The Power of Old Hats: Rediscovering Inosine-EpPCR to Create Starting Libraries for Whole-Cell-SELEX

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
Cell Pretreatment
2.2. PCR Conditions
2.2.1. Error-Prone PCR
2.2.2. PCR Conditions
2.2.3. PCR Cleanup
2.2.4. Strand Separation
2.2.5. Primers
2.2.6. SELEX Library Control
2.3. Binding Assays
2.4. Next-Generation Sequencing
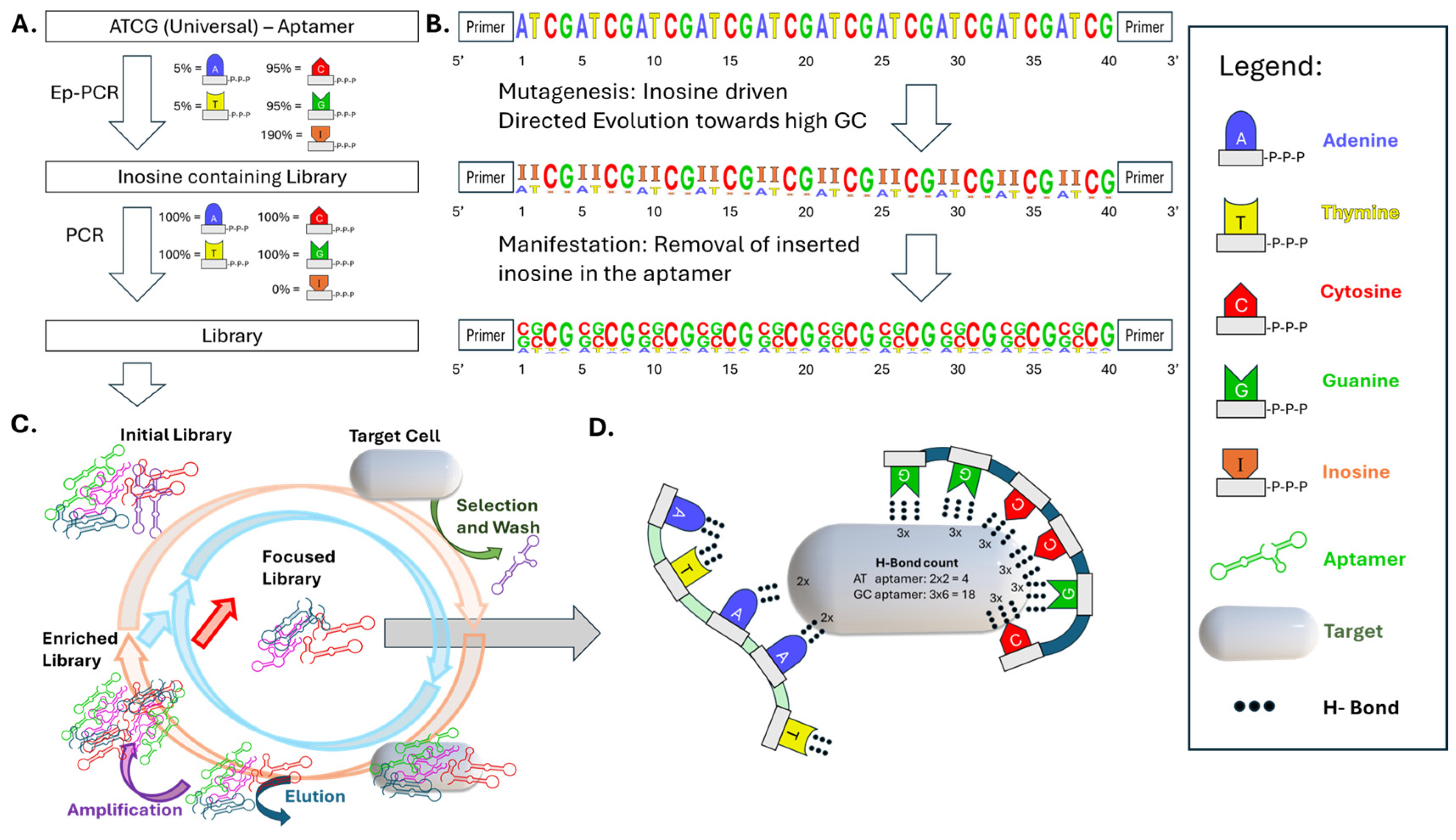
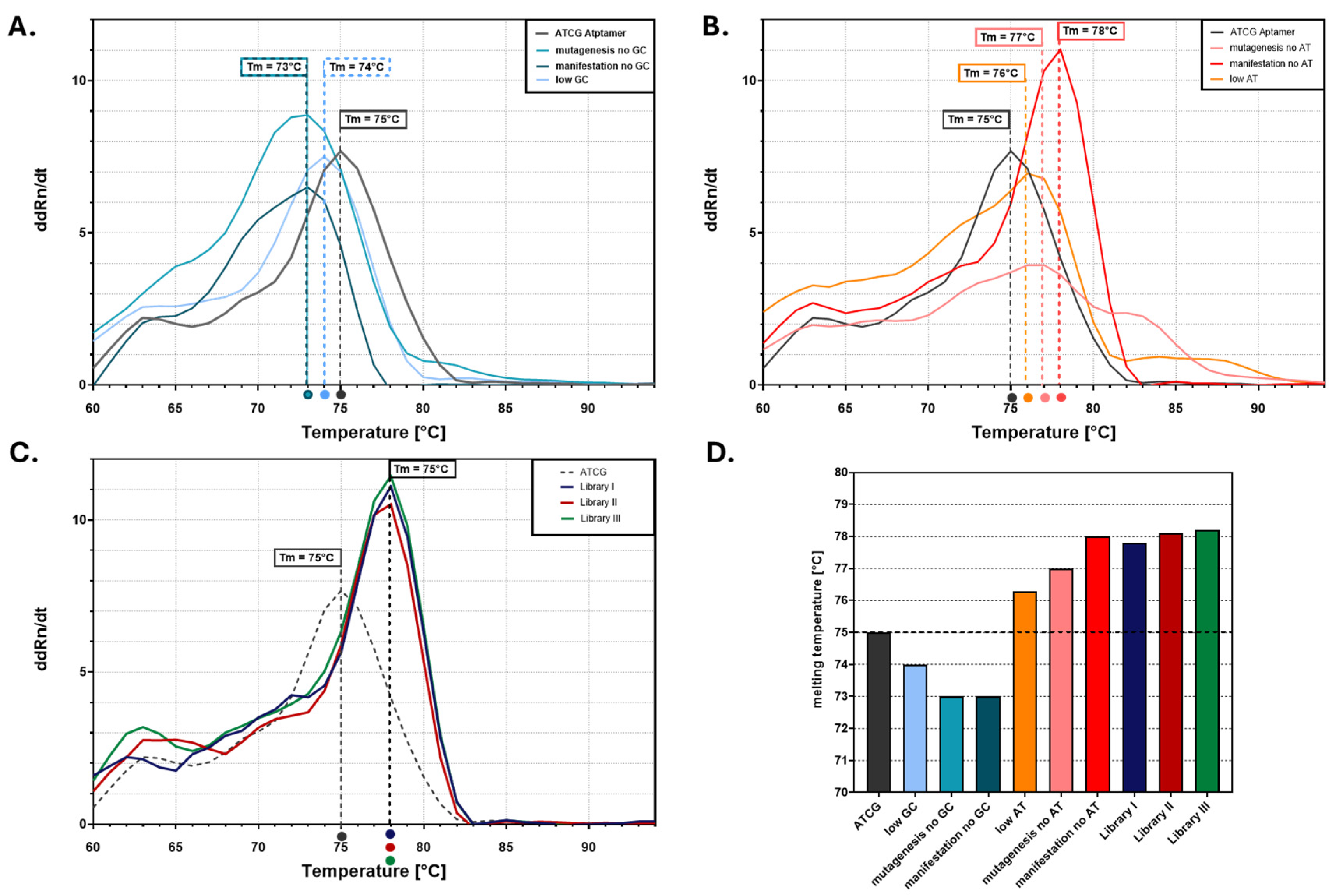
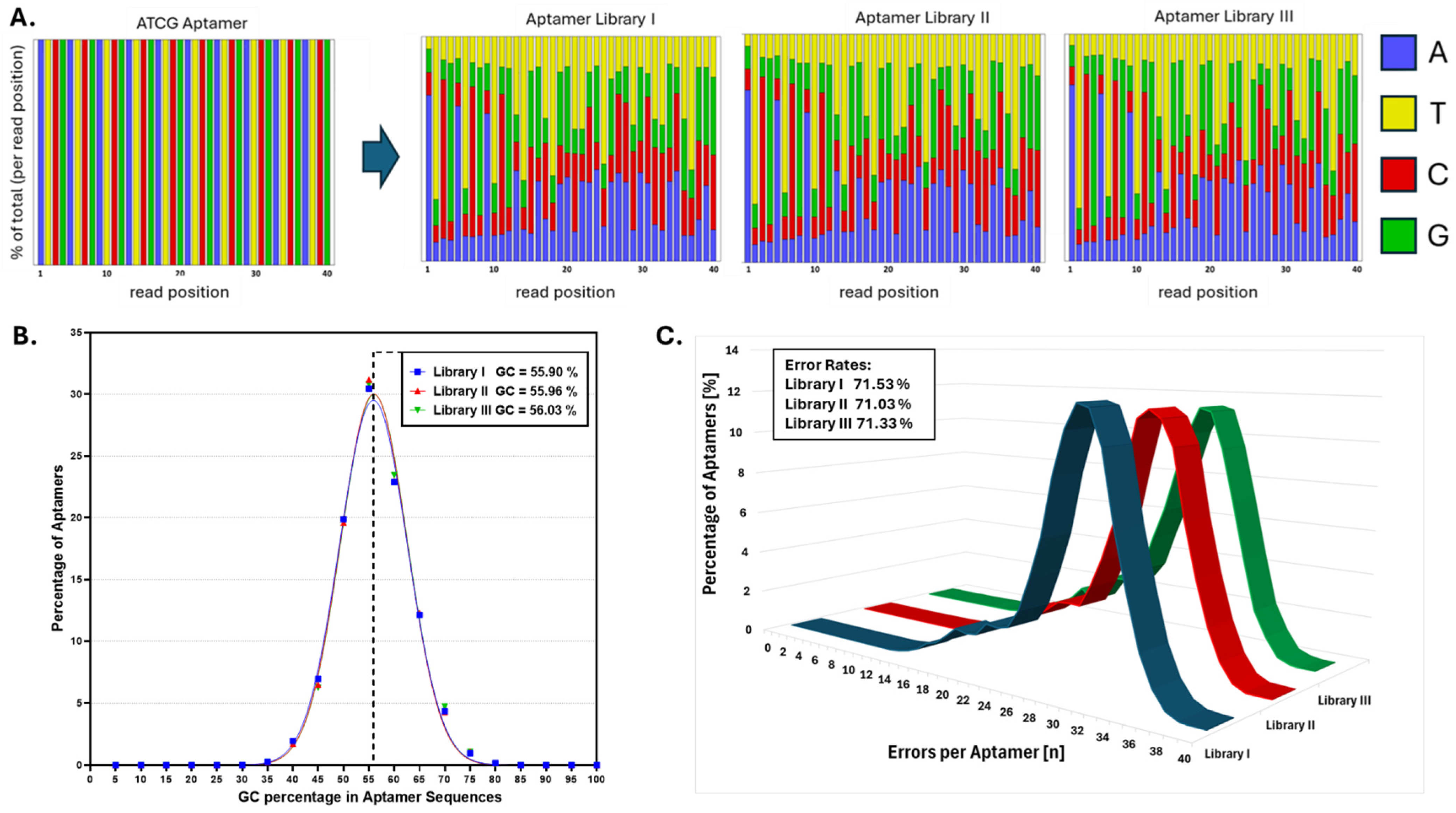
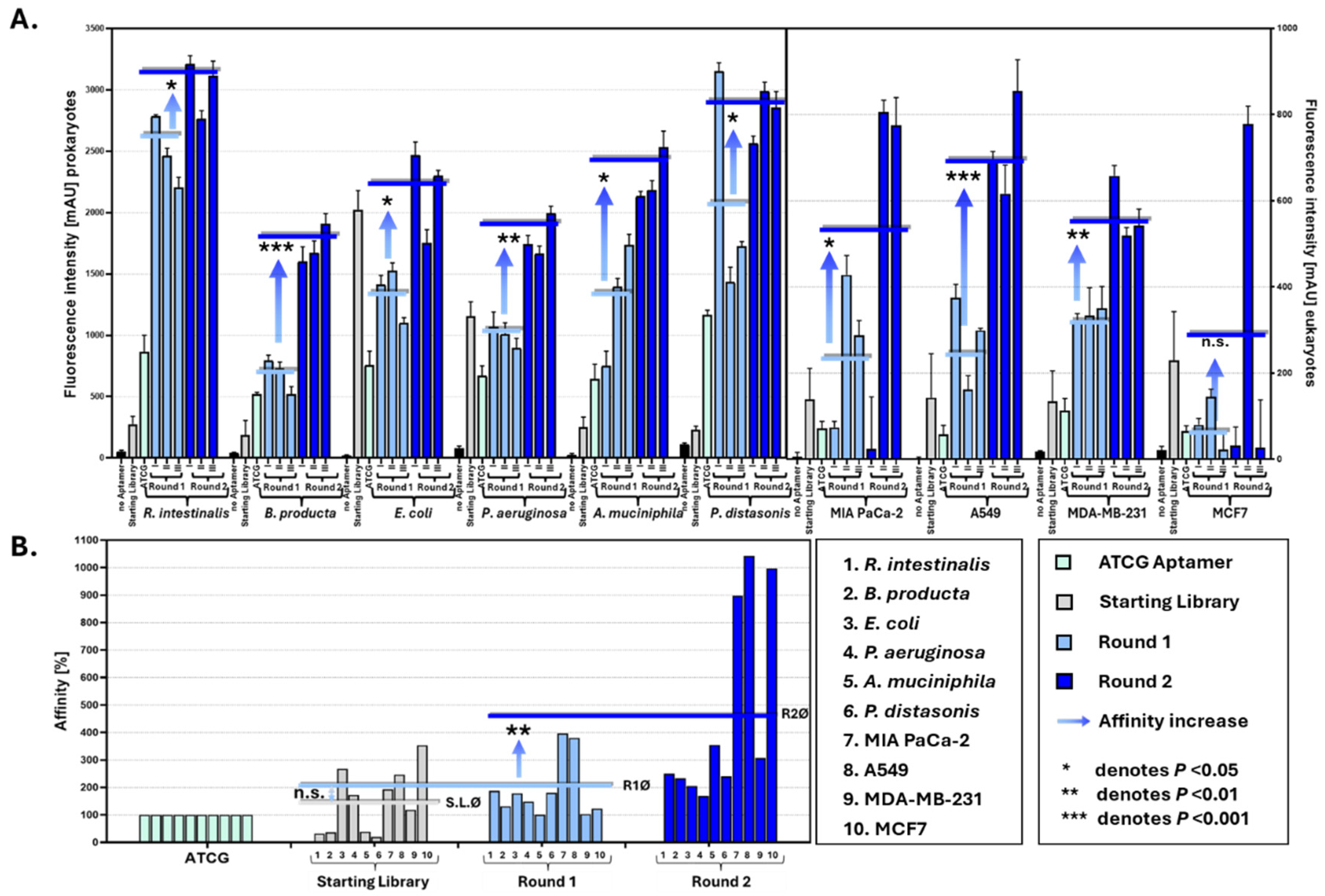
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific Enzymatic Amplification of DNA In Vitro: The Polymerase Chain Reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, R.; Wallace, R.; Volpato, F.; Hu, S.P.; Trent, R.J. Utility of the Polymerase Chain Reaction (PCR) for Prenatal Diagnosis of Genetic Disease. Pathology 1991, 23, 158–163. [Google Scholar] [CrossRef]
- Navidi, W.; Arnheim, N. Using PCR in Preimplantation Genetic Disease Diagnosis. Hum. Reprod. 1991, 6, 836–849. [Google Scholar] [CrossRef]
- Khot, P.D.; Fredricks, D.N. PCR-Based Diagnosis of Human Fungal Infections. Expert. Rev. Anti-Infect. Ther. 2009, 7, 1201–1221. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.B. DNA Probes and PCR for Diagnosis of Parasitic Infections. Clin. Microbiol. Rev. 1995, 8, 113–130. [Google Scholar] [CrossRef]
- Avni, T.; Leibovici, L.; Paul, M. PCR Diagnosis of Invasive Candidiasis: Systematic Review and Meta-Analysis. J. Clin. Microbiol. 2011, 49, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.S.; Wittwer, C.T. Real-Time PCR Technology for Cancer Diagnostics. Clin. Chem. 2002, 48, 1178–1185. [Google Scholar] [CrossRef]
- Elnifro, E.M.; Ashshi, A.M.; Cooper, R.J.; Klapper, P.E. Multiplex PCR: Optimization and Application in Diagnostic Virology. Clin. Microbiol. Rev. 2000, 13, 559–570. [Google Scholar] [CrossRef]
- Alaeddini, R. Forensic Implications of PCR Inhibition—A Review. Forensic Sci. Int. Genet. 2012, 6, 297–305. [Google Scholar] [CrossRef]
- Lipp, M.; Shillito, R.; Giroux, R.; Spiegelhalter, F.; Charlton, S.; Pinero, D.; Song, P. Polymerase Chain Reaction Technology as Analytical Tool in Agricultural Biotechnology. J. AOAC Int. 2005, 88, 136–155. [Google Scholar] [CrossRef]
- Levin, R.E. The Application of Real-Time PCR to Food and Agricultural Systems. A Review. Food Biotechnol. 2004, 18, 97–133. [Google Scholar] [CrossRef]
- Henry, R.; Schang, C.; Chandrasena, G.I.; Deletic, A.; Edmunds, M.; Jovanovic, D.; Kolotelo, P.; Schmidt, J.; Williamson, R.; McCarthy, D. Environmental Monitoring of Waterborne Campylobacter: Evaluation of the Australian Standard and a Hybrid Extraction-Free MPN-PCR Method. Front. Microbiol. 2015, 6, 74. [Google Scholar] [CrossRef]
- Santo Domingo, J.W.; Bambic, D.G.; Edge, T.A.; Wuertz, S. Quo Vadis Source Tracking? Towards a Strategic Framework for Environmental Monitoring of Fecal Pollution. Water Res. 2007, 41, 3539–3552. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, F.P.; Courtenay, O.; Hibberd, V.; Hewinson, R.G.; Reilly, L.A.; Gaze, W.H.; Wellington, E.M.H. Environmental Monitoring of Mycobacterium Bovis in Badger Feces and Badger Sett Soil by Real-Time PCR, as Confirmed by Immunofluorescence, Immunocapture, and Cultivation. Appl. Environ. Microbiol. 2007, 73, 7471–7473. [Google Scholar] [CrossRef]
- Mordor Intelligence. Polymerase Chain Reaction (PCR) Market-Growth, Trends, COVID-19 Impact, and Forecasts (2024–2030); Mordor Intelligence: Telangana, India, 2024. [Google Scholar]
- Eckert, K.A.; Kunkel, T.A. DNA Polymerase Fidelity and the Polymerase Chain Reaction. Genome Res. 1991, 1, 17–24. [Google Scholar] [CrossRef]
- Rattray, A.J.; Strathern, J.N. Error-Prone DNA Polymerases: When Making a Mistake Is the Only Way to Get Ahead1. Annu. Rev. Genet. 2003, 37, 31–66. [Google Scholar] [CrossRef]
- Cadwell, R.C.; Joyce, G.F. Randomization of Genes by PCR Mutagenesis. PCR Methods Appl. 1992, 2, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Spee, J.H.; de Vos, W.M.; Kuipers, O.P. Efficient Random Mutagenesis Method with Adjustable Mutation Frequency by Use of PCR and DITP. Nucleic Acids Res. 1993, 21, 777–778. [Google Scholar] [CrossRef]
- Chen, K.; Arnold, F.H. Tuning the Activity of an Enzyme for Unusual Environments: Sequential Random Mutagenesis of Subtilisin E for Catalysis in Dimethylformamide. Proc. Natl. Acad. Sci. USA 1993, 90, 5618–5622. [Google Scholar] [CrossRef]
- Stemmer, W.P.C. Rapid Evolution of a Protein in Vitro by DNA Shuffling. Nature 1994, 370, 389–391. [Google Scholar] [CrossRef]
- Chapman, K.B.; Szostak, J.W. In Vitro Selection of Catalytic RNAs. Curr. Opin. Struct. Biol. 1994, 4, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Ostermeier, M.; Nixon, A.E.; Shim, J.H.; Benkovic, S.J. Combinatorial Protein Engineering by Incremental Truncation. Proc. Natl. Acad. Sci. USA 1999, 96, 3562–3567. [Google Scholar] [CrossRef]
- Liebeton, K.; Zonta, A.; Schimossek, K.; Nardini, M.; Lang, D.; Dijkstra, B.W.; Reetz, M.T.; Jaeger, K.E. Directed Evolution of an Enantioselective Lipase. Chem. Biol. 2000, 7, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.S.; Tee, K.L.; Hauer, B.; Schwaneberg, U. Sequence Saturation Mutagenesis (SeSaM): A Novel Method for Directed Evolution. Nucleic Acids Res. 2004, 32, e26. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as Targeted Therapeutics: Current Potential and Challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef]
- Bozza, M.; Sheardy, R.D.; Dilone, E.; Scypinski, S.; Galazka, M. Characterization of the Secondary Structure and Stability of an RNA Aptamer That Binds Vascular Endothelial Growth Factor. Biochemistry 2006, 45, 7639–7643. [Google Scholar] [CrossRef]
- Chopra, A.; Shukla, R.; Sharma, T. Aptamers as an Emerging Player in Biology. Aptamers Synth. Antibodies 2014, 1, 1–11. [Google Scholar]
- Ku, T.-H.; Zhang, T.; Luo, H.; Yen, T.M.; Chen, P.-W.; Han, Y.; Lo, Y.-H. Nucleic Acid Aptamers: An Emerging Tool for Biotechnology and Biomedical Sensing. Sensors 2015, 15, 16281–16313. [Google Scholar] [CrossRef]
- Kratschmer, C.; Levy, M. Effect of Chemical Modifications on Aptamer Stability in Serum. Nucleic Acid. Ther. 2017, 27, 335–344. [Google Scholar] [CrossRef]
- Brown, A.; Brill, J.; Amini, R.; Nurmi, C.; Li, Y. Development of Better Aptamers: Structured Library Approaches, Selection Methods, and Chemical Modifications. Angew. Chem. Int. Ed. Engl. 2024, 63, e202318665. [Google Scholar] [CrossRef]
- Vorobyeva, M.A.; Davydova, A.S.; Vorobjev, P.E.; Venyaminova, A.G. Key Aspects of Nucleic Acid Library Design for in Vitro Selection. Int. J. Mol. Sci. 2018, 19, 470. [Google Scholar] [CrossRef] [PubMed]
- Kneißle, K.; Krämer, M.; Kissmann, A.-K.; Xing, H.; Müller, F.; Amann, V.; Noschka, R.; Gottschalk, K.-E.; Bozdogan, A.; Andersson, J.; et al. A Polyclonal SELEX Aptamer Library Allows Differentiation of Candida albicans, C. auris and C. parapsilosis Cells from Human Dermal Fibroblasts. J. Fungi 2022, 8, 856. [Google Scholar] [CrossRef]
- Kissmann, A.-K.; Bolotnikov, G.; Li, R.; Müller, F.; Xing, H.; Krämer, M.; Gottschalk, K.-E.; Andersson, J.; Weil, T.; Rosenau, F. IMPATIENT-QPCR: Monitoring SELEX Success during in Vitro Aptamer Evolution. Appl. Microbiol. Biotechnol. 2024, 108, 284. [Google Scholar] [CrossRef]
- Raber, H.F.; Kubiczek, D.H.; Bodenberger, N.; Kissmann, A.-K.; D’souza, D.; Xing, H.; Mayer, D.; Xu, P.; Knippschild, U.; Spellerberg, B.; et al. FluCell-SELEX Aptamers as Specific Binding Molecules for Diagnostics of the Health Relevant Gut Bacterium Akkermansia Muciniphila. Int. J. Mol. Sci. 2021, 22, 10425. [Google Scholar] [CrossRef]
- Kissmann, A.-K.; Wolf, D.; Krämer, M.; Müller, F.; Amann, V.; Xing, H.; Gottschalk, K.-E.; Weil, T.; Eichmann, R.; Schäfer, P.; et al. Polyclonal Aptamer Libraries from a FluRoot-SELEX for the Specific Labeling of the Apical and Elongation/Differentiation Zones of Arabidopsis Thaliana Roots. Int. J. Mol. Sci. 2022, 23, 12220. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Zhang, Y.; Krämer, M.; Kissmann, A.-K.; Henkel, M.; Weil, T.; Knippschild, U.; Rosenau, F. A Polyclonal Selex Aptamer Library Directly Allows Specific Labelling of the Human Gut Bacterium Blautia Producta without Isolating Individual Aptamers. Molecules 2022, 27, 5693. [Google Scholar] [CrossRef]
- Kubiczek, D.; Raber, H.; Bodenberger, N.; Oswald, T.; Sahan, M.; Mayer, D.; Wiese, S.; Stenger, S.; Weil, T.; Rosenau, F. The Diversity of a Polyclonal FluCell-SELEX Library Outperforms Individual Aptamers as Emerging Diagnostic Tools for the Identification of Carbapenem Resistant Pseudomonas Aeruginosa. Chemistry 2020, 26, 14536–14545. [Google Scholar] [CrossRef]
- Zhang, Y.; Xing, H.; Bolotnikov, G.; Krämer, M.; Gotzmann, N.; Knippschild, U.; Kissmann, A.-K.; Rosenau, F. Enriched Aptamer Libraries in Fluorescence-Based Assays for Rikenella Microfusus-Specific Gut Microbiome Analyses. Microorganisms 2023, 11, 2266. [Google Scholar] [CrossRef]
- Xing, H.; Kissmann, A.-K.; Raber, H.F.; Krämer, M.; Amann, V.; Kohn, K.; Weil, T.; Rosenau, F. Polyclonal Aptamers for Specific Fluorescence Labeling and Quantification of the Health Relevant Human Gut Bacterium Parabacteroides Distasonis. Microorganisms 2021, 9, 2284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xing, H.; Bolotnikov, G.; Krämer, M.; Bozdogan, A.; Kissmann, A.-K.; Weil, T.; Spellerberg, B.; Stenger, S.; Rosenau, F. Robust Fluorometric Aptamer Assay for Direct and Rapid Detection of Clinical Isolates of Candida Spec. Int. J. Mol. Sci. 2024, 25, 3444. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Zhang, Y.; Krämer, M.; Kissmann, A.-K.; Amann, V.; Raber, H.F.; Weil, T.; Stieger, K.R.; Knippschild, U.; Henkel, M.; et al. A Polyclonal Aptamer Library for the Specific Binding of the Gut Bacterium Roseburia Intestinalis in Mixtures with Other Gut Microbiome Bacteria and Human Stool Samples. Int. J. Mol. Sci. 2022, 23, 7744. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Y.; Reddy, K.; Vangaveti, S.; Sheng, J. Inosine-Induced Base Pairing Diversity during Reverse Transcription. ACS Chem. Biol. 2024, 19, 348–356. [Google Scholar] [CrossRef]
- Kohlberger, M.; Gadermaier, G. SELEX: Critical Factors and Optimization Strategies for Successful Aptamer Selection. Biotechnol. Appl. Biochem. 2022, 69, 1771–1792. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Ning, Z.; Duan, M.; Lin, X.; Duan, N.; Wang, Z.; Wu, S. Strategies for Improving Aptamer Screening Efficiency: Library Design, Awareness Innovation, and Instrument Assistance. Crit. Rev. Anal. Chem. 2024, 47, 1–21. [Google Scholar] [CrossRef]
- Komarova, N.; Kuznetsov, A. Inside the Black Box: What Makes SELEX Better? Molecules 2019, 24, 3598. [Google Scholar] [CrossRef]
- Kinghorn, A.B.; Fraser, L.A.; Lang, S.; Shiu, S.C.-C.; Tanner, J.A. Aptamer Bioinformatics. Int. J. Mol. Sci. 2017, 18, 2516. [Google Scholar] [CrossRef]
- Alam, K.K.; Chang, J.L.; Burke, D.H. FASTAptamer: A Bioinformatic Toolkit for High-Throughput Sequence Analysis of Combinatorial Selections. Mol. Ther. Nucleic Acids 2015, 4, e230. [Google Scholar] [CrossRef]
- Kramer, S.T.; Gruenke, P.R.; Alam, K.K.; Xu, D.; Burke, D.H. FASTAptameR 2.0: A Web Tool for Combinatorial Sequence Selections. Mol. Ther. Nucleic Acids 2022, 29, 862–870. [Google Scholar] [CrossRef]
- Xing, H.; Zhang, Y.; Li, R.; Ruzicka, H.-M.; Hain, C.; Andersson, J.; Bozdogan, A.; Henkel, M.; Knippschild, U.; Hasler, R.; et al. A Blautia Producta Specific GFET-Based Aptasensor for Quantitative Monitoring of Microbiome Quality. Nanoscale Horiz. 2024, 10, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xing, H.; Li, R.; Andersson, J.; Bozdogan, A.; Strassl, R.; Draphoen, B.; Lindén, M.; Henkel, M.; Knippschild, U.; et al. Specific GFET-Based Aptasensors for Monitoring of Microbiome Quality: Quantification of the Enteric Health-Relevant Bacterium Roseburia Intestinalis. Adv. Healthc. Mater. 2025, 14, e2403827. [Google Scholar] [CrossRef] [PubMed]
- Kissmann, A.-K.; Andersson, J.; Bozdogan, A.; Amann, V.; Krämer, M.; Xing, H.; Raber, H.F.; Kubiczek, D.H.; Aspermair, P.; Knoll, W.; et al. Polyclonal Aptamer Libraries as Binding Entities on a Graphene FET Based Biosensor for the Discrimination of Apo- and Holo-Retinol Binding Protein 4. Nanoscale Horiz. 2022, 7, 770–778. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolotnikov, G.; Kissmann, A.-K.; Gruber, D.; Bellmann, A.; Hasler, R.; Kleber, C.; Knoll, W.; Rosenau, F. The Power of Old Hats: Rediscovering Inosine-EpPCR to Create Starting Libraries for Whole-Cell-SELEX. Biosensors 2025, 15, 448. https://doi.org/10.3390/bios15070448
Bolotnikov G, Kissmann A-K, Gruber D, Bellmann A, Hasler R, Kleber C, Knoll W, Rosenau F. The Power of Old Hats: Rediscovering Inosine-EpPCR to Create Starting Libraries for Whole-Cell-SELEX. Biosensors. 2025; 15(7):448. https://doi.org/10.3390/bios15070448
Chicago/Turabian StyleBolotnikov, Grigory, Ann-Kathrin Kissmann, Daniel Gruber, Andreas Bellmann, Roger Hasler, Christoph Kleber, Wolfgang Knoll, and Frank Rosenau. 2025. "The Power of Old Hats: Rediscovering Inosine-EpPCR to Create Starting Libraries for Whole-Cell-SELEX" Biosensors 15, no. 7: 448. https://doi.org/10.3390/bios15070448
APA StyleBolotnikov, G., Kissmann, A.-K., Gruber, D., Bellmann, A., Hasler, R., Kleber, C., Knoll, W., & Rosenau, F. (2025). The Power of Old Hats: Rediscovering Inosine-EpPCR to Create Starting Libraries for Whole-Cell-SELEX. Biosensors, 15(7), 448. https://doi.org/10.3390/bios15070448