Labeled Bovine Serum Albumin as a Fluorescent Biosensor to Monitor the Stability of Lipid-Based Formulations

 , , , , , ,
, , , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Bovine Serum Albumin Conjugation
2.3. Fluorescence Measurements on Acrylodan-Conjugated BSA (Albuminodan)
2.4. Preparation of DPPC/POPG Formulation
2.5. PLA2 Hydrolysis Experiment
2.6. Spectral Analysis
3. Results and Discussion
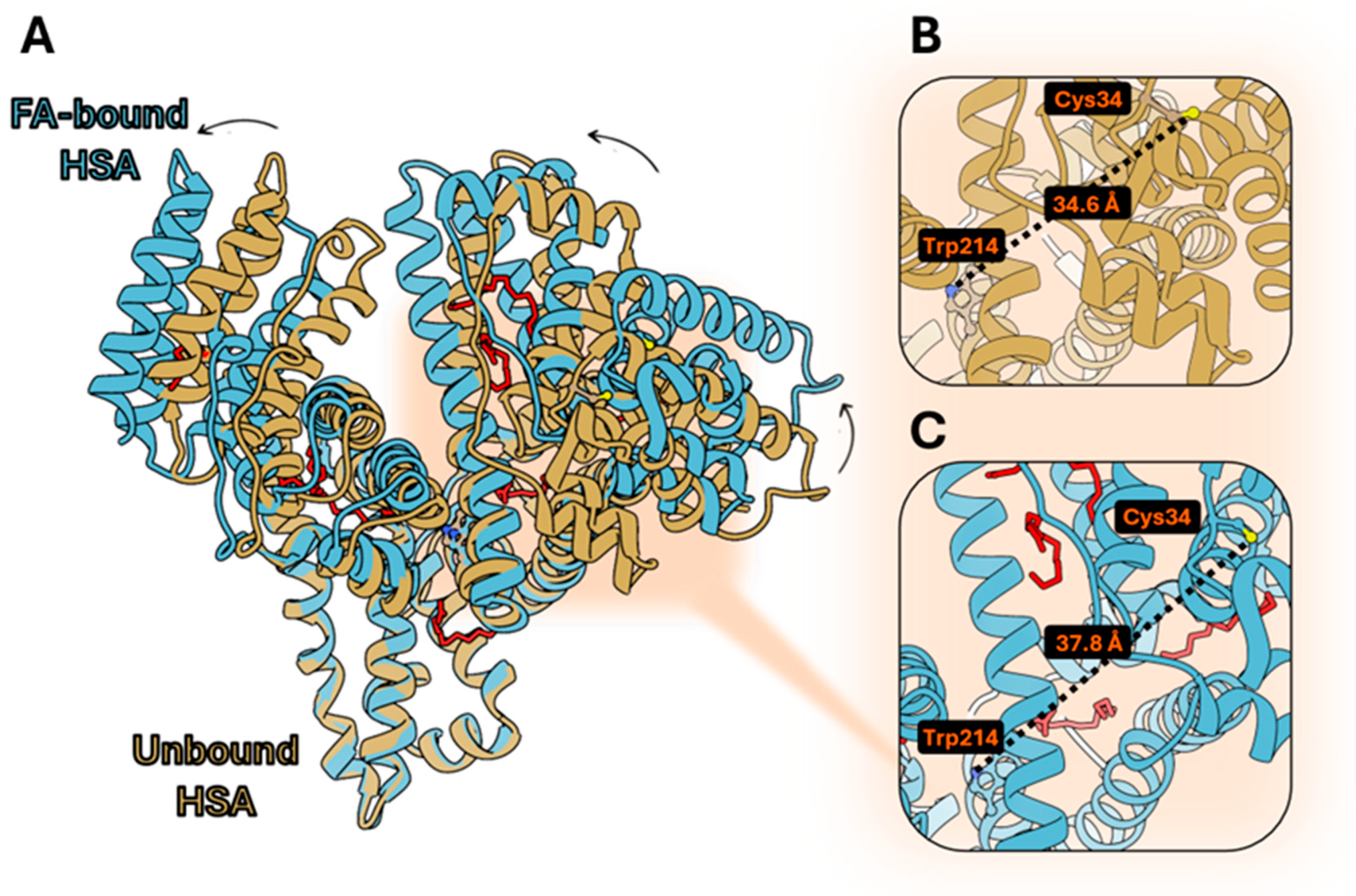
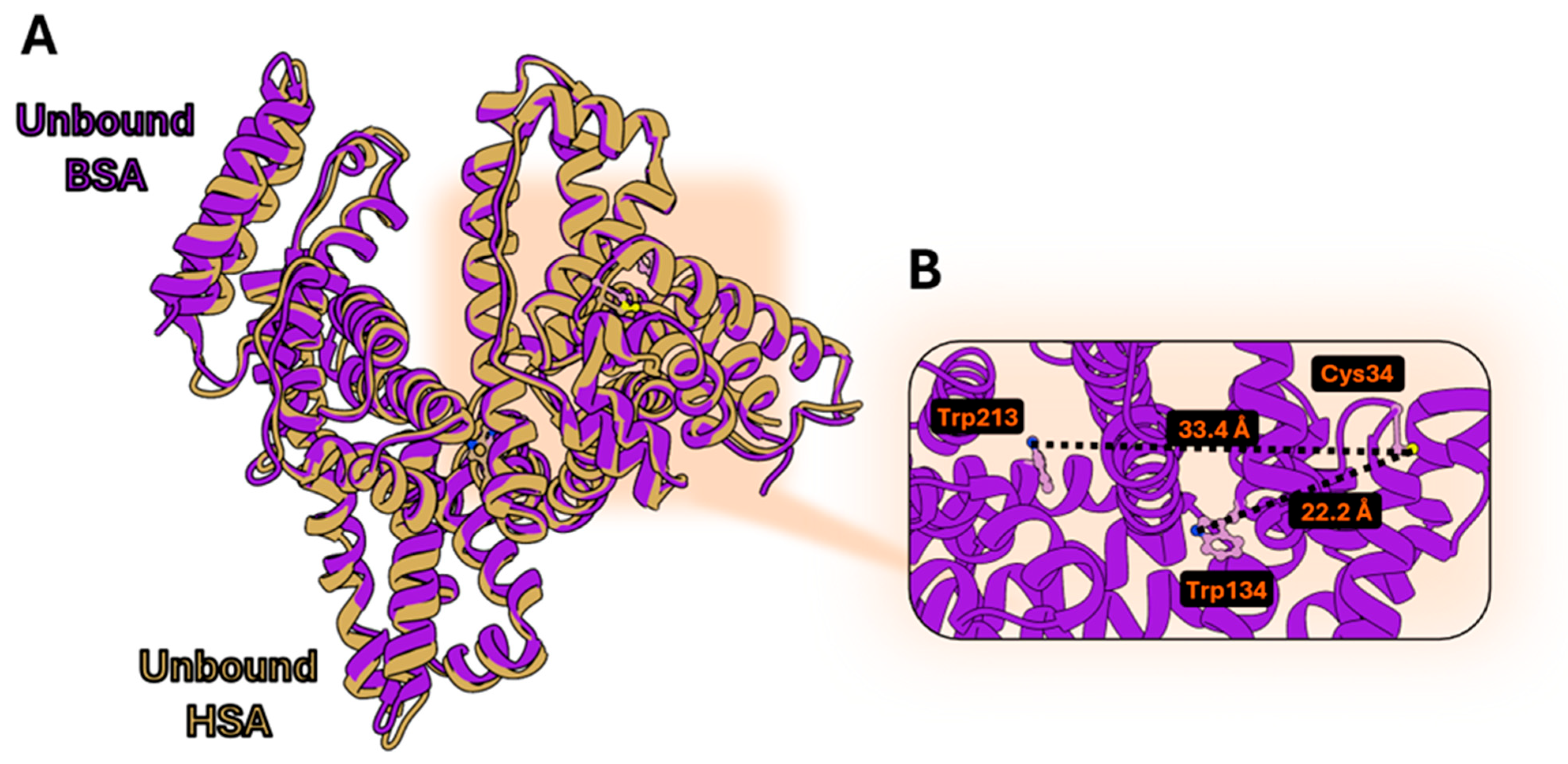
3.1. Structural Comparison Between Human and Bovine Serum Albumin
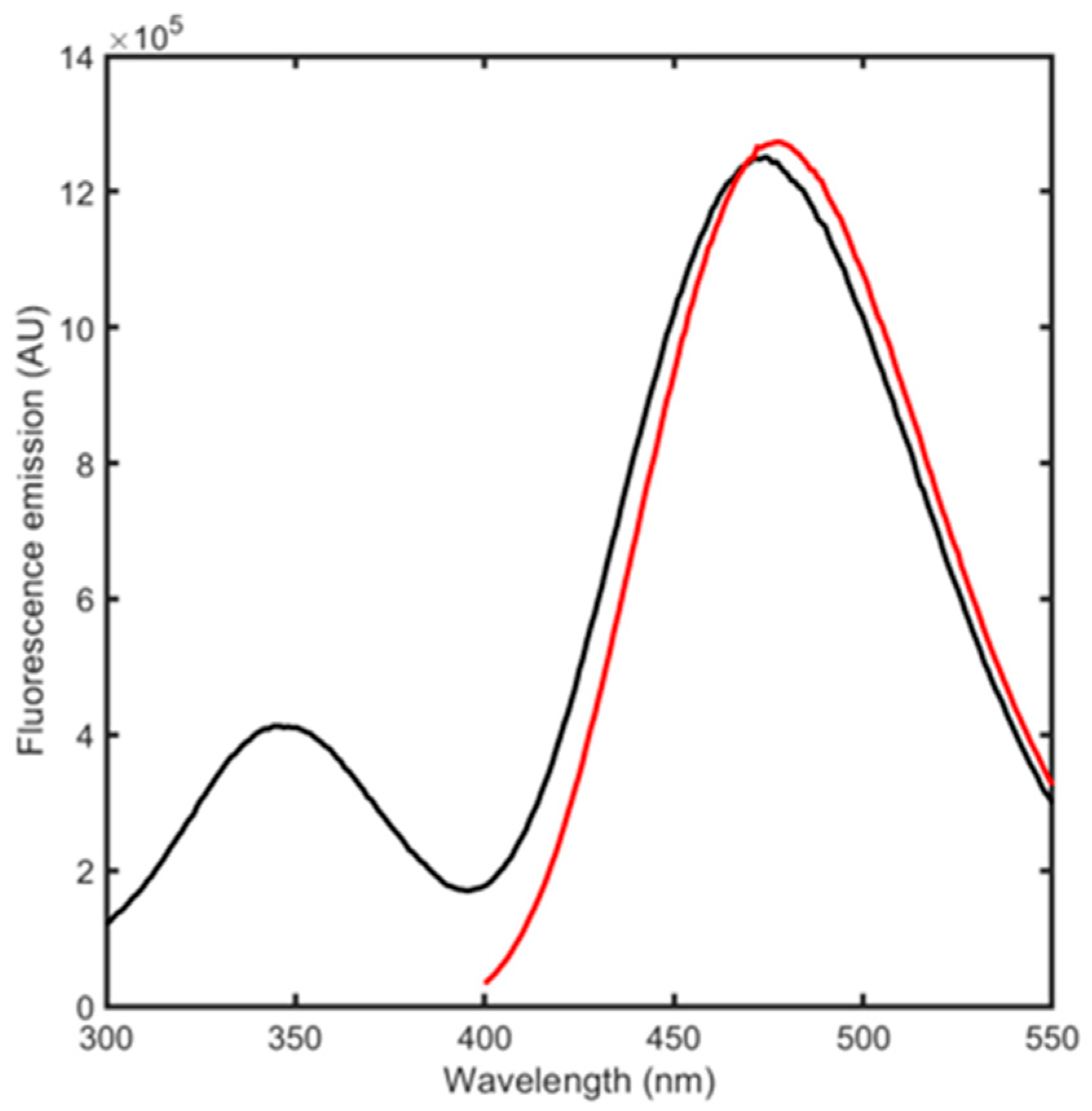
3.2. BSA Spectroscopic Evaluation
3.3. BSA Conjugation to Acrylodan
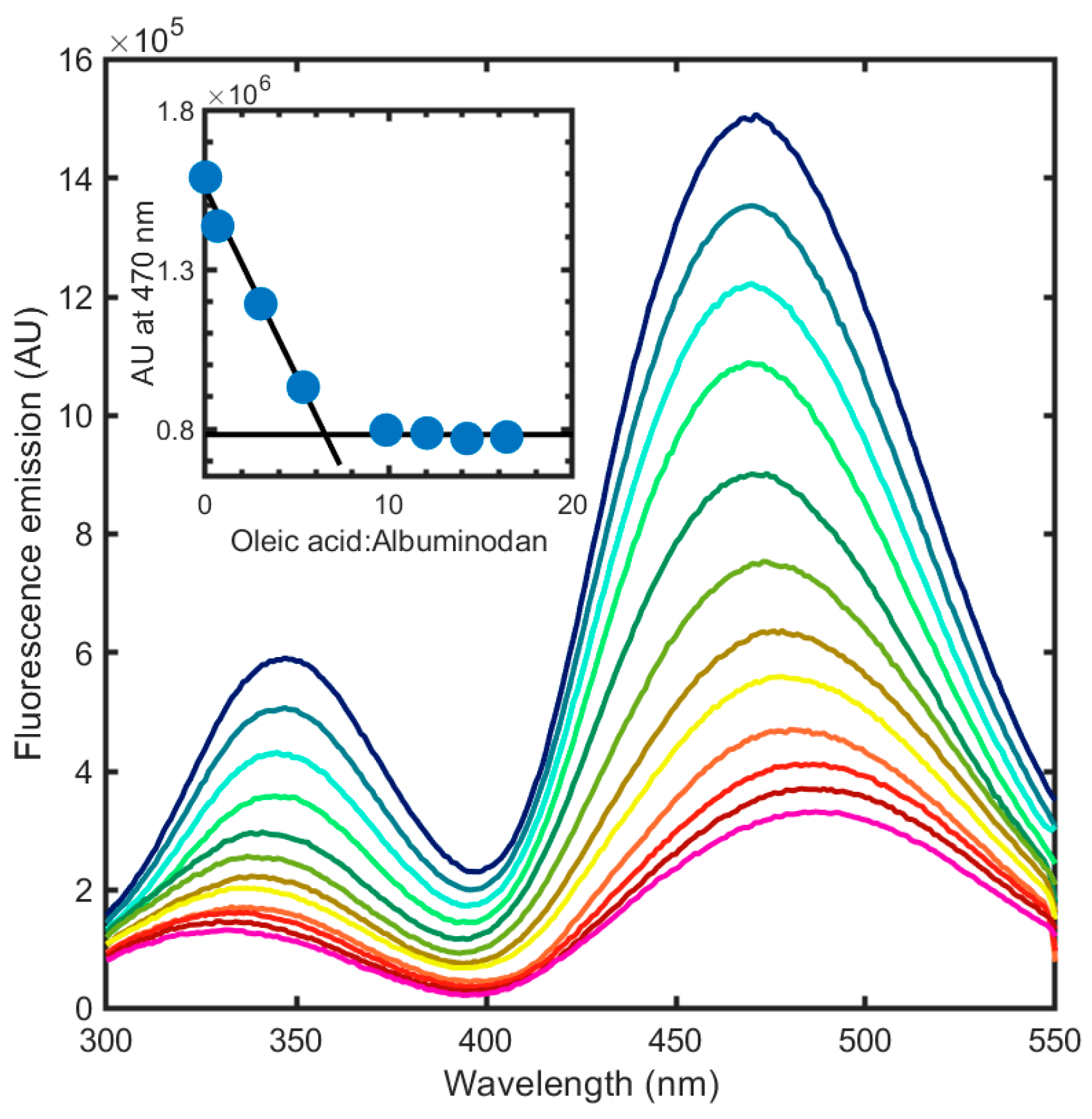
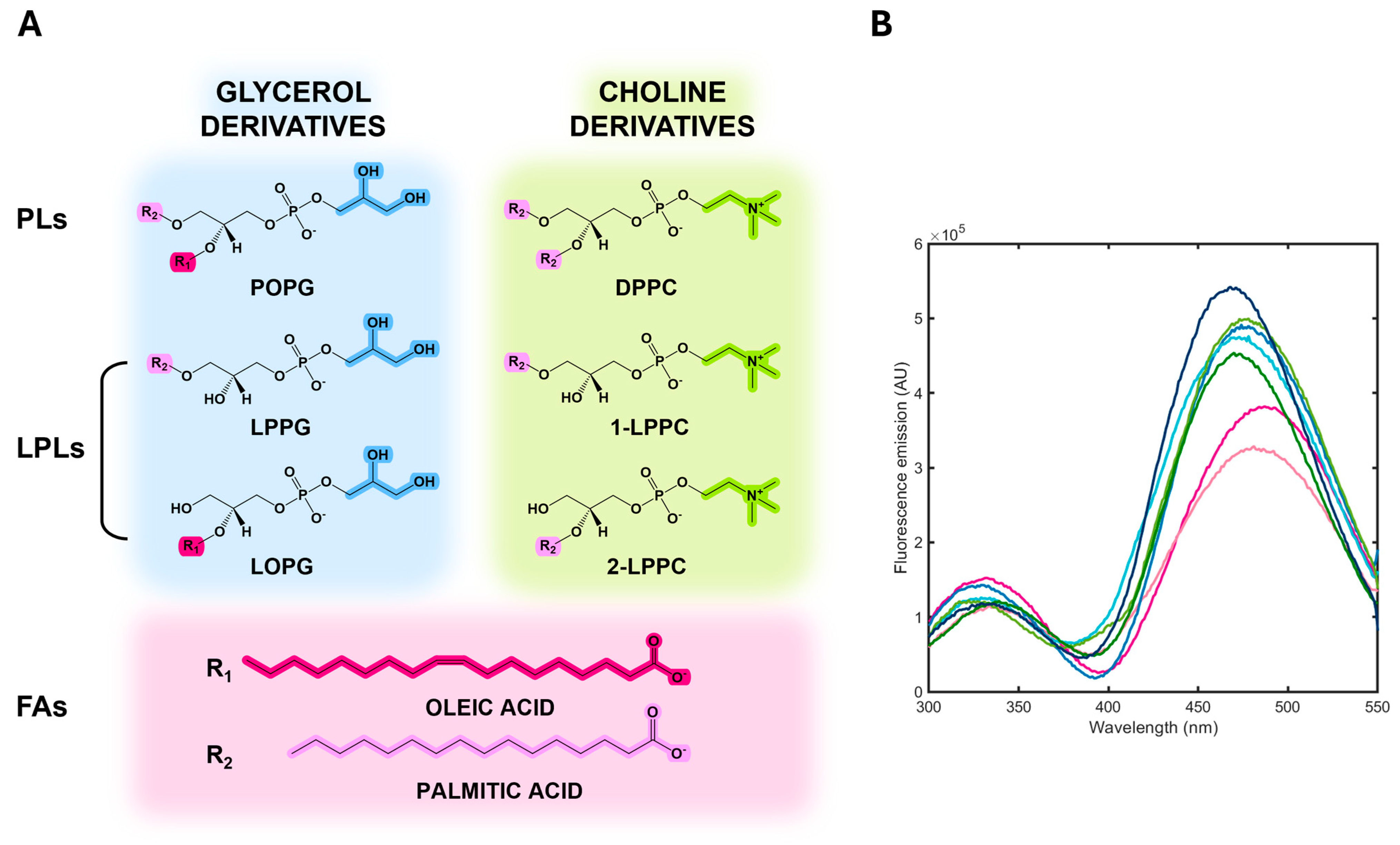
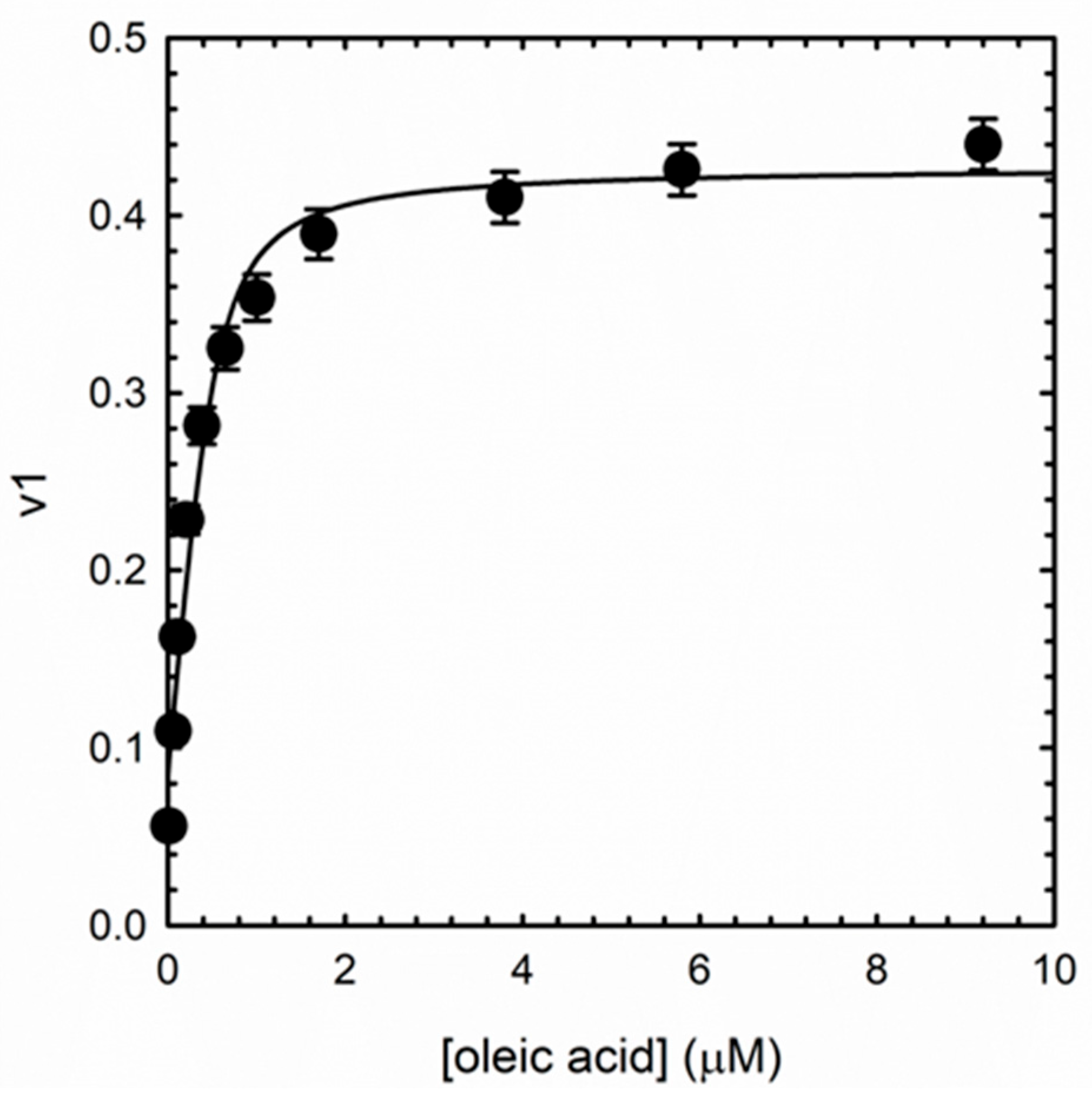
3.4. Albuminodan Binding to PLs, LPLs, and FAs
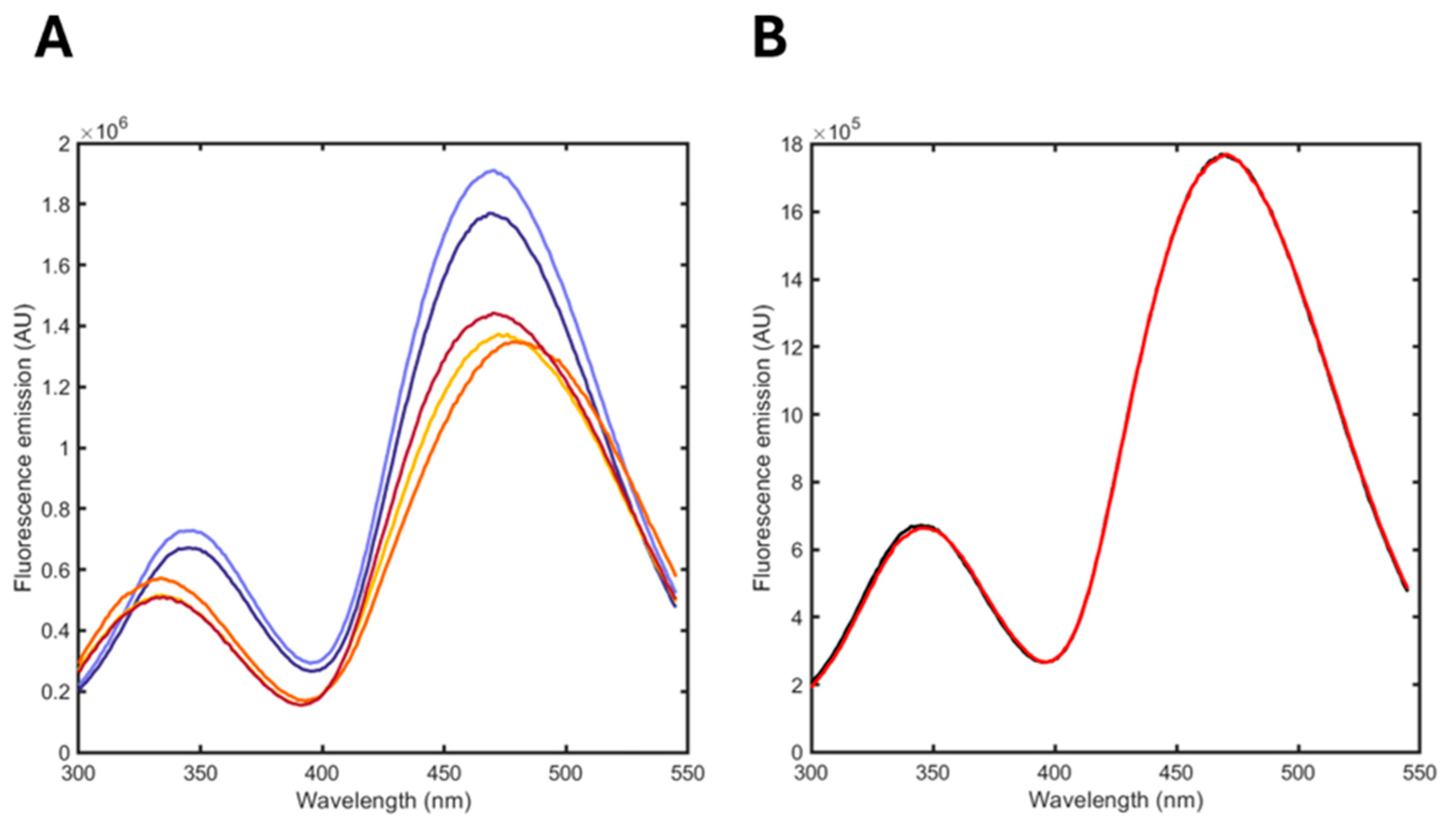
3.5. Monitoring the Hydrolysis of a DPPC/POPG Formulation Induced by Phospholipase A2 (PLA2)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LPNs | Lipid nanoparticles |
| DSPC | 1,2-distearoyl-sn-glycero-3-phosphocholine |
| PEG | Polyethylene glycol |
| FAs | Fatty acids |
| LPLs | Lysophospholipids |
| ELSD | Evaporative light scattering detection |
| CAD | Charged aerosol detection |
| iFABP | Intestinal fatty acid-binding protein |
| FFA | Free fatty acid |
| PLs | Phospholipids |
| BSA | Bovine serum albumin |
| FRET | Fluorescence resonance energy transfer |
| DMSO | Dimethyl sulfoxide |
| PLA2 | Phospholipase A2 |
| DPPC | Dipalmitoyl phosphatidylcholine |
| POPG | 1-palmitoyl-2-oleoyl-sn-glycero-3-(phospho-rac-(1-glycerol)) |
| LPPC | Lysopalmitoylphosphatidylcholin |
| LOPG | Lysooleilphosphatidylglycerol |
| LPPG | Lysopalmitoylphosphatidylglycerol |
| SUV | Small unilamellar vesicles |
| HSA | Human serum albumin |
| DOL | Degree of labeling |
| SVD | Singular value decomposition |
References
- Liu, Y.; Bravo, K.M.C.; Liu, J. Targeted liposomal drug delivery: A nanoscience and biophysical perspective. Nanoscale Horiz. 2021, 6, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles─From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef] [PubMed]
- Curstedt, T.; Halliday, H.L.; Speer, C.P. A unique story in neonatal research: The development of a porcine surfactant. Neonatology 2015, 107, 321–329. [Google Scholar] [CrossRef]
- Alfutaimani, A.S.; Alharbi, N.K.; Alahmari, A.S.; Alqabbani, A.A.; Aldayel, A.M. Exploring the landscape of Lipid Nanoparticles (LNPs): A comprehensive review of LNPs types and biological sources of lipids. Int. J. Pharm. 2024, 8, 100305. [Google Scholar] [CrossRef]
- Hald Albertsen, C.; Kulkarni, J.A.; Witzigmann, D.; Lind, M.; Petersson, K.; Simonsen, J.B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar] [CrossRef]
- Giordani, S.; Marassi, V.; Zattoni, A.; Roda, B.; Reschiglian, P. Liposomes characterization for market approval as pharmaceutical products: Analytical methods, guidelines and standardized protocols. J. Pharm. Biomed. Anal. 2023, 236, 115751. [Google Scholar] [CrossRef] [PubMed]
- Administration, F.A.D. Liposome Drug Products: Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/liposome-drug-products-chemistry-manufacturing-and-controls-human-pharmacokinetics-and (accessed on 1 March 2025).
- Singh, R.; Ajagbe, M.; Bhamidipati, S.; Ahmad, Z.; Ahmad, I. A rapid isocratic high-performance liquid chromatography method for determination of cholesterol and 1,2-dioleoyl-sn-glycero-3-phosphocholine in liposome-based drug formulations. J. Chromatogr. A 2005, 1073, 347–353. [Google Scholar] [CrossRef]
- Shimizu, Y.; Nakata, M.; Matsunuma, J.; Mizuochi, T. Simultaneous quantification of components of neoglycolipid-coated liposomes using high-performance liquid chromatography with evaporative light scattering detection. J. Chromatogr. B Biomed. Sci. Appl. 2001, 754, 127–133. [Google Scholar] [CrossRef]
- Jeschek, D.; Lhota, G.; Wallner, J.; Vorauer-Uhl, K. A versatile, quantitative analytical method for pharmaceutical relevant lipids in drug delivery systems. J. Pharm. Biomed. Anal. 2016, 119, 37–44. [Google Scholar] [CrossRef]
- Kiełbowicz, G.; Smuga, D.; Gładkowski, W.; Chojnacka, A.; Wawrzeńczyk, C. An LC method for the analysis of phosphatidylcholine hydrolysis products and its application to the monitoring of the acyl migration process. Talanta 2012, 94, 22–29. [Google Scholar] [CrossRef]
- Damnjanović, J.; Nakano, H.; Iwasaki, Y. Simple and Efficient Profiling of Phospholipids in Phospholipase D-modified Soy Lecithin by HPLC with Charged Aerosol Detection. J. Am. Oil Chem. Soc. 2013, 90, 951–957. [Google Scholar] [CrossRef]
- Reis, A.; Domingues, M.R.M.; Amado, F.M.L.; Ferrer-Correia, A.J.; Domingues, P. Radical peroxidation of palmitoyl-lineloyl-glycerophosphocholine liposomes: Identification of long-chain oxidised products by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 855, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Marioli, M.; Zhang, K. Analytical characterization of liposomes and other lipid nanoparticles for drug delivery. J. Pharm. Biomed. Anal. 2021, 192, 113642. [Google Scholar] [CrossRef] [PubMed]
- Visnovitz, T.; Osteikoetxea, X.; Sódar, B.W.; Mihály, J.; Lőrincz, P.; Vukman, K.V.; Tóth, E.; Koncz, A.; Székács, I.; Horváth, R.; et al. An improved 96 well plate format lipid quantification assay for standardisation of experiments with extracellular vesicles. J. Extracell. Vesicles 2019, 8, 1565263. [Google Scholar] [CrossRef]
- London, E.; Feligenson, G.W. A convenient and sensitive fluorescence assay for phospholipid vesicles using diphenylhexatriene. Anal. Biochem. 1978, 88, 203–211. [Google Scholar] [CrossRef]
- Gil, B.; Keshavarz, M.; Wales, D.; Darzi, A.; Yeatman, E. Orthogonal Surface-Enhanced Raman Scattering/Field-Effect Transistor Detection of Breast and Colorectal Cancer-Derived Exosomes using Graphene as a Tag-Free Diagnostic Template. Adv. NanoBiomed Res. 2023, 3, 2300055. [Google Scholar] [CrossRef]
- Yasuda, T.; Watanabe, H.; Hirosawa, K.M.; Suzuki, K.G.N.; Suga, K.; Hanashima, S. Fluorescence Spectroscopic Analysis of Lateral and Transbilayer Fluidity of Exosome Membranes. Langmuir 2022, 38, 14695–14703. [Google Scholar] [CrossRef]
- Domecq, A.; Disalvo, E.A.; Bernik, D.L.; Florenzano, F.; Politi, M.J. A Stability Test of Liposome Preparations Using Steady-State Fluorescent Measurements. Drug Deliv. 2001, 8, 155–160. [Google Scholar] [CrossRef]
- Chandra, A.; Datta, A. A Peptide-Based Fluorescent Sensor for Anionic Phospholipids. ACS Omega 2022, 7, 10347–10354. [Google Scholar] [CrossRef]
- Richieri, G.V.; Ogata, R.T.; Kleinfeld, A.M. The measurement of free fatty acid concentration with the fluorescent probe ADIFAB: A practical guide for the use of the ADIFAB probe. Mol. Cell. Biochem. 1999, 192, 87–94. [Google Scholar] [CrossRef]
- Richieri, G.V.; Ogata, R.T.; Kleinfeld, A.M. A fluorescently labeled intestinal fatty acid binding protein. Interactions with fatty acids and its use in monitoring free fatty acids. J. Biol. Chem. 1992, 267, 23495–23501. [Google Scholar] [CrossRef] [PubMed]
- Faggiano, S.; Ronda, L.; Raboni, S.; Mozzarelli, A. ADIFAB fluorescence data used for the quantification of free fatty acids in media at different pH. Data Brief 2019, 22, 158–163. [Google Scholar] [CrossRef]
- Faggiano, S.; Ronda, L.; Raboni, S.; Sartor, F.; Cavatorta, V.; Sgarbi, E.; Caivano, G.; Pertile, M.; Mozzarelli, A. Phospholipid components of the synthetic pulmonary surfactant CHF5633 probed by fluorescence spectroscopy. Int. J. Pharm. 2018, 553, 290–297. [Google Scholar] [CrossRef]
- Singh, J.; Ranganathan, R. Quantitation of lysolipids, fatty acids, and phospholipase A2 activity and correlation with membrane polarity. J. Lipid Res. 2012, 53, 1993–2001. [Google Scholar] [CrossRef]
- Spector, A.A.; John, K.; Fletcher, J.E. Binding of long-chain fatty acids to bovine serum albumin. J. Lipid Res. 1969, 10, 56–67. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Era, S.; Bhamidipati, S.P.; Reed, R.G. Locations of the three primary binding sites for long-chain fatty acids on bovine serum albumin. Proc. Natl Acad. Sci. USA 1991, 88, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Sun, S.; Bekos, E.J.; Bright, F.V. Dynamics surrounding Cys-34 in native, chemically denatured, and silica-adsorbed bovine serum albumin. Anal. Chem. 1995, 67, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, J.H. Morrissey Lab Protocol for Preparing Phospholipid Vesicles (SUV) by Sonication. Available online: https://tf7.org/protocols/ (accessed on 25 November 2024).
- The MathWorks Inc. MATLAB, version: 9.13.0 (R2022b); The MathWorks Inc.: Natick, MA, USA, 2022. [Google Scholar]
- Curry, S.; Mandelkow, H.; Brick, P.; Franks, N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Mol. Biol. 1998, 5, 827–835. [Google Scholar] [CrossRef]
- Pomyalov, S.; Minetti, C.A.; Remeta, D.P.; Bonala, R.; Johnson, F.; Zaitseva, I.; Iden, C.; Golebiewska, U.; Breslauer, K.J.; Shoham, G.; et al. Structural and mechanistic insights into the transport of aristolochic acids and their active metabolites by human serum albumin. J. Biol. Chem. 2024, 300, 107358. [Google Scholar] [CrossRef]
- Narazaki, R.; Maruyama, T.; Otagiri, M. Probing the cysteine 34 residue in human serum albumin using fluorescence techniques. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 1997, 1338, 275–281. [Google Scholar] [CrossRef]
- Hagag, N.; Birnbaum, E.R.; Darnall, D.W. Resonance energy transfer between cysteine-34, tryptophan-214, and tyrosine-411 of human serum albumin. Biochemistry 1983, 22, 2420–2427. [Google Scholar] [CrossRef]
- Suzukida, M.; Le, H.P.; Shahid, F.; McPherson, R.A.; Birnbaum, E.R.; Darnall, D.W. Resonance energy transfer between cysteine-34 and tryptophan-214 in human serum albumin. Distance measurements as a function of pH. Biochemistry 1983, 22, 2415–2420. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Lai, A.J.; Alaee, Y.; Ranganathan, R. Partitioning of lysolipids, fatty acids and their mixtures in aqueous lipid bilayers: Solute concentration/composition effects. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Tayeh, N.; Rungassamy, T.; Albani, J.R. Fluorescence spectral resolution of tryptophan residues in bovine and human serum albumins. J. Pharm. Biomed. Anal. 2009, 50, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Taketomi, Y.; Sato, H.; Yamamoto, K. Secreted phospholipase A2 revisited. J. Biochem. 2011, 150, 233–255. [Google Scholar] [CrossRef]
- Astudillo, A.M.; Balboa, M.A.; Balsinde, J. Selectivity of phospholipid hydrolysis by phospholipase A2 enzymes in activated cells leading to polyunsaturated fatty acid mobilization. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2019, 1864, 772–783. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | KD (μM) |
|---|---|
| DPPC | 0.137 ± 0.070 |
| POPG | 0.243 ± 0.087 |
| LPPG | 0.218 ± 0.079 |
| LOPG | 0.187 ± 0.050 |
| LPPC | 0.183 ± 0.057 |
| oleic acid | 0.104 ± 0.046 |
| palmitic acid | 0.395 ± 0.105 |
| Spontaneous Hydrolysis (%) | Enzymatic Hydrolysis (%) | ||||
|---|---|---|---|---|---|
| t = 0 | t = 2 mos | t = 2 min | t = 1 h | t = 6 h | |
| ligand-free albuminodan | 71 | 66 | 4 | 4 | 6 |
| oleic acid | 0 | 0 | 54 | 83 | 11 |
| palmitic acid | 0 | 0 | 0 | 0 | 0 |
| LPPG | 0 | 0 | 4 | 0 | 51 |
| LPPC | 0 | 0 | 0 | 0 | 0 |
| LOPG | 0 | 0 | 0 | 0 | 0 |
| DPPC | 0 | 20 | 18 | 13 | 31 |
| POPG | 29 | 14 | 20 | 0 | 1 |
| R2 | 0.9996 | 0.9996 | 0.9978 | 0.9953 | 0.9969 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bova, S.; Faggiano, S.; De Bei, O.; Marchetti, M.; Bruno, S.; Campanini, B.; Bettati, S.; Ronda, L. Labeled Bovine Serum Albumin as a Fluorescent Biosensor to Monitor the Stability of Lipid-Based Formulations. Biosensors 2025, 15, 425. https://doi.org/10.3390/bios15070425
Bova S, Faggiano S, De Bei O, Marchetti M, Bruno S, Campanini B, Bettati S, Ronda L. Labeled Bovine Serum Albumin as a Fluorescent Biosensor to Monitor the Stability of Lipid-Based Formulations. Biosensors. 2025; 15(7):425. https://doi.org/10.3390/bios15070425
Chicago/Turabian StyleBova, Stefania, Serena Faggiano, Omar De Bei, Marialaura Marchetti, Stefano Bruno, Barbara Campanini, Stefano Bettati, and Luca Ronda. 2025. "Labeled Bovine Serum Albumin as a Fluorescent Biosensor to Monitor the Stability of Lipid-Based Formulations" Biosensors 15, no. 7: 425. https://doi.org/10.3390/bios15070425
APA StyleBova, S., Faggiano, S., De Bei, O., Marchetti, M., Bruno, S., Campanini, B., Bettati, S., & Ronda, L. (2025). Labeled Bovine Serum Albumin as a Fluorescent Biosensor to Monitor the Stability of Lipid-Based Formulations. Biosensors, 15(7), 425. https://doi.org/10.3390/bios15070425