A Novel Single-Layer Microfluidic Device for Dynamic Stimulation, Culture, and Imaging of Mammalian Cells

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
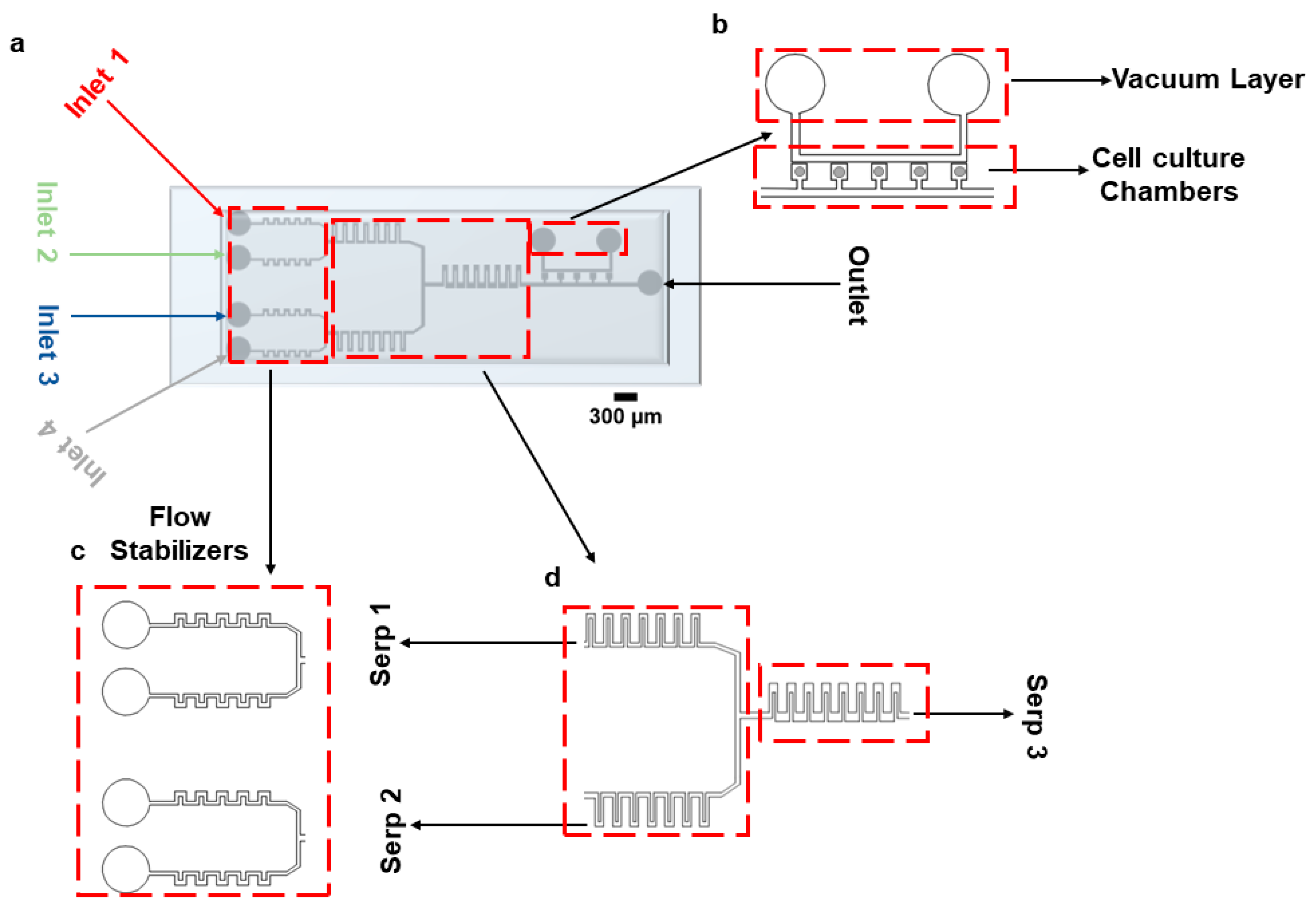
2.1. Device Design
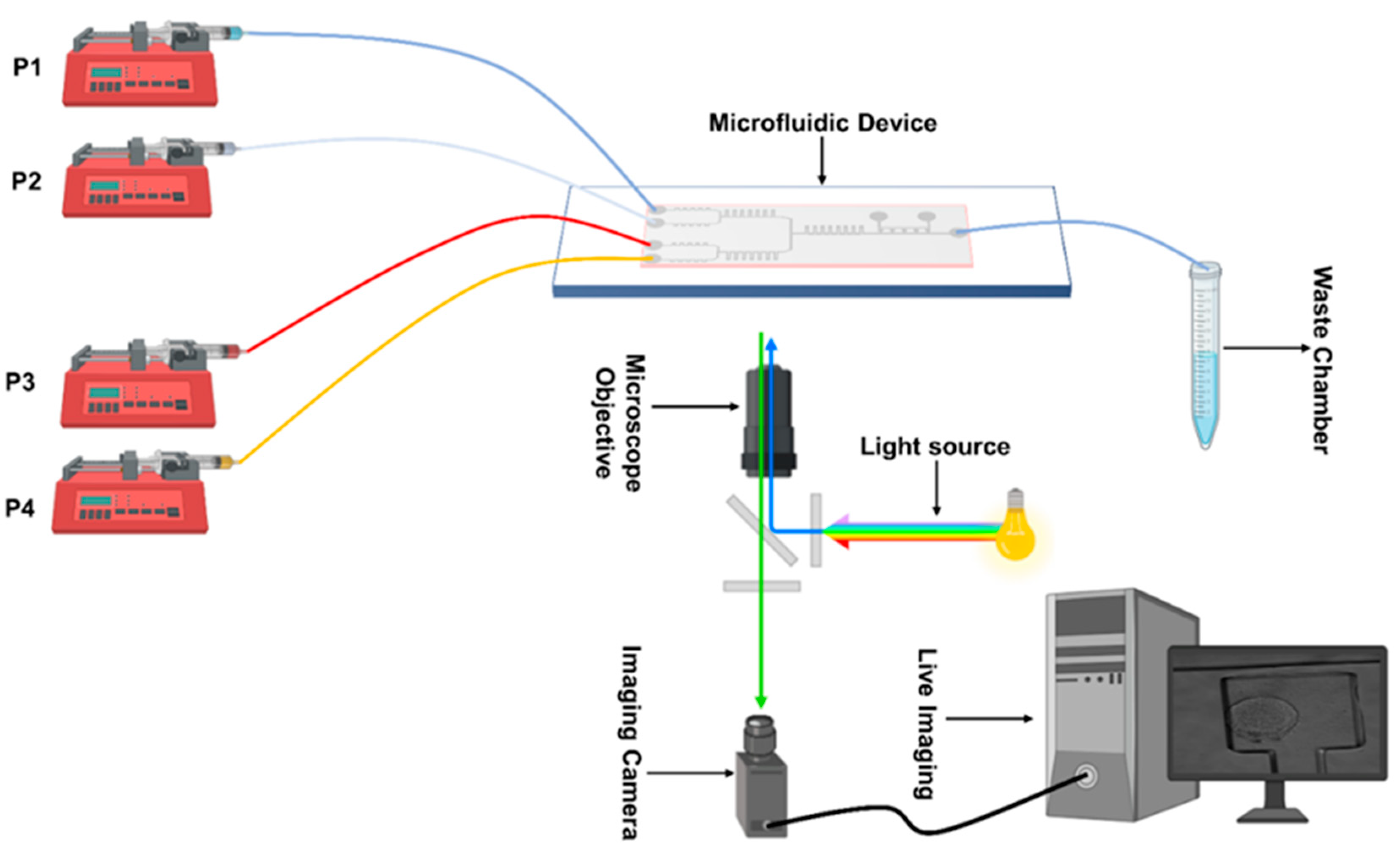
2.2. Mixing Experiments and Imaging
2.3. Diffusion and Flow Modeling
2.4. Cell Culture and Device Loading
2.5. Cell Culture and Device Loading for the Open Loop Experiment
3. Results
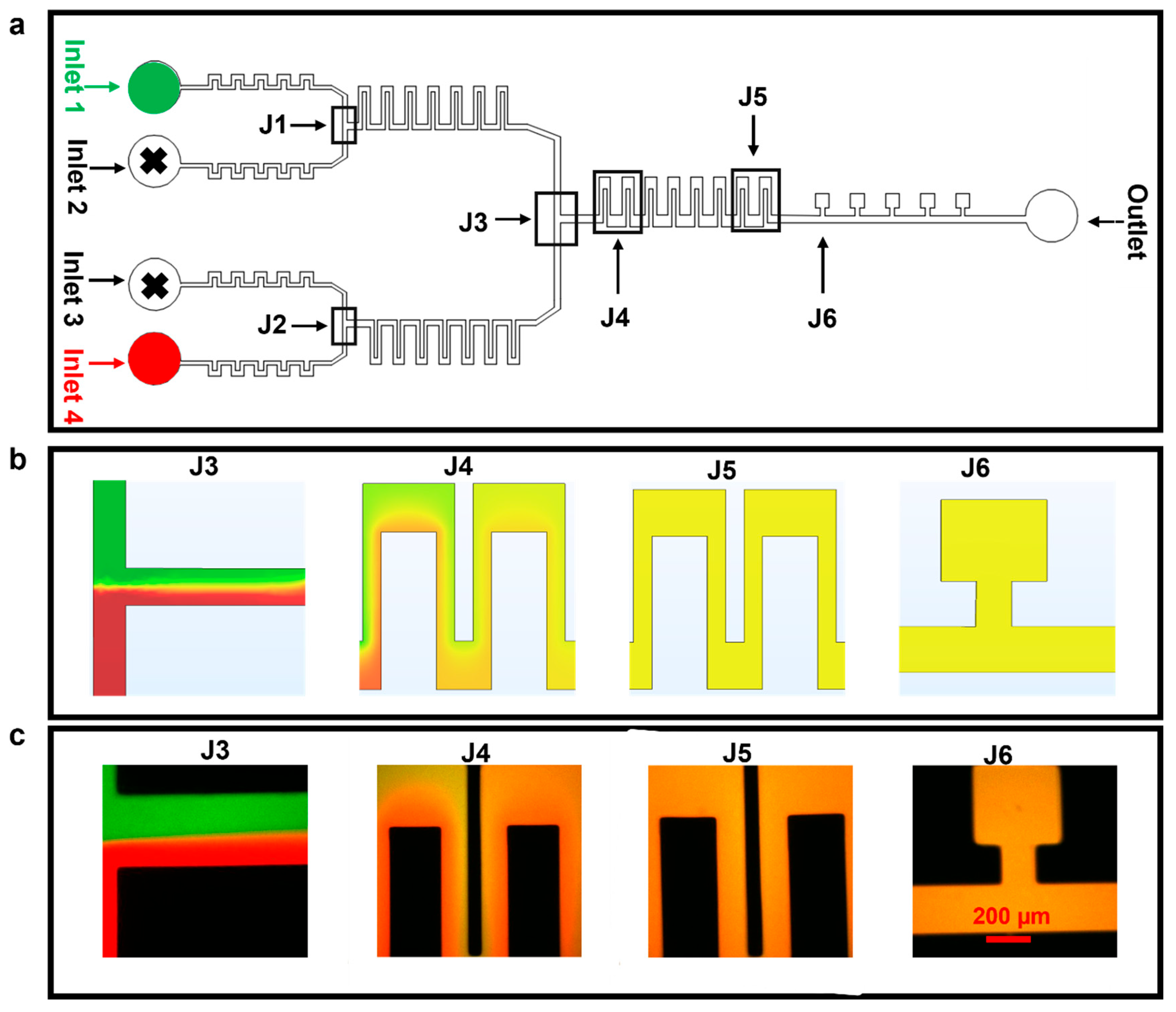
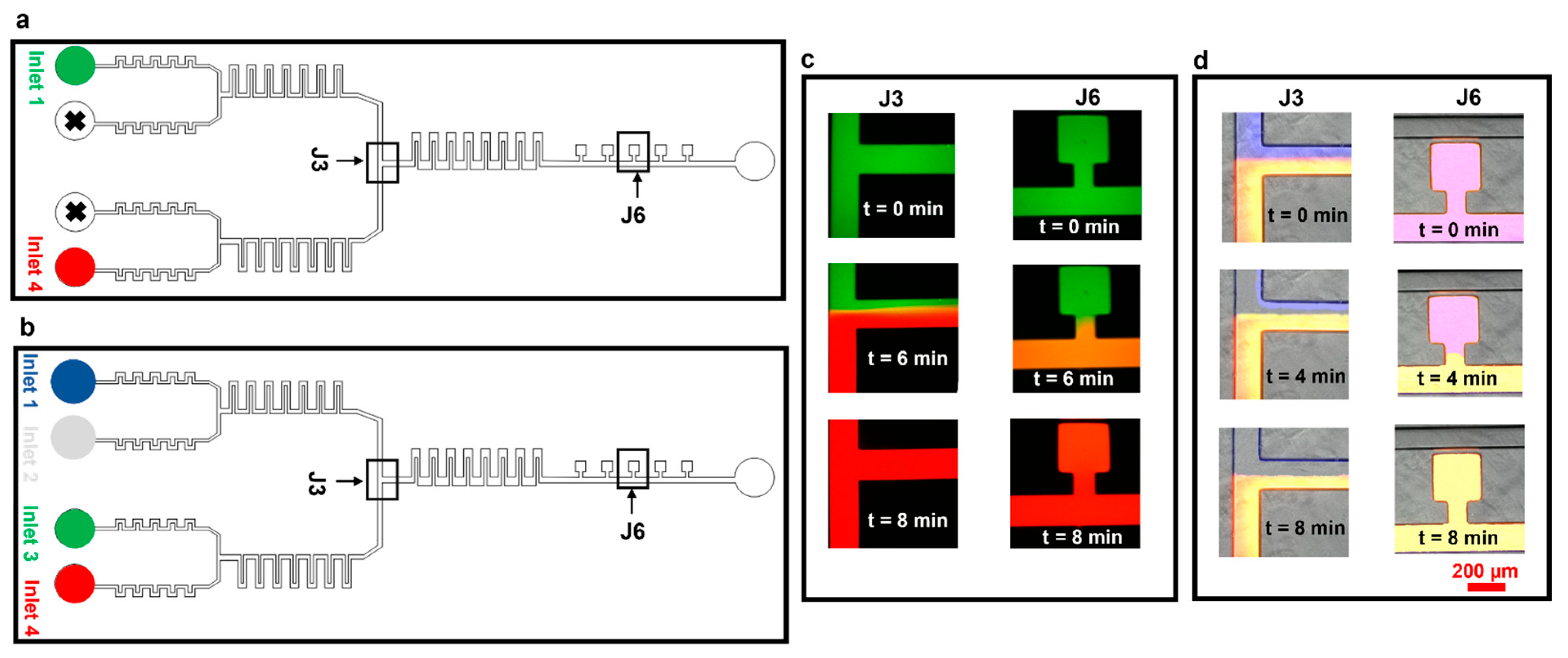
3.1. Diffusion Mixing Experiments
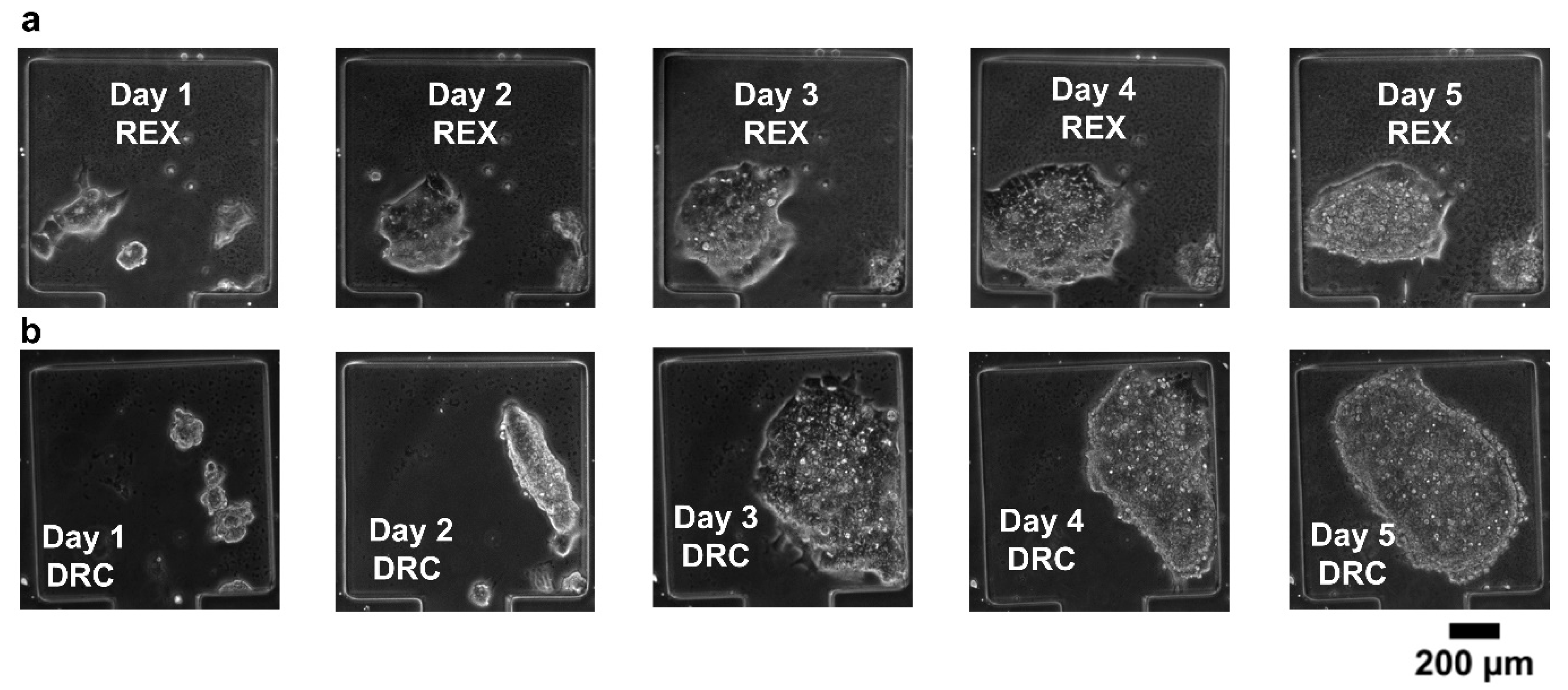
3.2. Cell Viability Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torino, S.; Corrado, B.; Iodice, M.; Coppola, G. PDMS-Based Microfluidic Devices for Cell Culture. Inventions 2018, 3, 65. [Google Scholar] [CrossRef]
- Bergmann, S.; Steinert, M. From Single Cells to Engineered and Explanted Tissues: New Perspectives in Bacterial Infection Biology. Int. Rev. Cell Mol. Biol. 2015, 319, 1–44. [Google Scholar]
- Ruppen, J.; Cortes-Dericks, L.; Marconi, E.; Karoubi, G.; Schmid, R.A.; Peng, R.; Marti, T.M.; Guenat, O.T. A microfluidic platform for chemoresistive testing of multicellular pleural cancer spheroids. Lab Chip 2014, 14, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Mehling, M.; Tay, S. Microfluidic cell culture. Curr. Opin. Biotechnol. 2014, 25, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Lenshof, A.; Laurell, T. Continuous separation of cells and particles in microfluidic systems. Chem. Soc. Rev. 2010, 39, 1203–1217. [Google Scholar] [CrossRef]
- Rothbauer, M.; Zirath, H.; Ertl, P. Recent advances in microfluidic technologies for cell-to-cell interaction studies. Lab Chip 2018, 18, 249–270. [Google Scholar] [CrossRef]
- Halldorsson, S.; Lucumi, E.; Gomez-Sjoberg, R.; Fleming, R.M.T. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens. Bioelectron. 2015, 63, 218–231. [Google Scholar] [CrossRef]
- Matsuura, K.; Hashioka, S.; Takata, K. Sorting differentiated mammalian cells using deterministic lateral displacement microfluidic devices. Anal. Sci. 2024, 40, 1801–1807. [Google Scholar] [CrossRef]
- Ward, K.; Fan, Z.H. Mixing in microfluidic devices and enhancement methods. J. Micromech. Microeng. 2015, 25, 094001. [Google Scholar] [CrossRef]
- Broeren, S.; Pereira, I.F.; Wang, T.; den Toonder, J.; Wang, Y. On-demand microfluidic mixing by actuating integrated magnetic microwalls. Lab Chip 2023, 23, 1524–1530. [Google Scholar] [CrossRef]
- Chatani, T.; Shiraishi, S.; Miyazako, H.; Onoe, H.; Hori, Y. L-2L ladder digital-to-analogue converter for dynamics generation of chemical concentrations. R. Soc. Open Sci. 2023, 10, 230085. [Google Scholar] [CrossRef] [PubMed]
- Schuster, B.; Junkin, M.; Kashaf, S.S.; Romero-Calvo, I.; Kirby, K.; Matthews, J.; Weber, C.R.; Rzhetsky, A.; White, K.P.; Tay, S. Automated microfluidic platform for dynamic and combinatorial drug screening of tumor organoids. Nat. Commun. 2020, 11, 5271. [Google Scholar] [CrossRef] [PubMed]
- Romero, C.A.; Lozano, L.M.; Perfecto, Y.; Santa Cruz, F.J.; Garcia-Varela, R.; Chairez, I. Multi-cell lines culturing system utilizing microfluidic three-dimensional additive manufactured polymeric scaffolds with controlled pumping. Microchem. J. 2025, 212, 113193. [Google Scholar] [CrossRef]
- Dettinger, P.; Frank, T.; Etzrodt, M.; Ahmed, N.; Reimann, A.; Trenzinger, C.; Loeffler, D.; Kokkaliaris, K.D.; Schroeder, T.; Tay, S. Automated Microfluidic System for Dynamic Stimulation and Tracking of Single Cells. Anal. Chem. 2018, 90, 10695–10700. [Google Scholar] [CrossRef]
- Jaberi, A.; Monemian Esfahani, A.; Aghabaglou, F.; Park, J.S.; Ndao, S.; Tamayol, A.; Yang, R. Microfluidic Systems with Embedded Cell Culture Chambers for High-Throughput Biological Assays. ACS Appl. Bio Mater. 2020, 3, 6661–6671. [Google Scholar] [CrossRef]
- Pitingolo, G.; He, Y.; Huang, B.; Wang, L.; Shi, J.; Chen, Y. An automatic cell culture platform for differentiation of human induced pluripotent stem cells. Microelectron. Eng. 2020, 231, 111371. [Google Scholar] [CrossRef]
- Kane, K.I.W.; Moreno, E.L.; Hachi, S.; Walter, M.; Jarazo, J.; Oliveira, M.A.P.; Hankemeier, T.; Vulto, P.; Schwamborn, J.C.; Thoma, M.; et al. Automated microfluidic cell culture of stem cell derived dopaminergic neurons. Sci. Rep. 2019, 9, 1796. [Google Scholar] [CrossRef]
- Wang, L.; Ni, X.F.; Luo, C.X.; Zhang, Z.L.; Pang, D.W.; Chen, Y. Self-loading and cell culture in one layer microfluidic devices. Biomed. Microdevices 2009, 11, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Zhu, X.; Yu, T.; Luo, X.; Ouyang, Q.; Ji, H.; Chen, Y. A fast cell loading and high-throughput microfluidic system for long-term cell culture in zero-flow environments. Biotechnol. Bioeng. 2008, 101, 190–195. [Google Scholar] [CrossRef]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Mustafa, A.; Pedone, E.; Marucci, L.; Moschou, D.; Lorenzo, M.D. A flow-through microfluidic chip for continuous dielectrophoretic separation of viable and non-viable human T-cells. Electrophoresis 2022, 43, 501–508. [Google Scholar] [CrossRef] [PubMed]
- ur Rehman, A.; Zabibah, R.S.; Kharratian, S.; Mustafa, A. Microfluidic Device for the Separation of Non-Metastatic (MCF-7) and Non-Tumor (MCF-10A) Breast Cancer Cells Using AC Dielectrophoresis. JoVE 2022, 186, e63850. [Google Scholar]
- Wu, W.; Zhang, S.; Zhang, T.; Mu, Y. Immobilized Droplet Arrays in Thermosetting Oil for Dynamic Proteolytic Assays of Single Cells. ACS Appl. Mater. Interfaces 2021, 13, 6081–6090. [Google Scholar] [CrossRef] [PubMed]
- Gharib, G.; Butun, I.; Muganli, Z.; Kozalak, G.; Namli, I.; Sarraf, S.S.; Ahmadi, V.E.; Toyran, E.; van Wijnen, A.J.; Kosar, A. Biomedical Applications of Microfluidic Devices: A Review. Biosensors 2022, 12, 1023. [Google Scholar] [CrossRef] [PubMed]
- Kolnik, M.; Tsimring, L.S.; Hasty, J. Vacuum-assisted cell loading enables shear-free mammalian microfluidic culture. Lab Chip 2012, 12, 4732–4737. [Google Scholar] [CrossRef]
- Nocera, G.M.; Viscido, G.; Criscuolo, S.; Brillante, S.; Carbone, F.; Staiano, L.; Carrella, S.; di Bernardo, D. The VersaLive platform enables microfluidic mammalian cell culture for versatile applications. Commun. Biol. 2022, 5, 1034. [Google Scholar] [CrossRef]
- Yazdian Kashani, S.; Keshavarz Moraveji, M.; Bonakdar, S. Computational and experimental studies of a cell-imprinted-based integrated microfluidic device for biomedical applications. Sci. Rep. 2021, 11, 12130. [Google Scholar] [CrossRef]
- Postiglione, L.; Napolitano, S.; Pedone, E.; Rocca, D.L.; Aulicino, F.; Santorelli, M.; Tumaini, B.; Marucci, L.; di Bernardo, D. Regulation of Gene Expression and Signaling Pathway Activity in Mammalian Cells by Automated Microfluidics Feedback Control. ACS Synth. Biol. 2018, 7, 2558–2565. [Google Scholar] [CrossRef]
- Pedone, E.; de Cesare, I.; Zamora-Chimal, C.G.; Haener, D.; Postiglione, L.; La Regina, A.; Shannon, B.; Savery, N.J.; Grierson, C.S.; di Bernardo, M.; et al. Cheetah: A Computational Toolkit for Cybergenetic Control. ACS Synth. Biol. 2021, 10, 979–989. [Google Scholar] [CrossRef]
- Pedone, E.; Postiglione, L.; Aulicino, F.; Rocca, D.L.; Montes-Olivas, S.; Khazim, M.; di Bernardo, D.; Pia Cosma, M.; Marucci, L. A tunable dual-input system for on-demand dynamic gene expression regulation. Nat. Commun. 2019, 10, 4481. [Google Scholar] [CrossRef]
- Khazim, M.; Pedone, E.; Postiglione, L.; di Bernardo, D.; Marucci, L. A microfluidic/microscopy-based platform for on-chip controlled gene expression in mammalian cells. Synth. Gene Circuits 2021, 2229, 205–219. [Google Scholar]
- Wray, J.; Kalkan, T.; Gomez-Lopez, S.; Eckardt, D.; Cook, A.; Kemler, R.; Smith, A. Inhibition of glycogen synthase kinase-3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat. Cell Biol. 2011, 13, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Parchem, R.J.; Ye, J.; Judson, R.L.; LaRussa, M.F.; Krishnakumar, R.; Blelloch, A.; Oldham, M.C.; Blelloch, R. Two miRNA clusters reveal alternative paths in late-stage reprogramming. Cell Stem Cell 2014, 14, 617–631. [Google Scholar] [CrossRef]
- Ying, Q.L.; Wray, J.; Nichols, J.; Batlle-Morera, L.; Doble, B.; Woodgett, J.; Cohen, P.; Smith, A. The ground state of embryonic stem cell self-renewal. Nature 2008, 453, 519–523. [Google Scholar] [CrossRef]
- de Cesare, I.; Zamora-Chimal, C.G.; Postiglione, L.; Khazim, M.; Pedone, E.; Shannon, B.; Fiore, G.; Perrino, G.; Napolitano, S.; di Bernardo, D.; et al. ChipSeg: An Automatic Tool to Segment Bacterial and Mammalian Cells Cultured in Microfluidic Devices. ACS Omega 2021, 6, 2473–2476. [Google Scholar] [CrossRef]
- An, D.; Kim, K.; Kim, J. Microfluidic System Based High Throughput Drug Screening System for Curcumin/TRAIL Combinational Chemotherapy in Human Prostate Cancer PC3 Cells. Biomol. Ther. 2014, 22, 355–362. [Google Scholar] [CrossRef]
- Ruzycka, M.; Cimpan, M.R.; Rios-Mondragon, I.; Grudzinski, I.P. Microfluidics for studying metastatic patterns of lung cancer. J. Nanobiotechnol. 2019, 17, 71. [Google Scholar] [CrossRef]
- Marucci, L. Nanog Dynamics in Mouse Embryonic Stem Cells: Results from Systems Biology Approaches. Stem Cells Int. 2017, 2017, 7160419. [Google Scholar] [CrossRef]
- Marucci, L.; Santini, S.; di Bernardo, M.; di Bernardo, D. Derivation, identification and validation of a computational model of a novel synthetic regulatory network in yeast. J. Math. Biol. 2011, 62, 685–706. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustafa, A.; La Regina, A.; Pedone, E.; Erten, A.; Marucci, L. A Novel Single-Layer Microfluidic Device for Dynamic Stimulation, Culture, and Imaging of Mammalian Cells. Biosensors 2025, 15, 427. https://doi.org/10.3390/bios15070427
Mustafa A, La Regina A, Pedone E, Erten A, Marucci L. A Novel Single-Layer Microfluidic Device for Dynamic Stimulation, Culture, and Imaging of Mammalian Cells. Biosensors. 2025; 15(7):427. https://doi.org/10.3390/bios15070427
Chicago/Turabian StyleMustafa, Adil, Antonella La Regina, Elisa Pedone, Ahmet Erten, and Lucia Marucci. 2025. "A Novel Single-Layer Microfluidic Device for Dynamic Stimulation, Culture, and Imaging of Mammalian Cells" Biosensors 15, no. 7: 427. https://doi.org/10.3390/bios15070427
APA StyleMustafa, A., La Regina, A., Pedone, E., Erten, A., & Marucci, L. (2025). A Novel Single-Layer Microfluidic Device for Dynamic Stimulation, Culture, and Imaging of Mammalian Cells. Biosensors, 15(7), 427. https://doi.org/10.3390/bios15070427