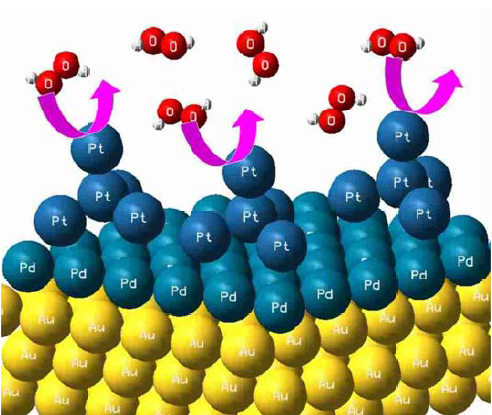
Trimetallic (Aurod-Pdshell-Ptcluster) Catalyst Used as Amperometric Hydrogen Peroxide Sensor




Abstract
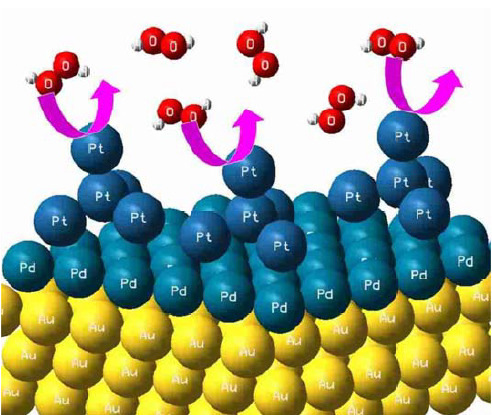
:
1. Introduction
2. Experimental Section
2.1. Apparatus and Reagents
2.2. Synthesis of Au Nanorods
2.3. Synthesis of Aurod-Pd Shell Structured Nanoparticles
2.4. Preparation of Au Rod-Pd Shell-Pt Cluster
2.5. Preparation of H2O2 Sensing Electrode
2.6. Preparation of H2O2 Solution
2.7. H2O2 Sensing
3. Results and Discussion
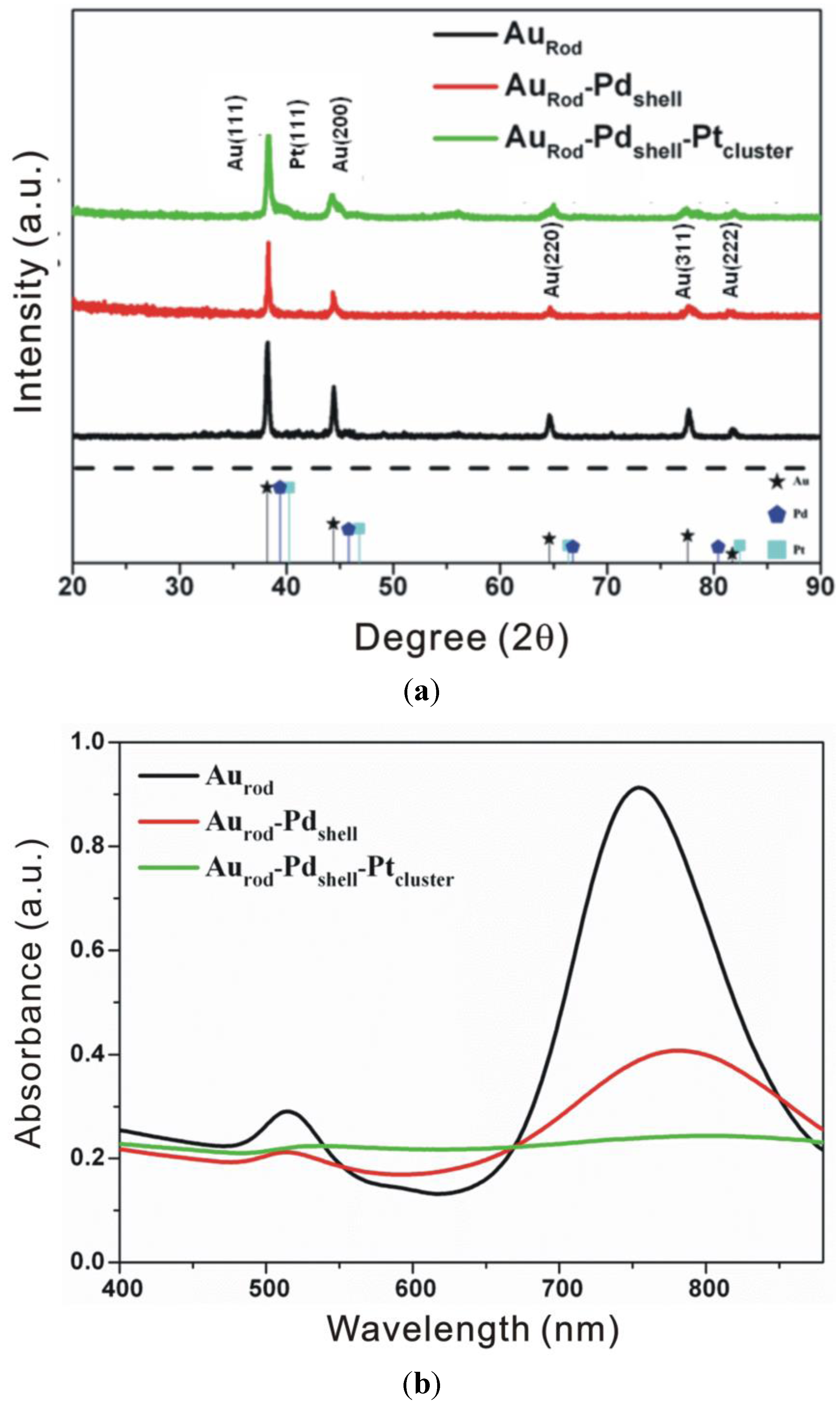
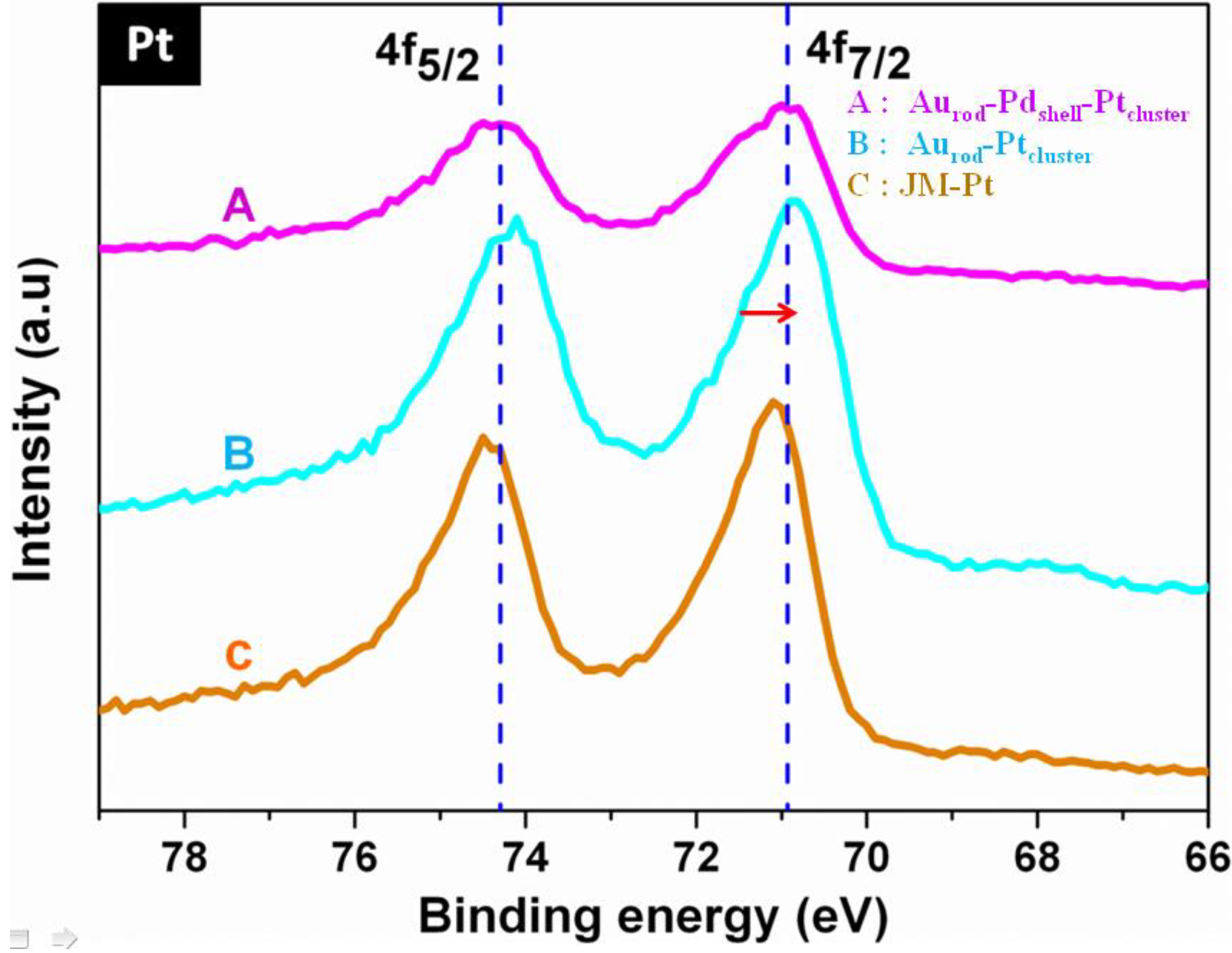
3.1. Characterization


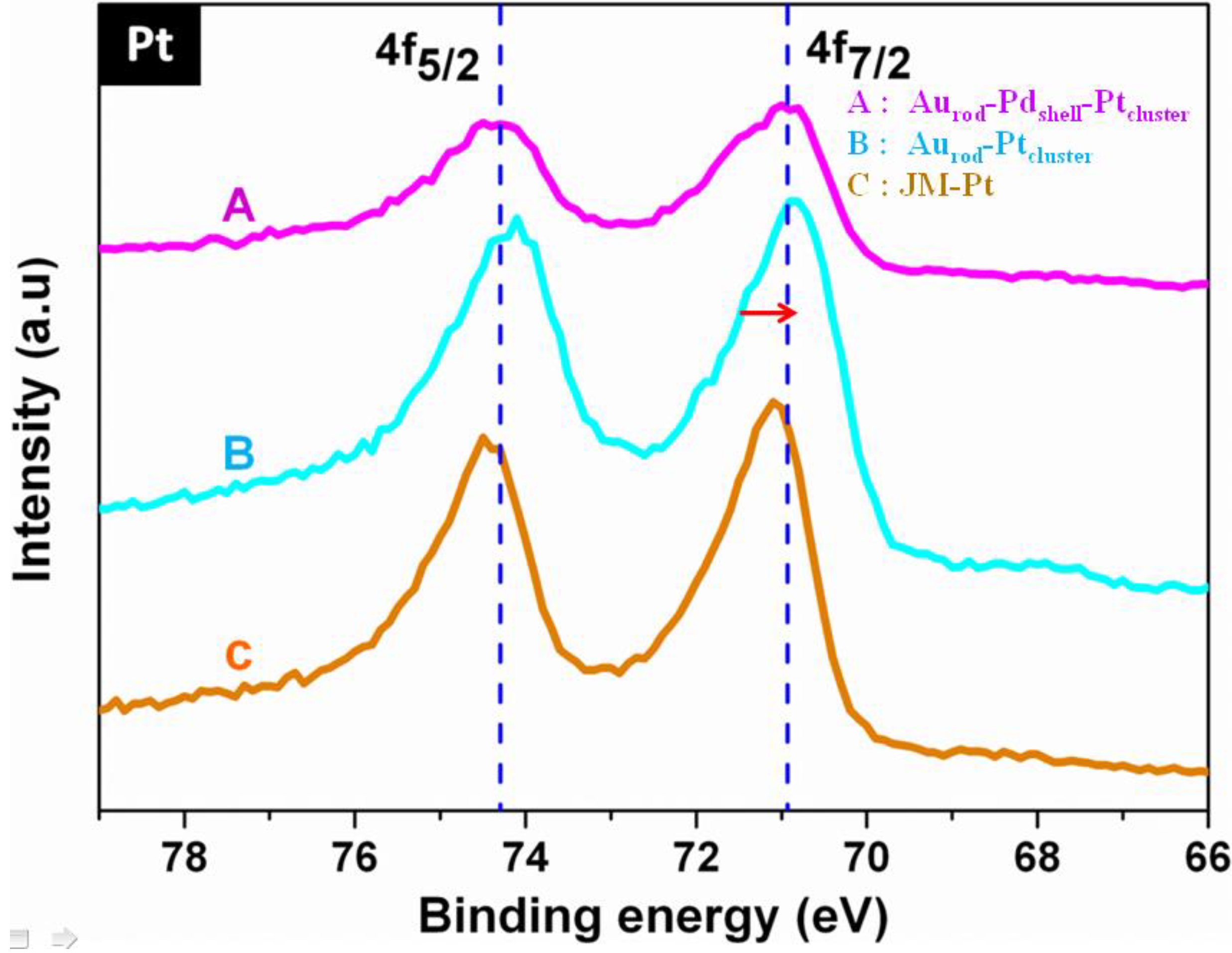
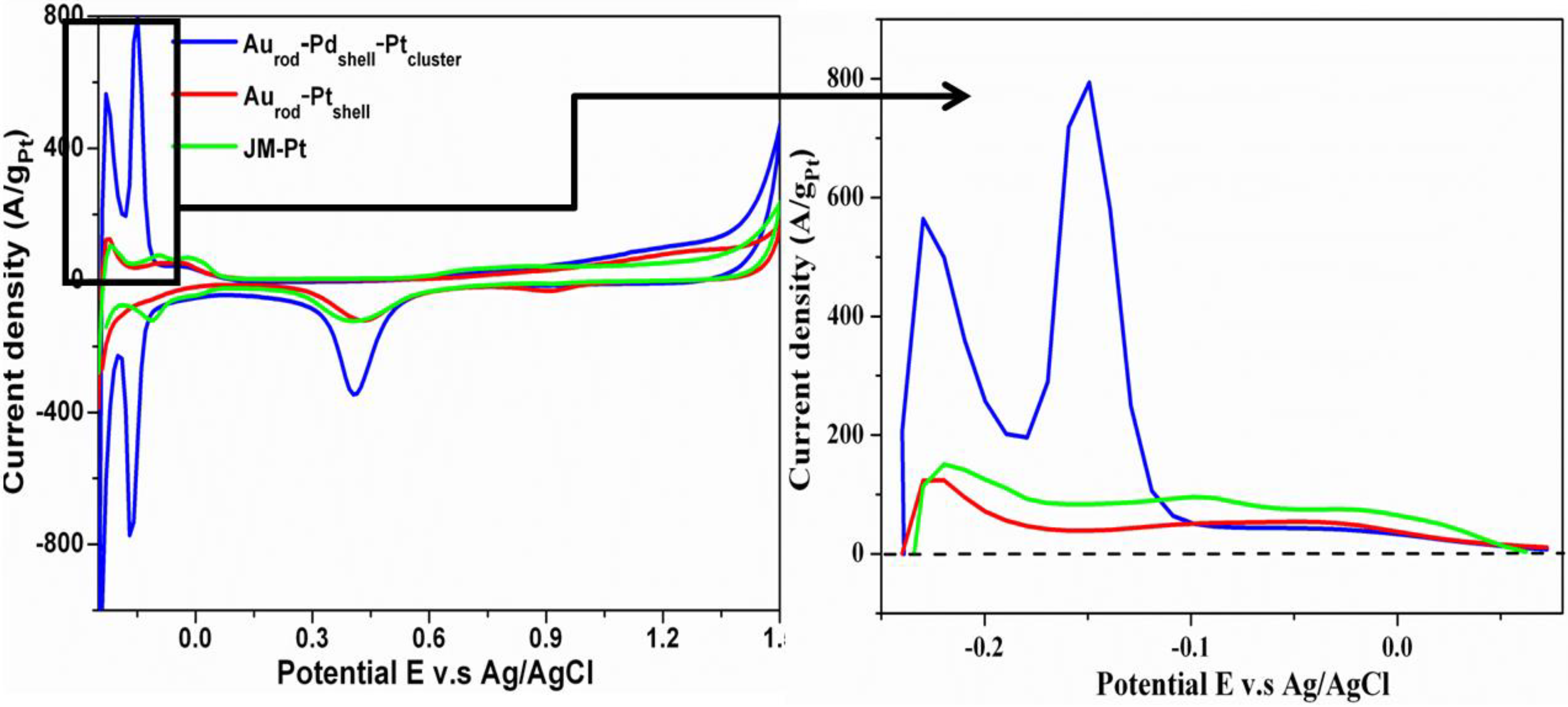
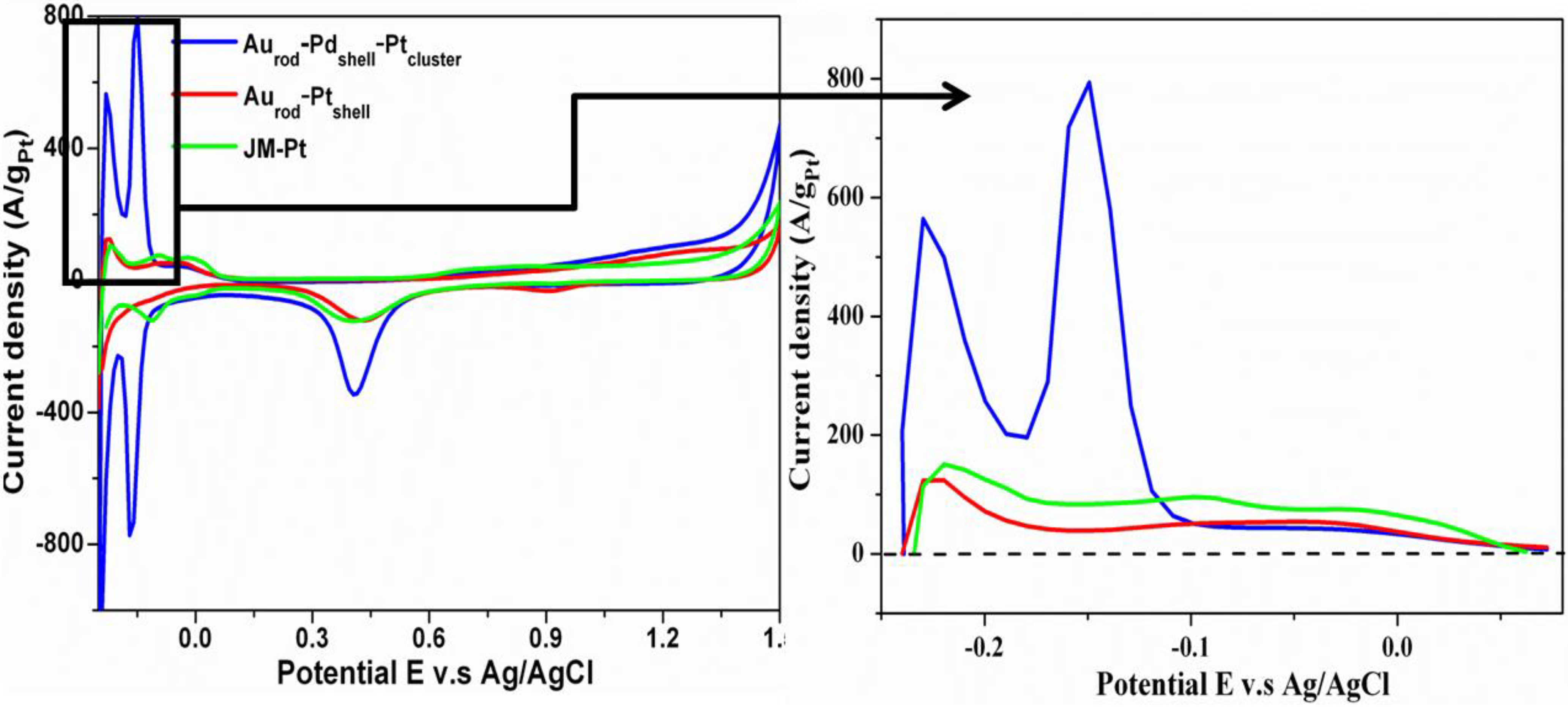
3.2. Cyclic Voltammograms (CVs)
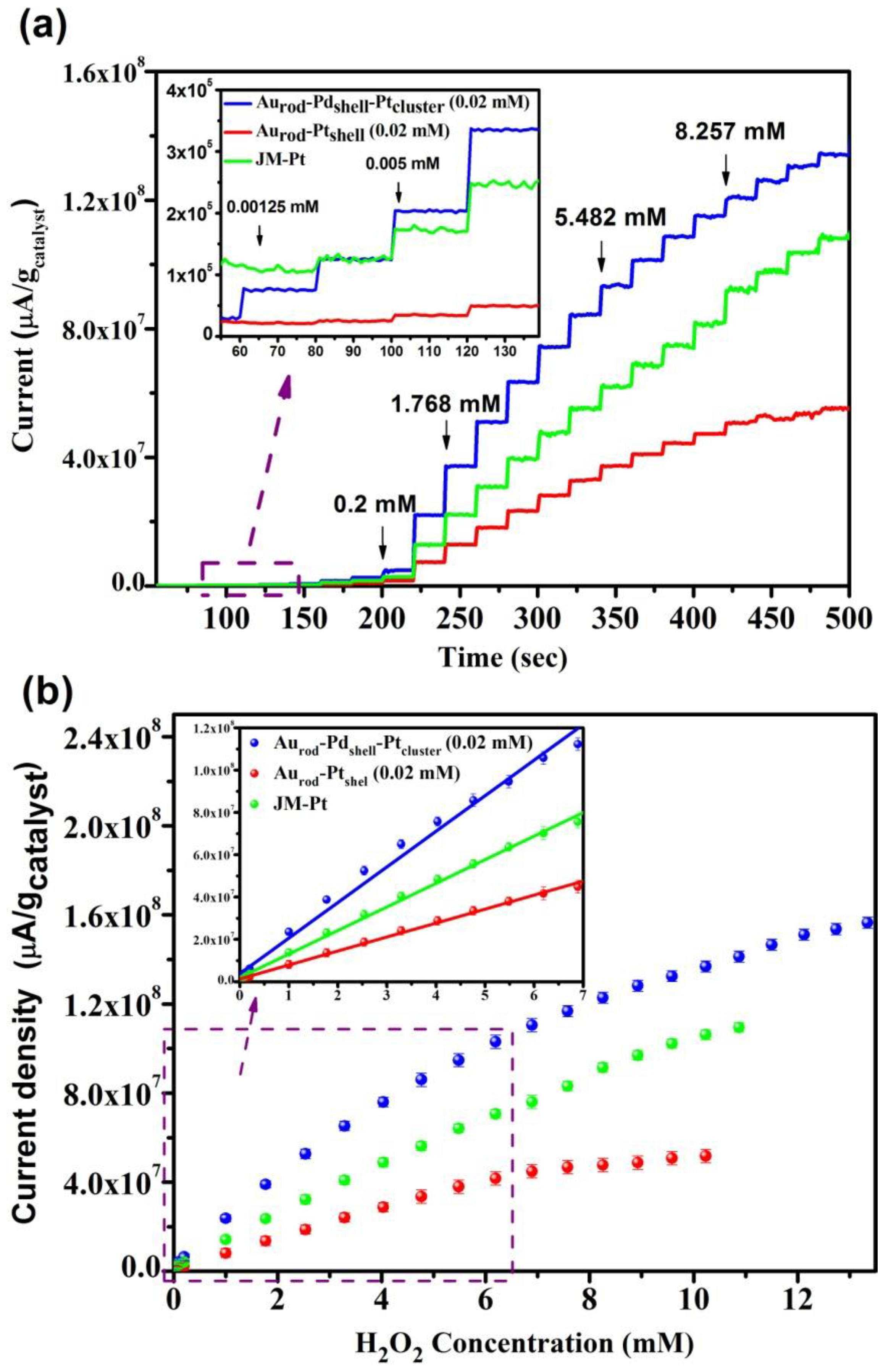
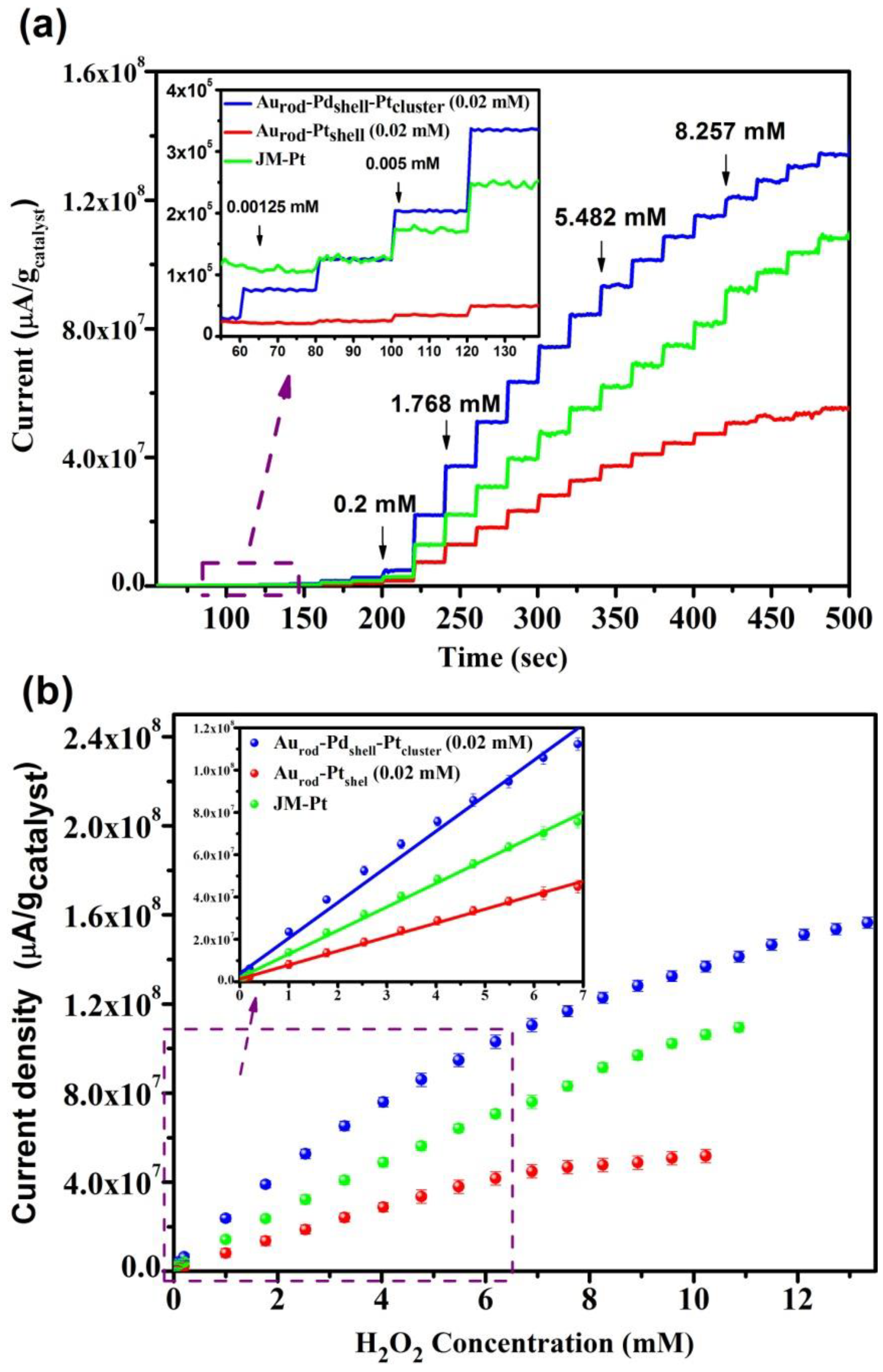
3.3. H2O2 Sensing Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Applied potential (0.4 V) vs. Ag/AgCl | Linear range (µM) | H2O2 sensitivity (µA/µM×gcat) | R2 |
|---|---|---|---|
| Aurod-Pdshell-Ptcluster | 0.00125–6.191 | 17,721 | 0.99364 |
| Aurod-Ptcluster | 0.005–5.482 | 6612 | 0.99765 |
| JM-Pt | 0.005–6.191 | 11,144 | 0.9978 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fang, P.P.; Duan, S.; Lin, X.D.; Anema, J.R.; Li, J.F.; Buriez, O.; Ding, Y.; Fan, F.R.; Wu, D.Y.; Ren, B.; Wang, Z.L.; Amatoreb, C.; Tian, Z.Q. Tailoring Au-core Pd-shell Pt-cluster nanoparticles for enhanced electrocatalytic activity. Chem. Sci. 2011, 2, 531–539. [Google Scholar] [CrossRef]
- Antolini, E. Composite materials: An emerging class of fuel cell catalyst supports. Appl. Catal. B 2010, 100, 413–426. [Google Scholar] [CrossRef]
- Campbell, C.T. Electronic perturbations. Nat. Chem. 2012, 4, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.L.; Lee, C.F.; Rick, J.; Wang, S.H.; Liu, C.C.; Hwang, B.J. Fabrication and application of amperometric glucose biosensor based on a novel PtPd bimetallic nanoparticle decorated multi-walled carbon nanotube catalyst. Biosens. Bioelectron. 2012, 33, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.L.; Pillai, C.; Rick, J.; Pan, C.J.; Wang, S.H.; Liu, C.C.; Hwang, B.J. Bimetallic PtM (M = Pd, Ir) nanoparticle decorated multi-walled carbon nanotube enzyme-free, mediator-less amperometric sensor for H2O2. Biosens. Bioelectron. 2012, 33, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Sarma, L.S.; Wang, G.R.; Chen, J.M.; Shih, S.C.; Tang, M.T.; Liu, D.G.; Lee, J.F.; Chen, J.M.; Hwang, B.J. Formation of bimetallic Ag-Pd nanoclusters via the reaction between Ag nanoclusters and Pd2+ ions. J. Phys. Chem. B 2006, 110, 10287–10295. [Google Scholar] [CrossRef] [PubMed]
- Tompos, A.; Margitfalvia, J.L.; Hegedsa, M.; Szegedia, Á.; Fierrob, J.L.G.; Rojasb, S. Characterization of trimetallic Pt-Pd-Au/CeO2 catalysts combinatorial designed for methane total oxidation. Comb. Chem. High Throughput Screen. 2007, 10, 71–82. [Google Scholar]
- Wu, Y.; Wang, D.; Chen, X.; Zhou, G.; Yu, R.; Li, Y. Defect-dominated shape recovery of nanocrystals: A new strategy for trimetallic catalysts. J. Am. Chem. Soc. 2013, 135, 12220–12223. [Google Scholar] [CrossRef] [PubMed]
- Song, H.M.; Anjum, D.H.; Sougrat, R.; Hedhili, M.N.; Khashab, N.M. Hollow Au@Pd and Au@Pt core-shell nanoparticles as electrocatalysts for ethanol oxidation reactions. J. Mater. Chem. 2012, 22, 25003–25010. [Google Scholar] [CrossRef]
- Zou, Y.; Xiang, C.; Sun, L.X.; Xu, F. Glucose biosensor based on electrodeposition of platinum nanoparticles onto carbon nanotubes and immobilizing enzyme with chitosan-SiO2 sol–gel. Biosens. Bioelectron. 2008, 23, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Debiemme, C. A very thin overoxidized polypyrrole membrane as coating for fast time response and selective H2O2 amperometric sensor. Biosens. Bioelectron. 2010, 25, 2454–2457. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liu, Y.; Su, F.; Liu, A.; Qiu, H. Nanoporous PtAg and PtCu alloys with hollow ligaments for enhanced electrocatalysis and glucose biosensing. Biosens. Bioelectron. 2011, 27, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.J.; Kumar, S.M.S.; Chen, C.H.; Chang, R.W.; Liu, D.G.; Lee, J.F. Size and alloying extent dependent physiochemical properties of Pt-Ag/C nanoparticles synthesized by the ethylene glycol method. J. Phys. Chem. C 2008, 112, 2370–2377. [Google Scholar] [CrossRef]
- Lai, F.J.; Sarma, L.S.; Chou, H.L.; Liu, D.G.; Hsieh, C.A.; Lee, J.F.; Hwang, B.J. Architecture of bimetallic PtxCo1-x electrocatalysts for oxygen reduction reaction as nvestigated by X-ray absorption spectroscopy. J. Phys. Chem. C 2009, 113, 12674–12681. [Google Scholar] [CrossRef]
- Ming, L.; Xi, X.; Liu, J. Electrochemically platinized carbon paste enzyme electrodes: A new design of amperometric glucose biosensors. Biotechnol. Lett. 2006, 28, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Hong, J.W.; Lee, Y.W.; Kim, M.; Kim, D.; Yun, W.S.; Han, S.W. Synthesis of AuPt heteronanostructures with enhanced electrocatalytic activity toward oxygen reduction. Angew. Chem. 2010, 49, 10197–10201. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.-I.; Rick, J.; Pan, C.-J.; Chou, H.-L.; Su, W.-N.; Chen, K.-J.; Liu, C.-C.; Yang, Y.-W.; Wang, C.-H.; Hwang, B.-J. Trimetallic (Aurod-Pdshell-Ptcluster) Catalyst Used as Amperometric Hydrogen Peroxide Sensor. Biosensors 2014, 4, 461-471. https://doi.org/10.3390/bios4040461
Cheng S-I, Rick J, Pan C-J, Chou H-L, Su W-N, Chen K-J, Liu C-C, Yang Y-W, Wang C-H, Hwang B-J. Trimetallic (Aurod-Pdshell-Ptcluster) Catalyst Used as Amperometric Hydrogen Peroxide Sensor. Biosensors. 2014; 4(4):461-471. https://doi.org/10.3390/bios4040461
Chicago/Turabian StyleCheng, Shou-I, John Rick, Chun-Jern Pan, Hung-Lung Chou, Wei-Nien Su, Kuan-Jung Chen, Chung-Chiun Liu, Yaw-Wen Yang, Chia-Hsin Wang, and Bing-Joe Hwang. 2014. "Trimetallic (Aurod-Pdshell-Ptcluster) Catalyst Used as Amperometric Hydrogen Peroxide Sensor" Biosensors 4, no. 4: 461-471. https://doi.org/10.3390/bios4040461