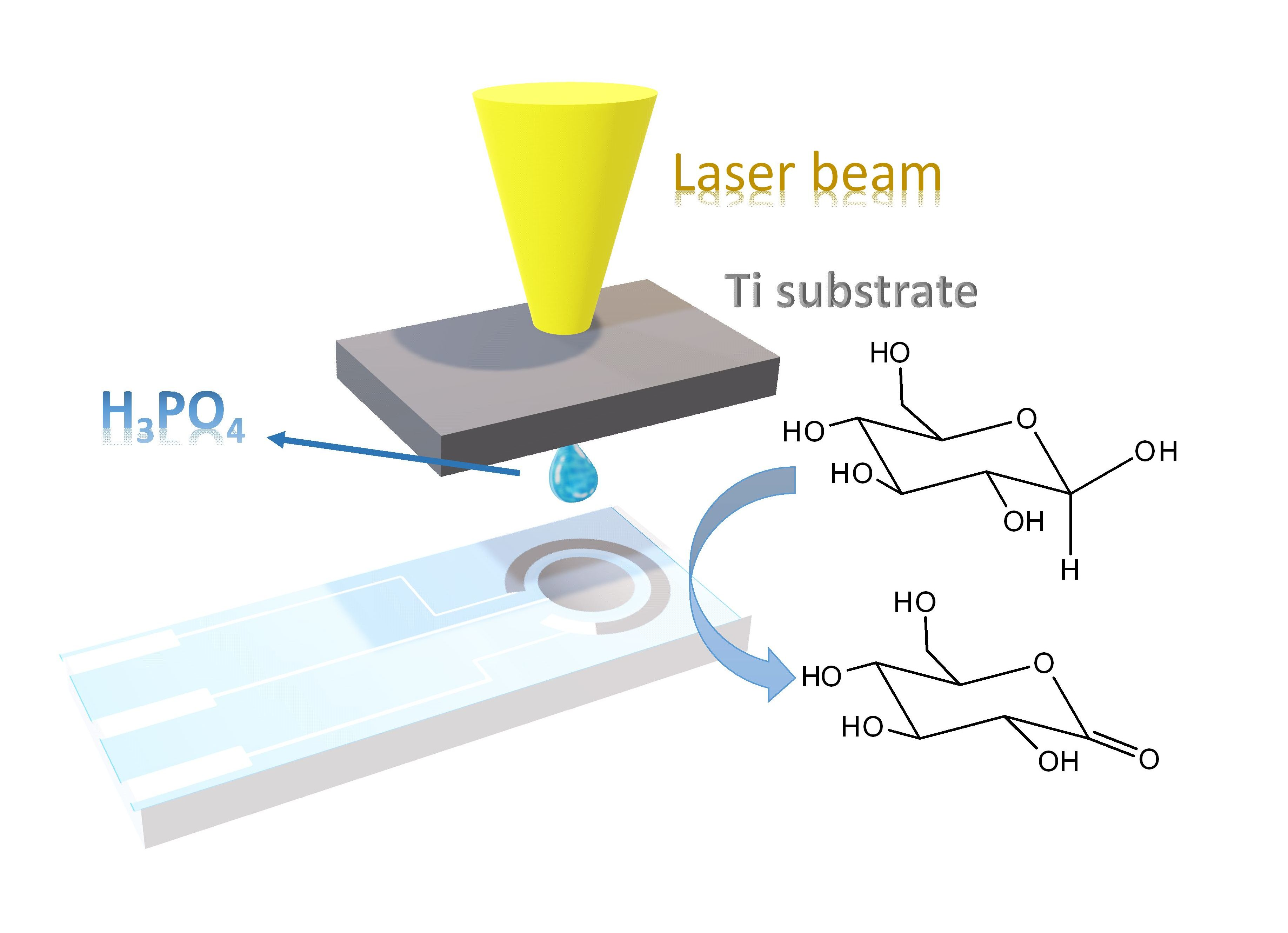
1. Introduction
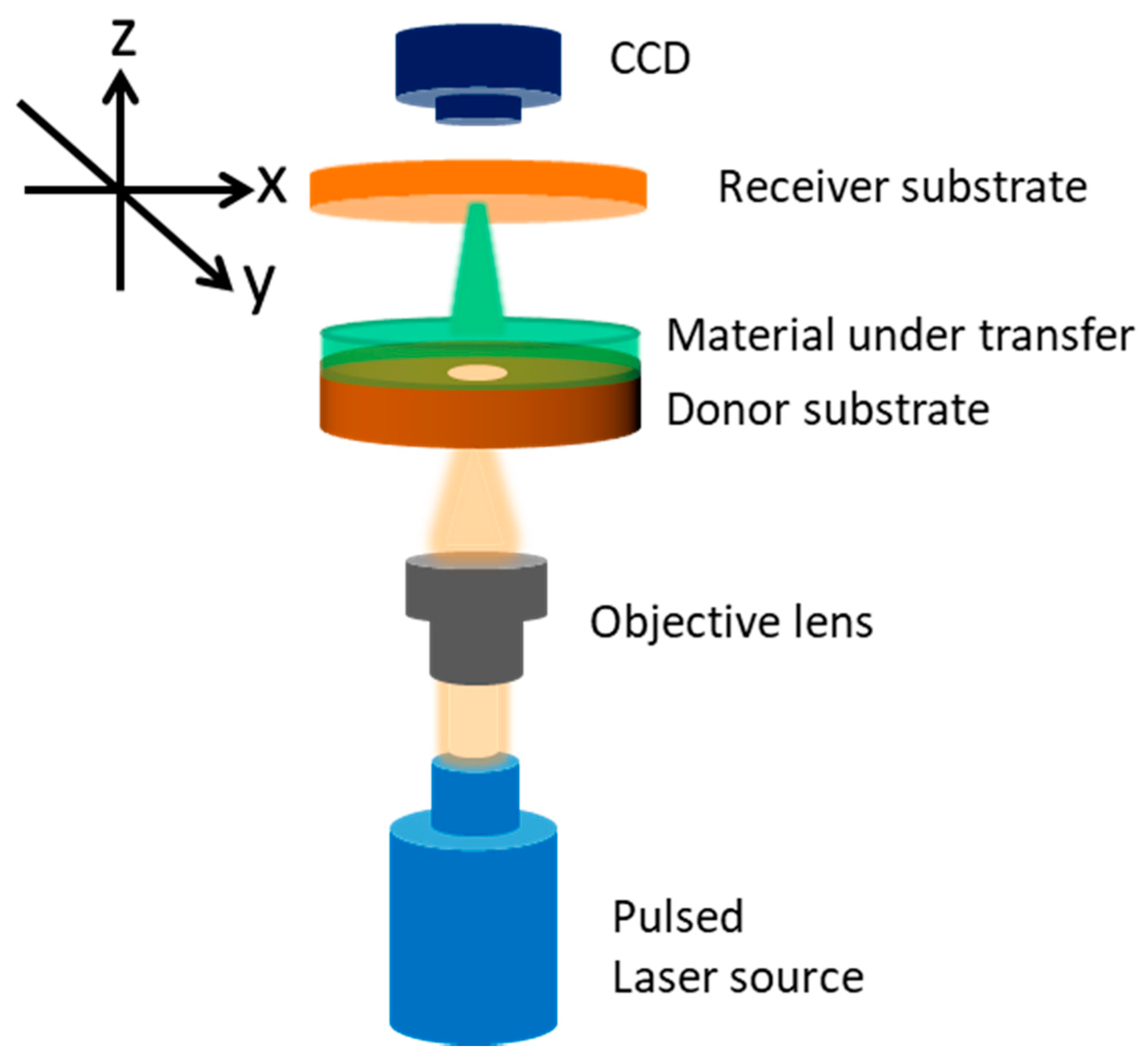
2. Materials and Methods
3. Results and Discussion
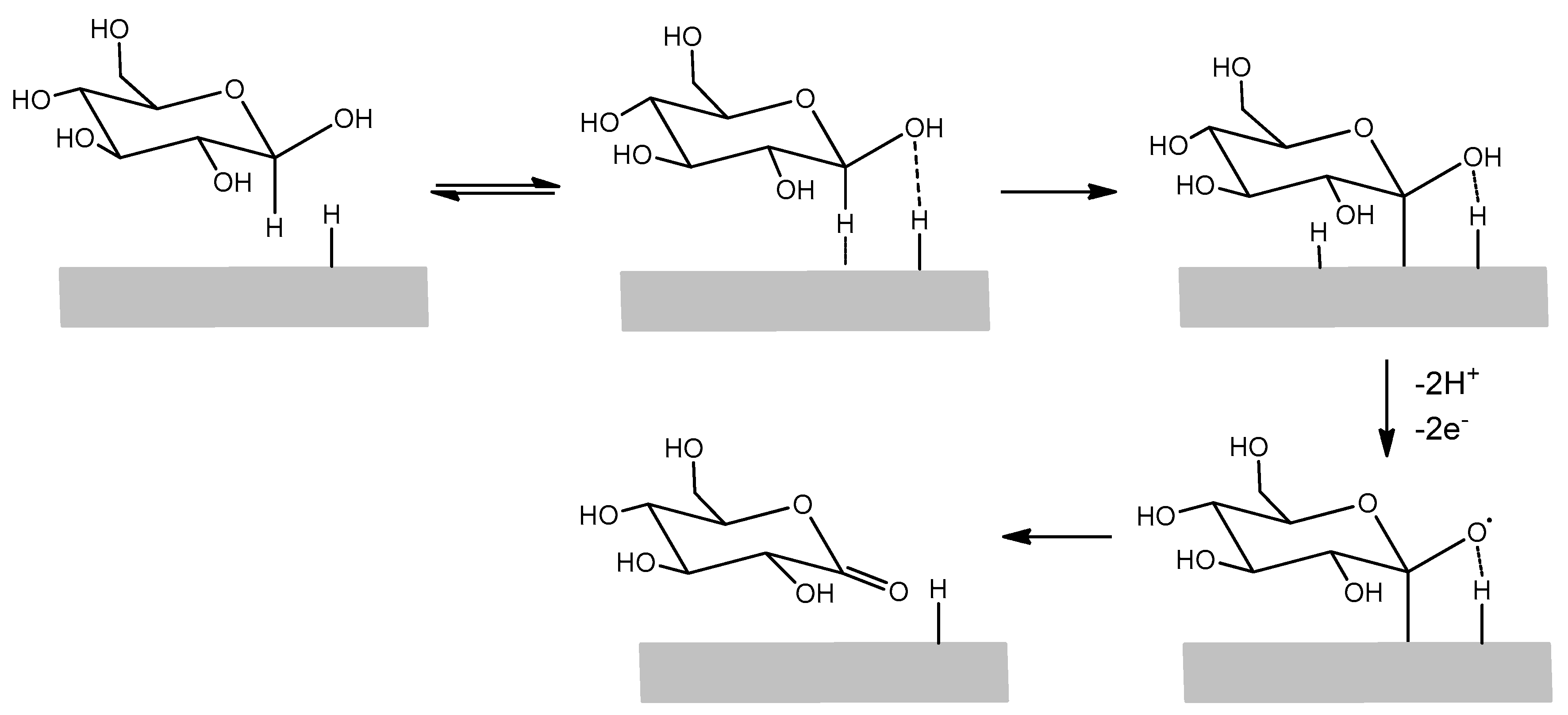
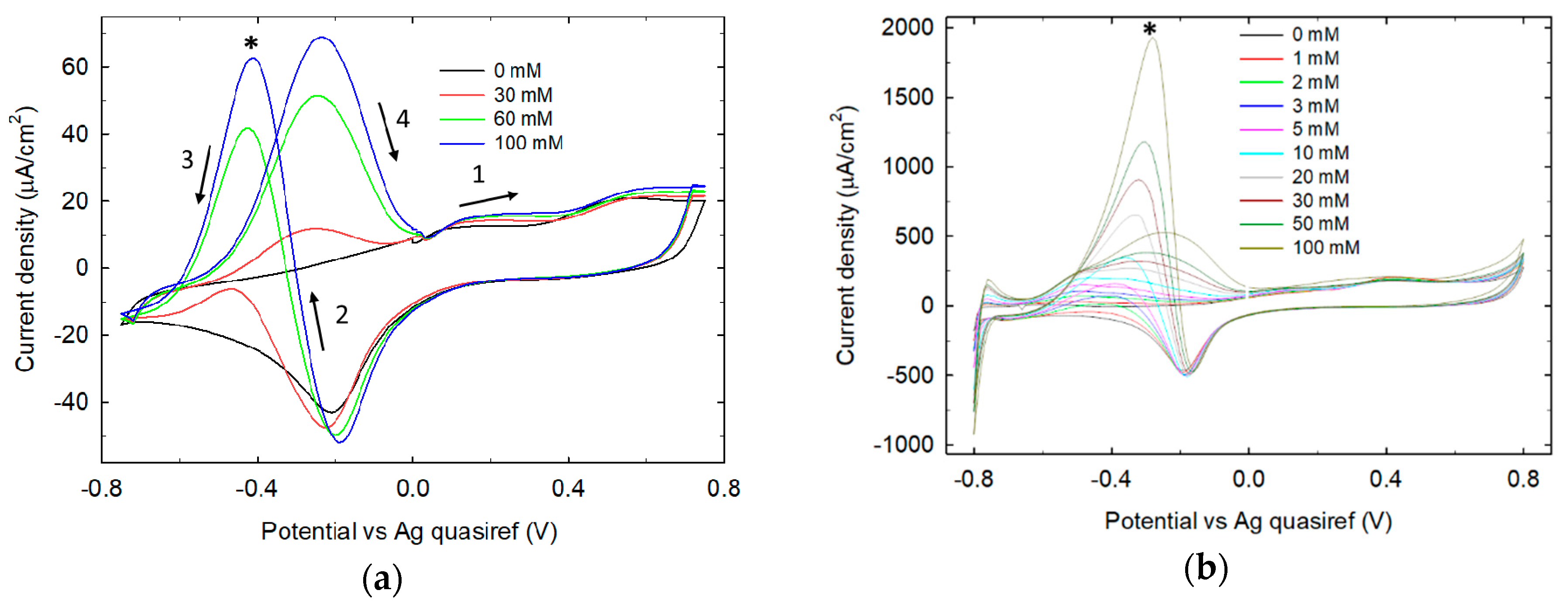
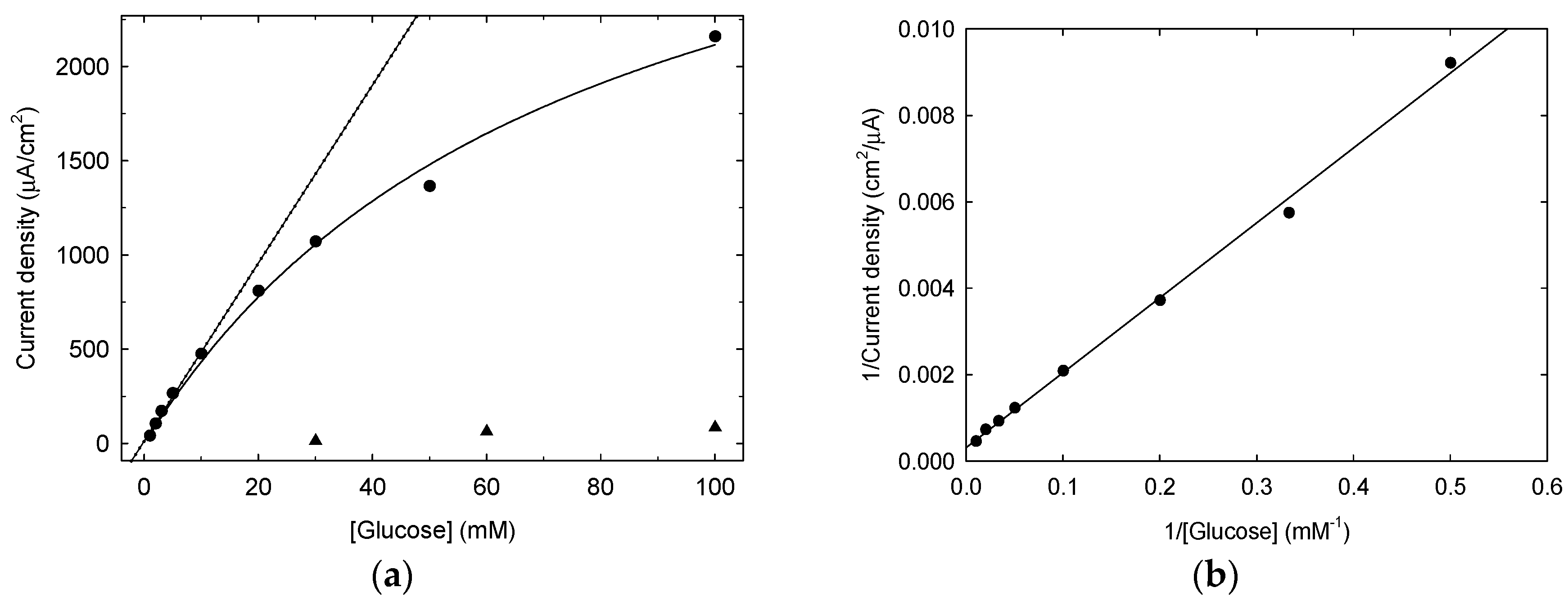
3.1. LIFT Treatment of Pt and Response to Glucose
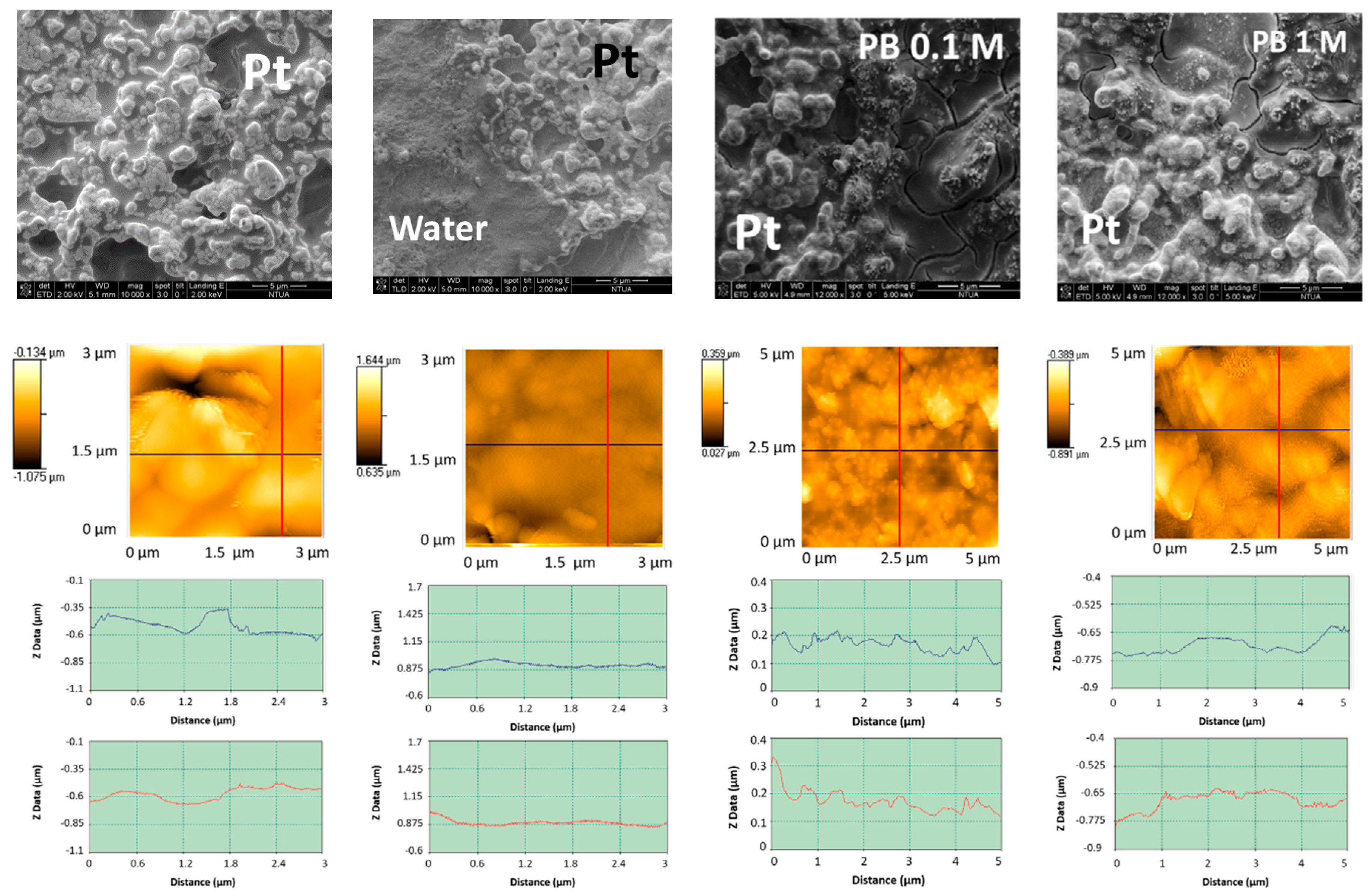
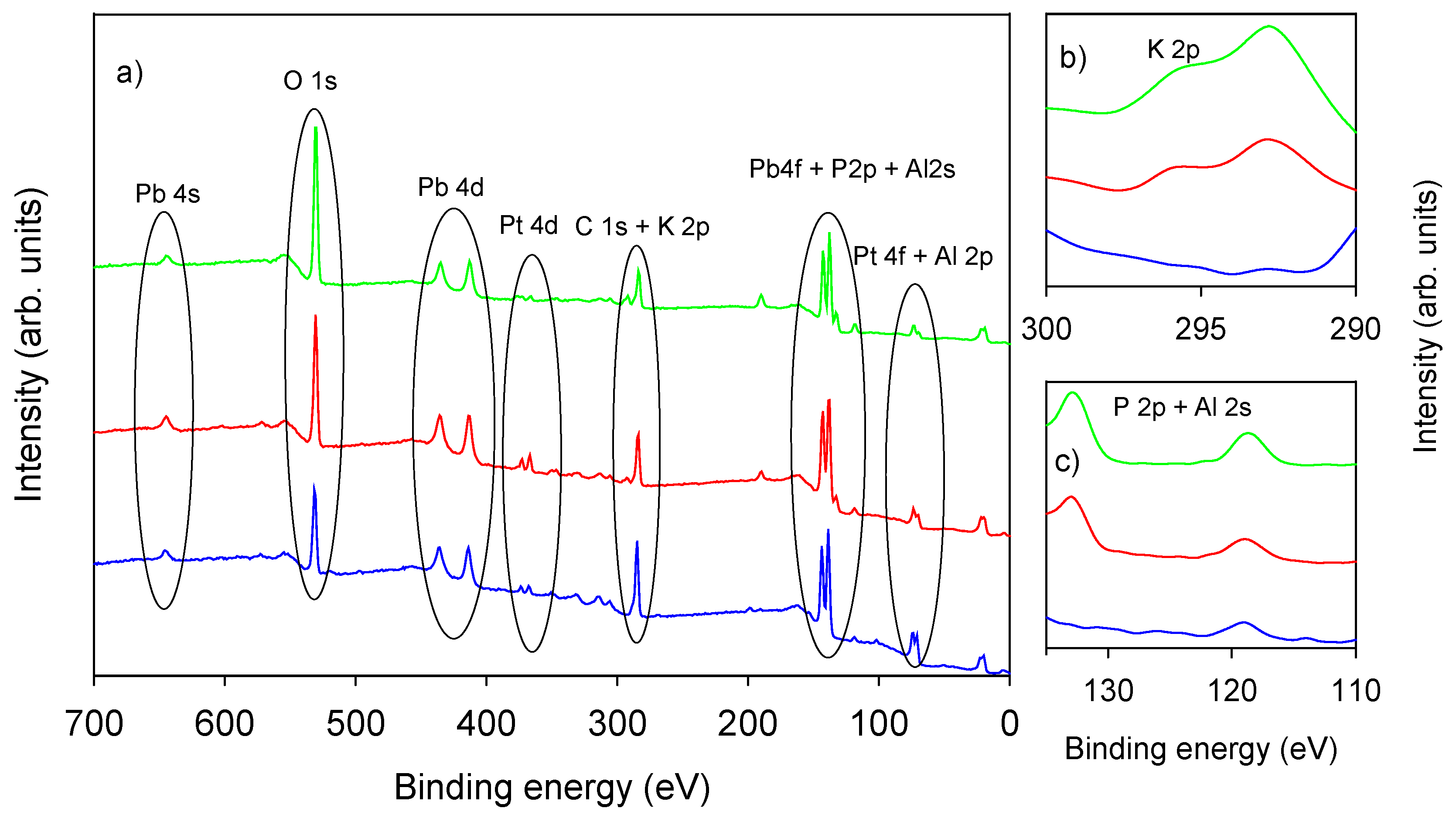
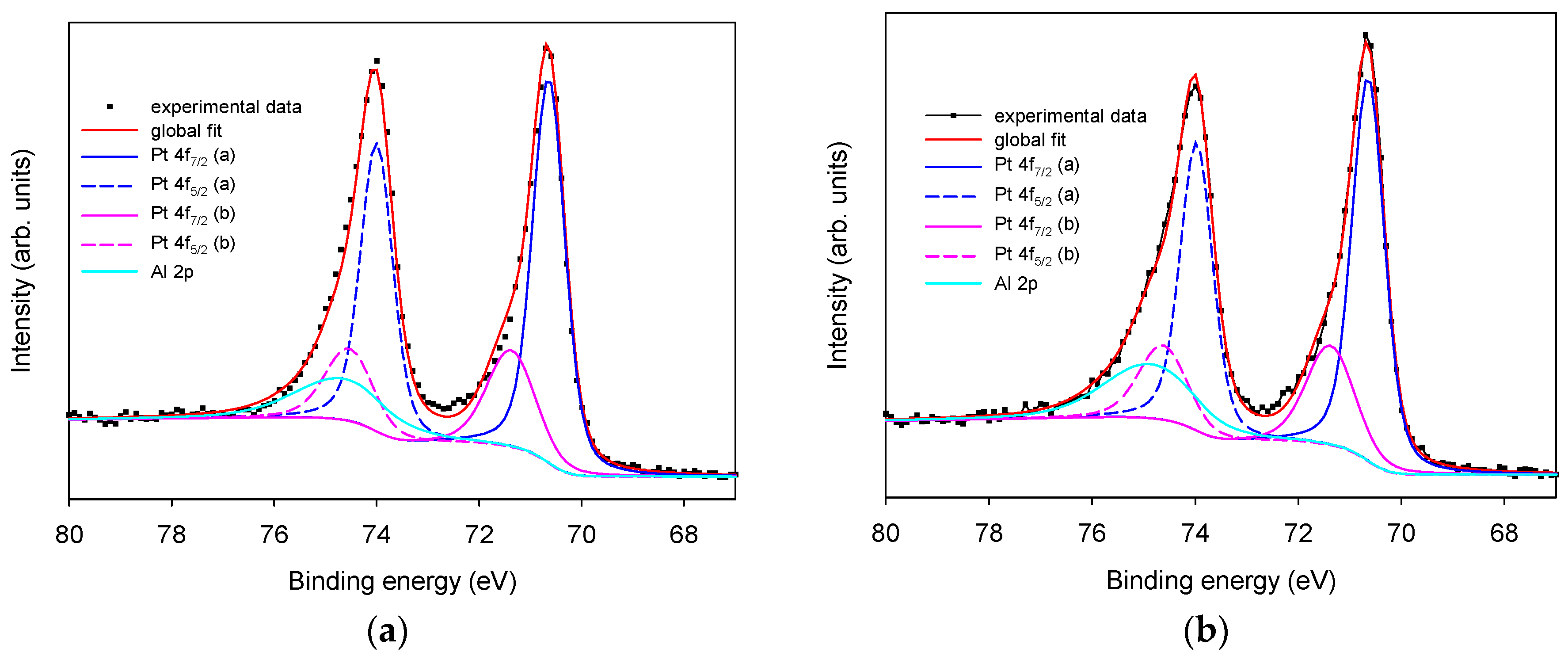
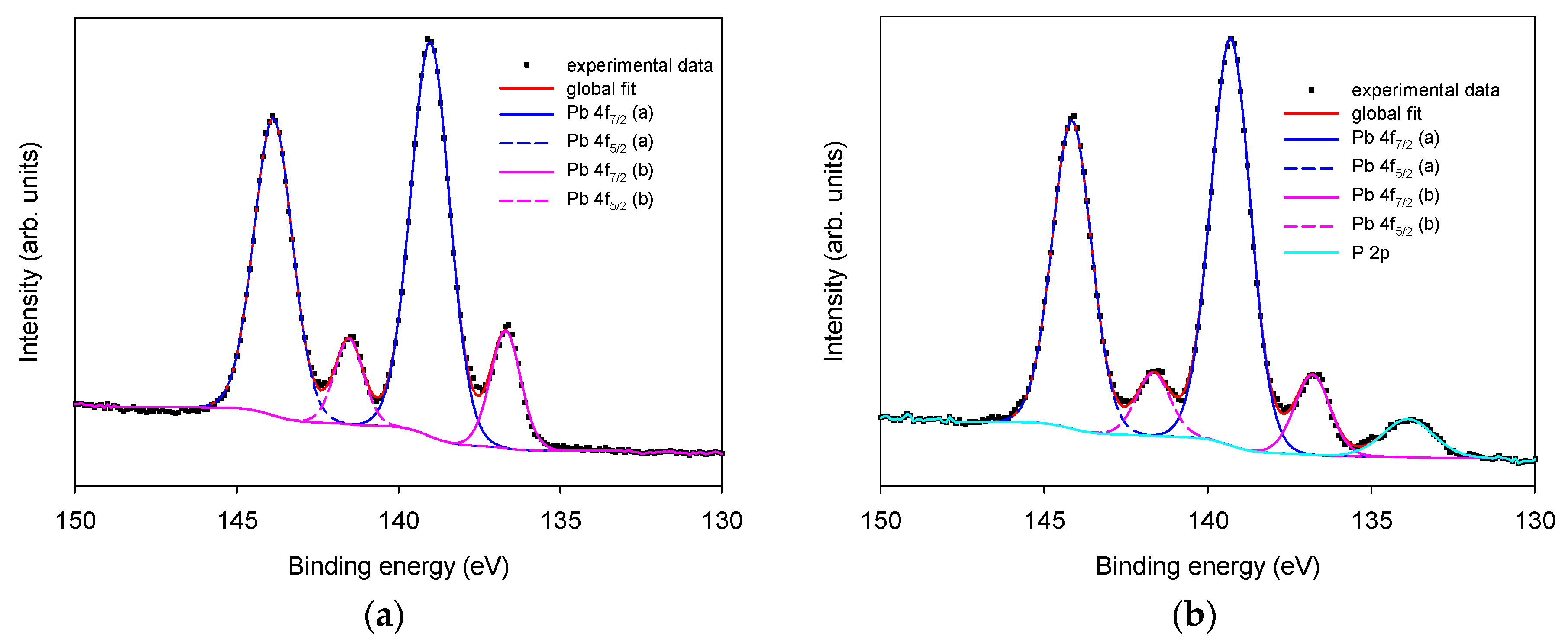
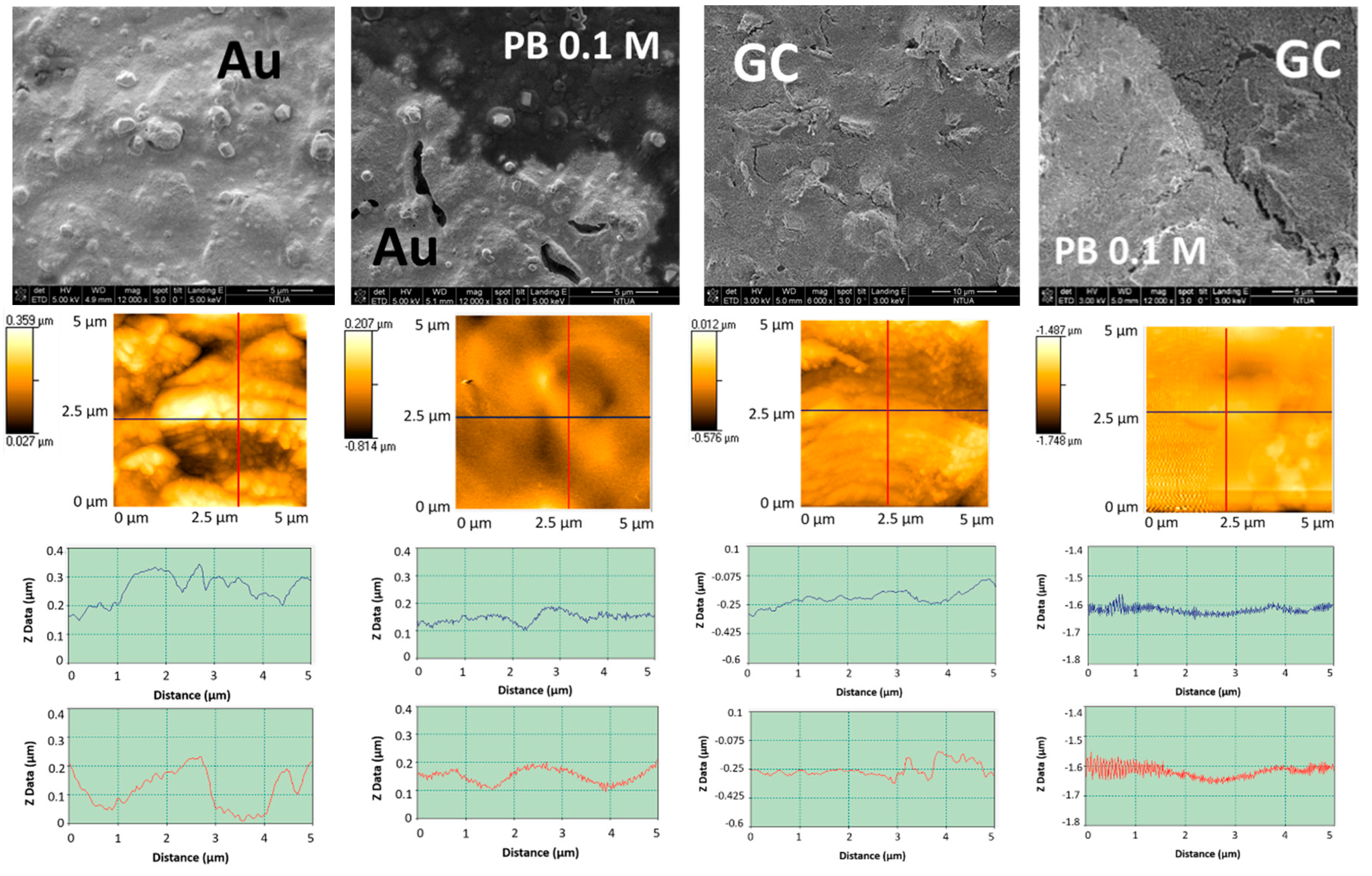
3.1.1. Morphological and Chemical Characterization
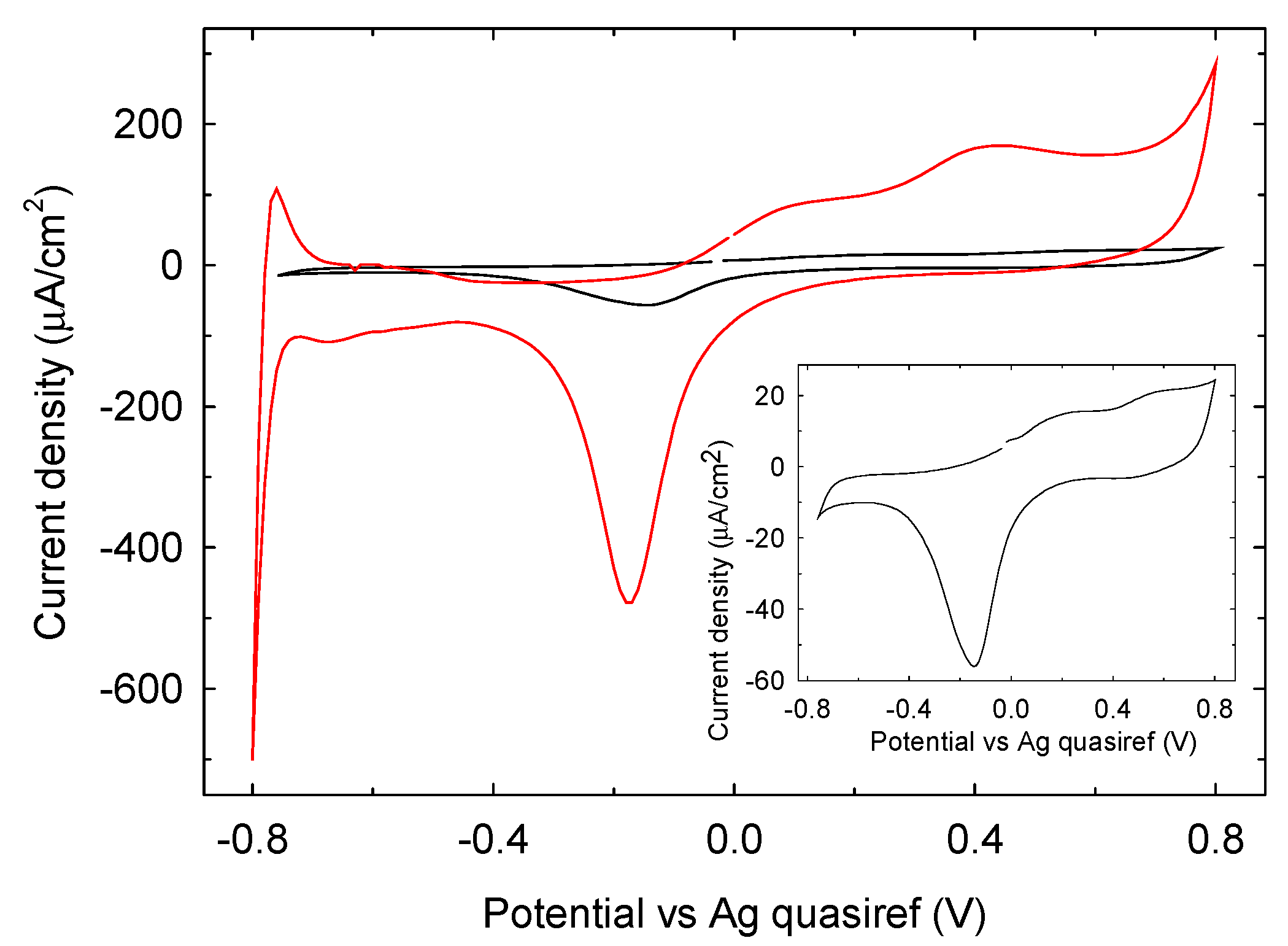
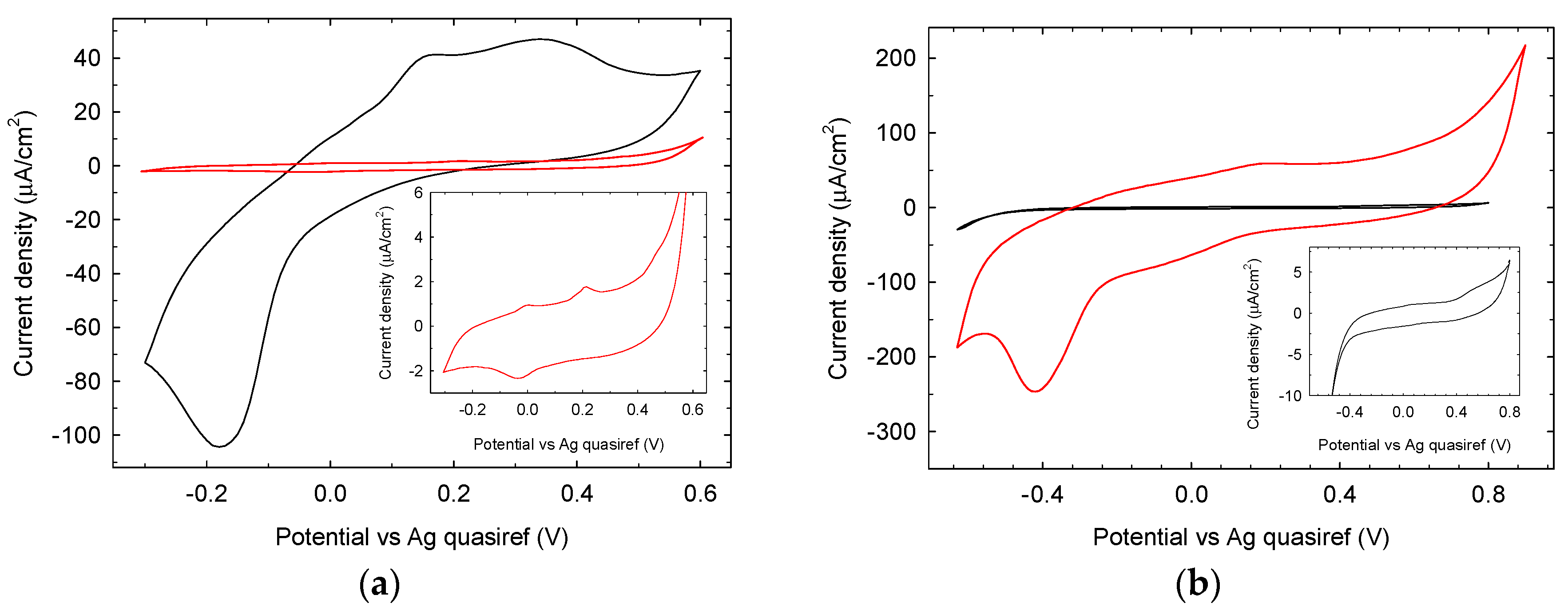
3.1.2. Electrochemical Characterization
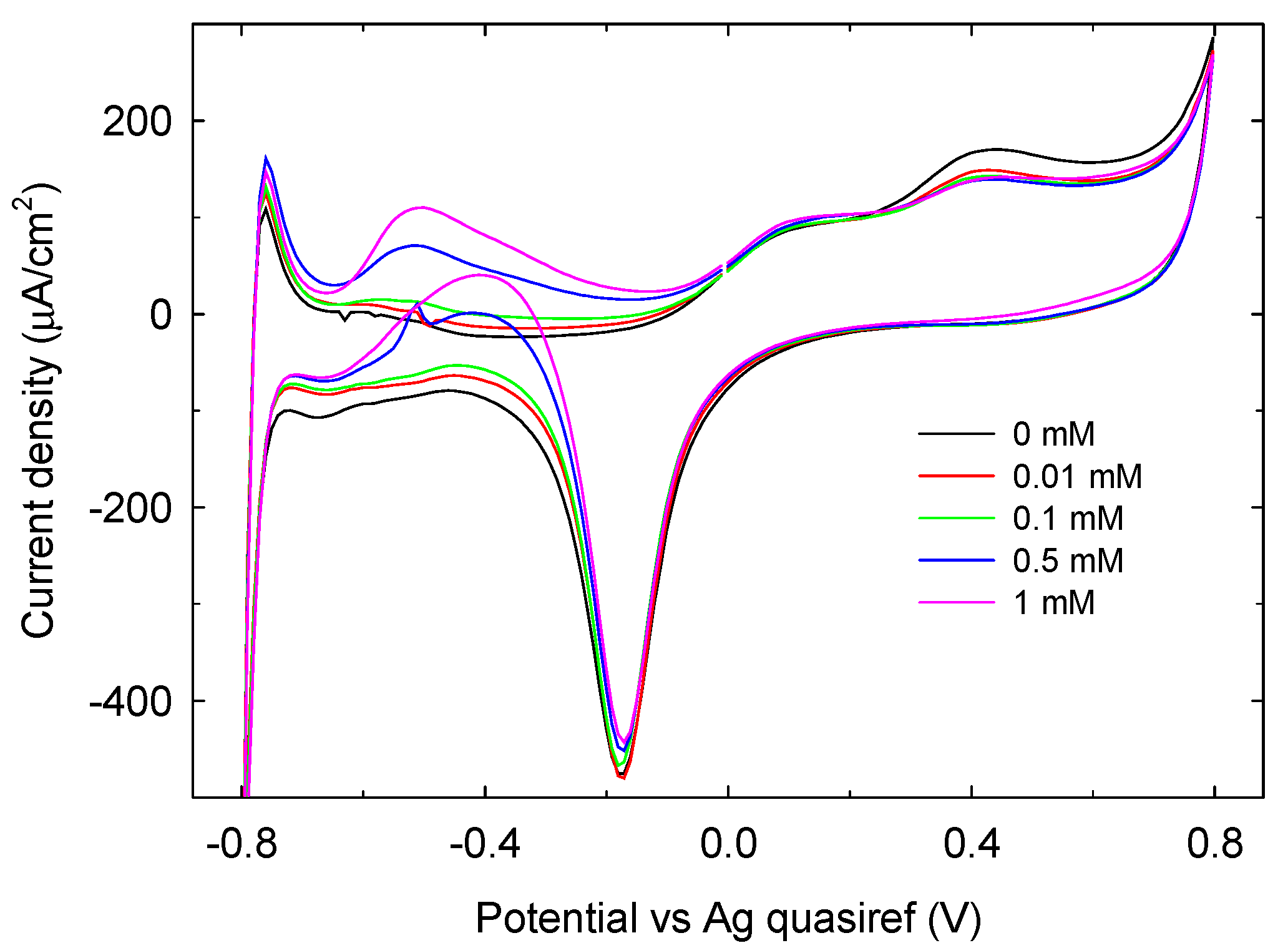
3.2. LIFT Treatment of Au and GC SPEs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aghahosseini, H.; Ramazani, A.; Azimzadeh Asiabi, P.; Gouranlou, F.; Hosseini, F.; Rezaei, A.; Min, B.-K.; Woo Joo, S. Glucose-based Biofuel Cells: Nanotechnology as a Vital Science in Biofuel Cells Performance. Nanochem. Res. 2016, 1, 183–204. [Google Scholar]
- Wang, J. Electrochemical Glucose Biosensors. In Sensors, Biosensors and Their Biomedical Applications, 1st ed.; Zhang, X., Ju, H., Wang, J., Eds.; Academic Press: Burlington, MA, USA, 2008; Volume 3, pp. 57–69. [Google Scholar]
- Chen, C.; Xie, Q.J.; Yang, D.W.; Xiao, H.L.; Fu, Y.C.; Tan, Y.M.; Yao, S.Z. Recent advances in electrochemical glucose biosensors: A review. RSC Adv. 2013, 3, 4473–4491. [Google Scholar] [CrossRef]
- Tian, K.; Prestgard, M.; Tiwari, A. A review of recent advances in nonenzymatic glucose sensors. Mater. Sci. Eng. C 2014, 41, 100–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; He, X.; Wang, L.; Gu, A.; Huang, Y.; Fang, B.; Geng, B.; Zhang, X. Non-enzymatic electrochemical sensing of glucose. Microchim. Acta 2013, 180, 161–186. [Google Scholar] [CrossRef]
- Rahman, M.M.; Ahammad, A.J.S.; Jin, J.-H.; Ahn, S.J.; Lee, J.-J. A Comprehensive Review of Glucose Biosensors Based on Nanostructured Metal-Oxides. Sensors 2010, 10, 4855–4886. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Foster, C.W.; Cumba, L.R.; do Carmo, D.R.; Banks, C.E. Can solvent induced surface modifications applied to screen-printed platforms enhance their electroanalytical performance? Analyst 2016, 141, 2783–2790. [Google Scholar] [CrossRef] [PubMed]
- Washe, A.P.; Lozano-Sánchez, P.; Bejarano-Nosas, D.; Katakis, I. Facile and versatile approaches to enhancing electrochemical performance of screen printed electrodes. Electrochim. Acta 2013, 91, 166–172. [Google Scholar] [CrossRef]
- Murugappan, K.; Lee, J.; Silvester, D.S. Comparative study of screen printed electrodes for ammonia gas sensing in ionic liquids. Electrochem. Commun. 2011, 13, 1435–1438. [Google Scholar] [CrossRef]
- Lee, J.; Arrigan, D.W.M.; Silvester, D.S. Mechanical polishing as an improved surface treatment for platinum screen-printed electrodes. Sens. Bio-Sens. Res. 2016, 9, 38–44. [Google Scholar] [CrossRef]
- Bohandy, J.; Kim, B.F.; Adrian, F.J. Metal-Deposition from a Supported Metal-Film Using an Excimer Laser. J. Appl. Phys. 1986, 60, 1538–1539. [Google Scholar] [CrossRef]
- Chatzipetrou, M.; Milano, F.; Giotta, L.; Chirizzi, D.; Trotta, M.; Massaouti, M.; Guascito, M.R.; Zergioti, I. Functionalization of gold screen printed electrodes with bacterial photosynthetic reaction centers by laser printing technology for mediatorless herbicide biosensing. Electrochem. Commun. 2016, 64, 46–50. [Google Scholar] [CrossRef]
- Guascito, M.R.; Malitesta, C.; Mazzotta, E.; Turco, A. Screen-Printed Glucose Oxidase-Based Biosensor for Inhibitive Detection of Heavy Metal Ions in a Flow Injection System. Sens. Lett. 2009, 7, 153–159. [Google Scholar] [CrossRef]
- Boutopoulos, C.; Touloupakis, E.; Pezzotti, I.; Giardi, M.T.; Zergioti, I. Direct laser immobilization of photosynthetic material on screen printed electrodes for amperometric biosensor. Appl. Phys. Lett. 2011, 98, 093703. [Google Scholar] [CrossRef]
- Touloupakis, E.; Boutopoulos, C.; Buonasera, K.; Zergioti, I.; Giardi, M.T. A photosynthetic biosensor with enhanced electron transfer generation realized by laser printing technology. Anal. Bioanal. Chem. 2012, 402, 3237–3244. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.E.; Salvi, A.M. Interpretation of the Shirley background in X-ray photoelectron spectroscopy analysis. J. Vac. Sci. Technol. A 2001, 19, 1170–1175. [Google Scholar] [CrossRef]
- NIST X-ray Photoelectron Spectroscopy Database. 2012. Available online: https://srdata.nist.gov/xps/Default.aspx (accessed on 10 September 2018).
- Guascito, M.R.; Ricciardi, G.; Rosa, A. Nickel-macrocycle interaction in nickel(II) porphyrins and porphyrazines bearing alkylthio β-substituents: A combined DFT and XPS study. J. Porphyr. Phthalocyanines 2017, 21, 371–380. [Google Scholar] [CrossRef]
- Boutopoulos, C.; Andreakou, P.; Kafetzopoulos, D.; Chatzandroulis, S.; Zergioti, I. Direct laser printing of biotin microarrays on low temperature oxide on Si substrates. Phys. Status Solidi 2008, 205, 2505–2508. [Google Scholar] [CrossRef]
- Björneholm, O.; Nilsson, A.; Tillborg, H.; Bennich, P.; Sandell, A.; Hernnäs, B.; Puglia, C.; Mårtensson, N. Overlayer structure from adsorbate and substrate core level binding energy shifts: CO, CCH3 and O on Pt(111). Surf. Sci. 1994, 315, L983–L989. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy; Perkin-Elmer Corp., Physical Electronics Division: Eden Prairie, MN, USA, 1995. [Google Scholar]
- Santos, M.C.; Machado, S.A.S. A voltammetric and nanogravimetric study of Te underpotential deposition on Pt in perchloric acid medium. Electrochim. Acta 2005, 50, 2289–2295. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: Hoboken, NJ, USA, 2000. [Google Scholar]
- Lim, B.; Jiang, M.; Camargo, P.H.C.; Cho, E.C.; Tao, J.; Lu, X.; Zhu, Y.; Xia, Y. Pd-Pt Bimetallic Nanodendrites with High Activity for Oxygen Reduction. Science 2009, 324, 1302. [Google Scholar] [CrossRef] [PubMed]
- Ernst, S.; Heitbaum, J.; Hamann, C.H. The electrooxidation of glucose in phosphate buffer solutions: Part I. Reactivity and kinetics below 350 mV/RHE. J. Electroanal. Chem. Interfacial Electrochem. 1979, 100, 173–183. [Google Scholar] [CrossRef]
- Bishop, G.W.; Ahiadu, B.K.; Smith, J.L.; Patterson, J.D. Use of Redox Probes for Characterization of Layer-by-Layer Gold Nanoparticle-Modified Screen-Printed Carbon Electrodes. J. Electrochem. Soc. 2017, 164, B23–B28. [Google Scholar] [CrossRef]
- Konopka, S.J.; McDuffie, B. Diffusion coefficients of ferri- and ferrocyanide ions in aqueous media, using twin-electrode thin-layer electrochemistry. Anal. Chem. 1970, 42, 1741–1746. [Google Scholar] [CrossRef]
- De Mele, M.F.L.; Videla, H.A.; Arvía, A.J. Potentiodynamic Study of Glucose Electro-Oxidation at Bright Platinum Electrodes. J. Electrochem. Soc. 1982, 129, 2207–2213. [Google Scholar] [CrossRef]
- Foo, K.Y.; Hameed, B.H. Insights into the modeling of adsorption isotherm systems. Chem. Eng. J. 2010, 156, 2–10. [Google Scholar] [CrossRef]
- Maaoui, H.; Teodoresu, F.; Wang, Q.; Pan, G.H.; Addad, A.; Chtourou, R.; Szunerits, S.; Boukherroub, R. Non-Enzymatic Glucose Sensing Using Carbon Quantum Dots Decorated with Copper Oxide Nanoparticles. Sensors 2016, 16, 1720. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhong, Y.; Zhang, Y.; Weng, W.; Li, S. Carbon quantum dots/octahedral Cu2O nanocomposites for non-enzymatic glucose and hydrogen peroxide amperometric sensor. Sens. Actuators B Chem. 2015, 206, 735–743. [Google Scholar] [CrossRef]
- Liu, M.; Liu, R.; Chen, W. Graphene wrapped Cu2O nanocubes: Non-enzymatic electrochemical sensors for the detection of glucose and hydrogen peroxide with enhanced stability. Biosens. Bioelectron. 2013, 45, 206–212. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt SPE At. % | Pt SPE LIFT PB 0.1 At. % | Pt SPE LIFT PB 1 At. % | |
|---|---|---|---|
| C 1s | 59.5 ± 0.6 | 37.7 ± 3.0 | 30.1 ± 2.3 |
| O 1s | 31.0 ± 0.8 | 44.5 ± 2.6 | 52.4 ± 1.0 |
| Pb 4f | 4.3 ± 0.3 | 5.3 ± 0.2 | 3.5 ± 0.3 |
| Pt 4f | 1.8 ± 0.3 | 1.6 ± 0.7 | 0.5 ± 0.2 |
| P 2p | 0.0 | 6.8 ± 1.3 | 8.2 ± 0.8 |
| Al 2s | 3.3 ± 0.4 | 3.6 ± 0.5 | 3.7 ± 3.2 |
| K 2p | 0.0 | 0.5 ± 0.1 | 1.6 ± 0.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milano, F.; Giotta, L.; Chirizzi, D.; Papazoglou, S.; Kryou, C.; De Bartolomeo, A.; De Leo, V.; Guascito, M.R.; Zergioti, I. Phosphate Modified Screen Printed Electrodes by LIFT Treatment for Glucose Detection. Biosensors 2018, 8, 91. https://doi.org/10.3390/bios8040091
Milano F, Giotta L, Chirizzi D, Papazoglou S, Kryou C, De Bartolomeo A, De Leo V, Guascito MR, Zergioti I. Phosphate Modified Screen Printed Electrodes by LIFT Treatment for Glucose Detection. Biosensors. 2018; 8(4):91. https://doi.org/10.3390/bios8040091
Chicago/Turabian StyleMilano, Francesco, Livia Giotta, Daniela Chirizzi, Simos Papazoglou, Christina Kryou, Annarita De Bartolomeo, Vincenzo De Leo, Maria Rachele Guascito, and Ioanna Zergioti. 2018. "Phosphate Modified Screen Printed Electrodes by LIFT Treatment for Glucose Detection" Biosensors 8, no. 4: 91. https://doi.org/10.3390/bios8040091
APA StyleMilano, F., Giotta, L., Chirizzi, D., Papazoglou, S., Kryou, C., De Bartolomeo, A., De Leo, V., Guascito, M. R., & Zergioti, I. (2018). Phosphate Modified Screen Printed Electrodes by LIFT Treatment for Glucose Detection. Biosensors, 8(4), 91. https://doi.org/10.3390/bios8040091