Fluorescence Lifetime Multiplexing with Fluorogen-Activating FAST Protein Variants and Red-Shifted Arylidene–Imidazolone Derivative as Fluorogen

 , , , , ,
, , , , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Plasmids
2.3. Primary Fluorescence Lifetime Screening In Vitro with Edinburgh Instruments Mini-Tau Spectrometer
2.4. Fluorescence-Lifetime Imaging Microscopy of HeLa Kyoto Live Cells
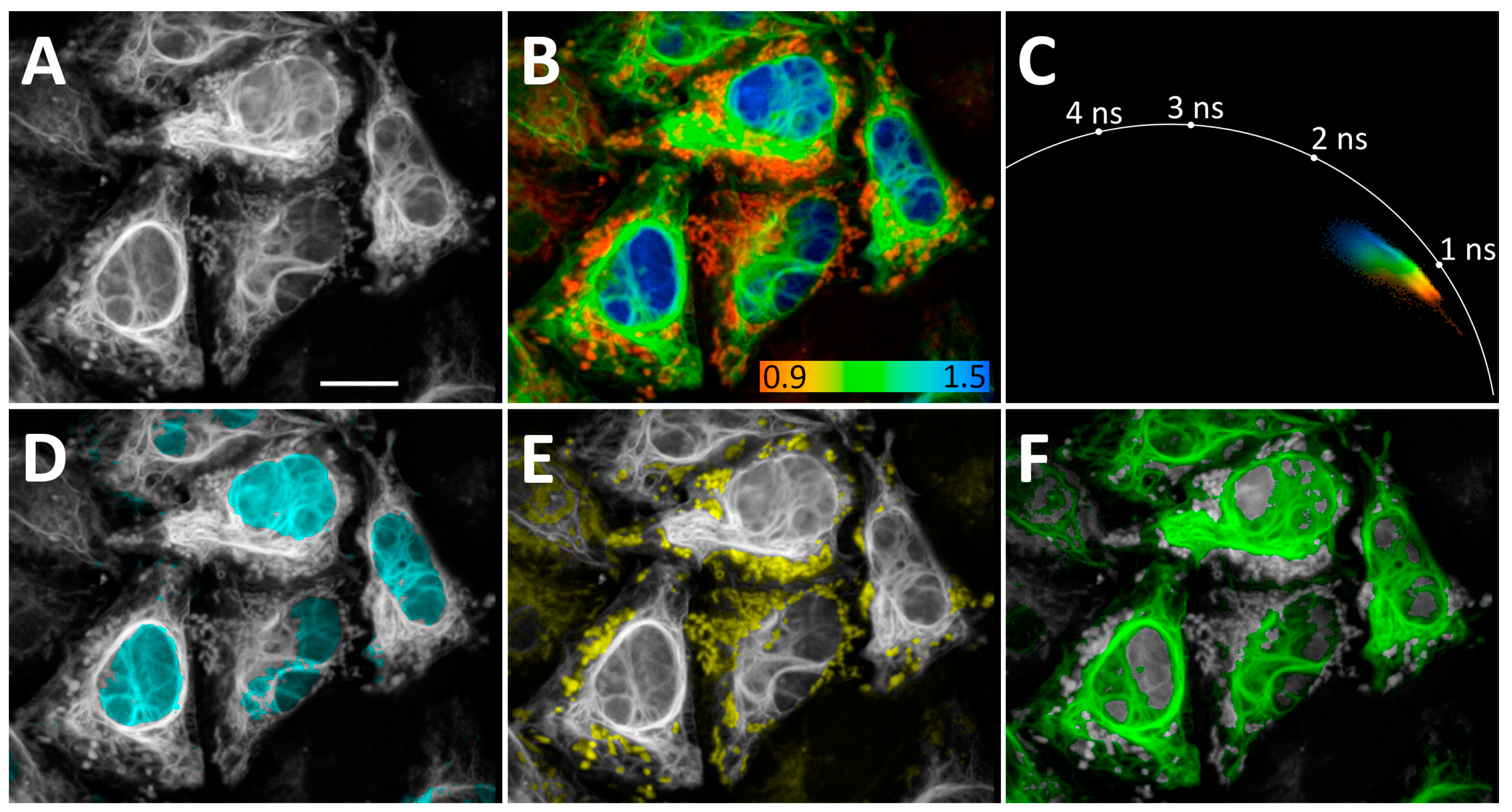
2.5. Fluorescence-Lifetime Imaging Microscopy Processing
- The blue crosshair was moved to the uniformly fluorescent region of the cell with medium signal intensity.
- The spatial binning factor (n) was set to 3–6 (“Bin” in the “Decay-Graph” window). The threshold parameter was set to 100–600. The time channel range used for fitting was adjusted with T1 and T2 values so the whole decay curve was included without the baseline regions. The number of exponential components was set to 2 or 3 in a “Multiexponential Decay” window.
- The decay matrix was calculated using the corresponding command (Calculate > Decay matrix).
- In each experiment, the lifetime data was collected from random cells in 4–6 fields of view.
- The color-coded and total intensity images were exported in .tiff format (File > Export…, “Color-coded image”, and “Gray-scale image”).
- Fitting results were analyzed using the Origin 2021 Software suite, and micrographs were processed with Fiji 1.54k.
- The blue crosshair was moved to the uniformly fluorescent region of the cell with medium signal intensity.
- The binning factor (n) was set to 3 (“Bin” in the “Decay-Graph” window). The threshold parameter was set to 100. The time channel range used for fitting was adjusted with T1 and T2 values to include the whole decay curve without the baseline regions. The number of exponential components was set to 2 in a “Multiexponential Decay” window.
- The color-coding was switched from continuous to discrete (Options > Color, “Mode discrete”), and the range of τi values was assigned to red, green, or blue color channels.
- The resulting FLIM images were exported as .tiff files and imported to FIJI 1.54k as RGB color-type files.
- RGB files were transformed to RGB stacks (Image > Type > RGB stack). The stacks were separated into three images (Image > Stacks > Stack to Images), one of which was empty. The resulting images contained pixels with a τi from an assigned range.
- Each image was transformed into 16-bit files (Image > Type > 16 bit); a colorblind-friendly palette was used for final figures representation (Image > LUT).
- Steps 1–2 are identical to those in the protocol for panel b.
- Phasor plot was calculated (“Phasor plot” button).
- The color-coding was switched from continuous to discrete mode (Options > Color, “Mode discrete”) and the range of red color was set from 1 to 2000 ps; green and blue channels were left empty.
- The box “Select cluster” was marked and the cluster for vimentin was selected.
- The image was exported in .tiff format.
- The range of the green channel was set from 1 to 2000 ps, and the red and blue channels were left empty.
- Steps 4–5 were repeated for the H2B cluster.
- FIJI 1.54k was used to prepare figures for publication.
- Steps 1–2 are identical to those in the protocol for panel c of Figure 2.
- The color-coding was switched from continuous to discrete mode (Options > Color, “Mode discrete”) and the ranges of the blue and green colors were set from 1 to 2000 ps, the red channel was left empty.
- The box “Select cluster” was marked, and the cluster for nuclei was selected.
- The image was exported in .tiff format.
- The color-coding was set to discrete mode (Options > Color, “Mode discrete”) and the ranges of the red and green colors were set from 1 to 2000 ps, the Blue channel was left empty.
- Steps 3–4 were repeated for the mitochondria cluster.
- The range of the green channel was set from 1 to 2000 ps, and the red and blue channels were left empty.
- Steps 4–5 were repeated for vimentin cluster.
- FIJI 1.54k was used to prepare figures for publication.
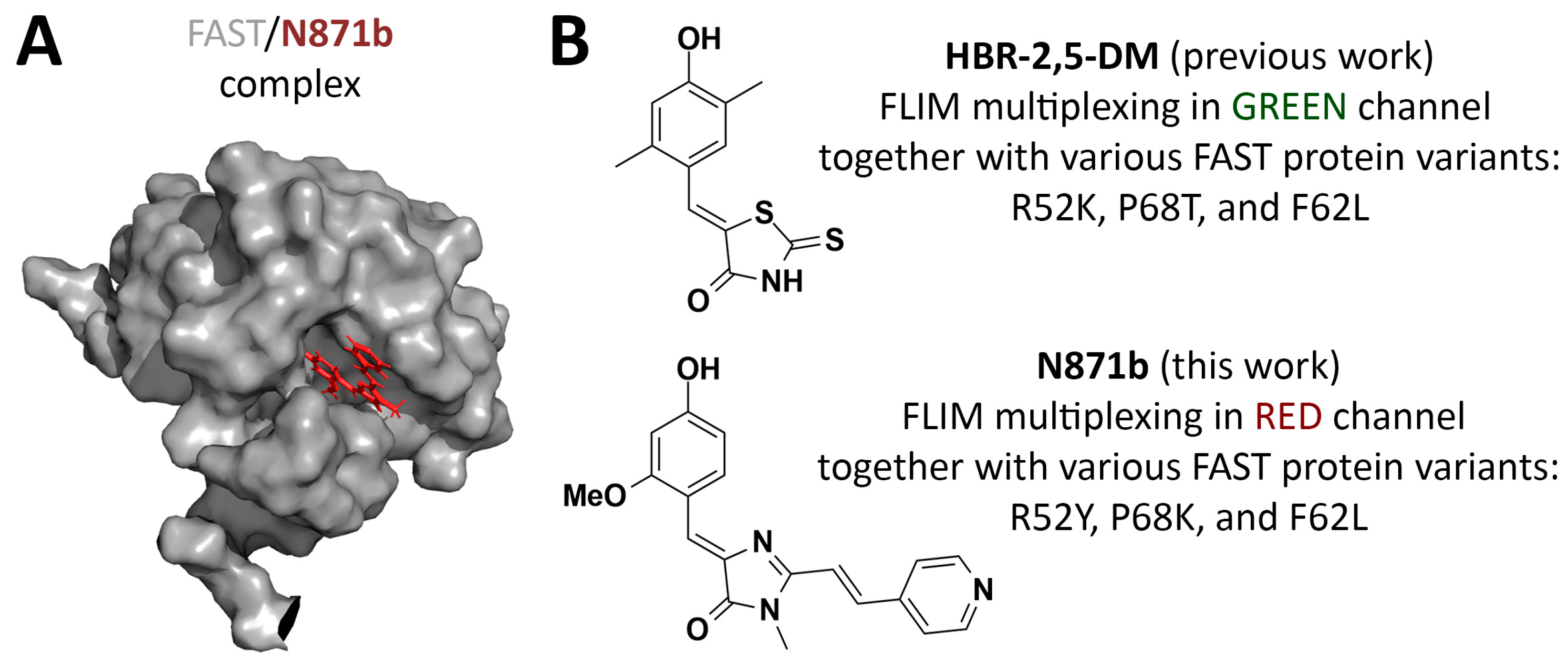
3. Results and Discussion
3.1. Primary Fluorescence-Lifetime Screening In Vitro
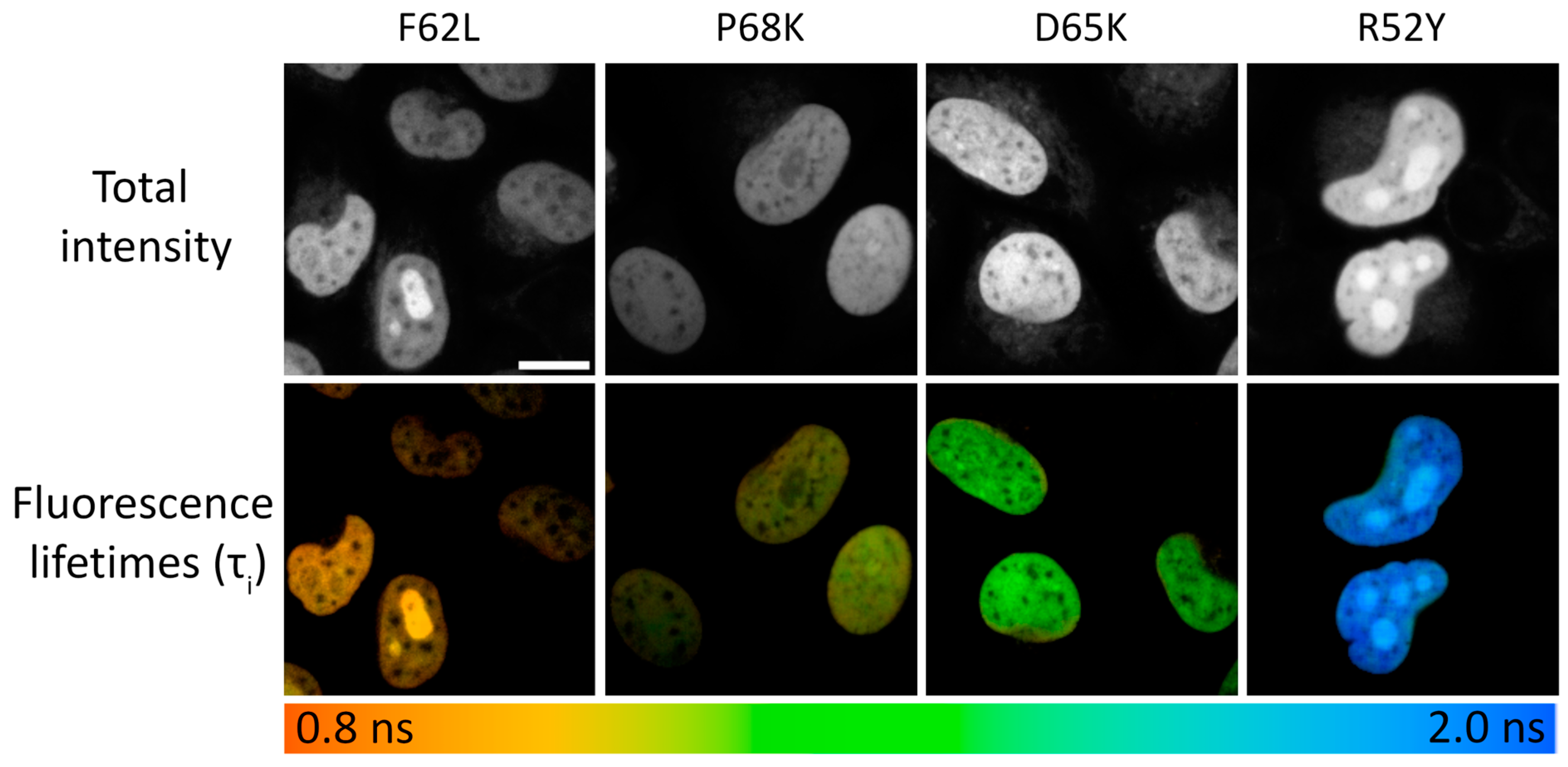
3.2. Fluorescence-Lifetime Measurements in Cells
3.3. FLIM-Based Visualization of Multiple Intracellular Targets
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FLIM | Fluorescence-lifetime imaging microscopy |
| SLP | Self-labeling protein |
| FAST | Fluorescence-Activating and Absorption-Shifting Tag |
| TCSPC | Time-correlated single-photon counting |
References
- Datta, R.; Heaster, T.M.; Sharick, J.T.; Gillette, A.A.; Skala, M.C. Fluorescence Lifetime Imaging Microscopy: Fundamentals and Advances in Instrumentation, Analysis, and Applications. J. Biomed. Opt. 2020, 25, 1. [Google Scholar] [CrossRef]
- Frei, M.S.; Koch, B.; Hiblot, J.; Johnsson, K. Live-Cell Fluorescence Lifetime Multiplexing Using Synthetic Fluorescent Probes. ACS Chem. Biol. 2022, 17, 1321–1327. [Google Scholar] [CrossRef]
- Starling, T.; Carlon-Andres, I.; Iliopoulou, M.; Kraemer, B.; Loidolt-Krueger, M.; Williamson, D.J.; Padilla-Parra, S. Multicolor Lifetime Imaging and Its Application to HIV-1 Uptake. Nat. Commun. 2023, 14, 4994. [Google Scholar] [CrossRef]
- Frei, M.S.; Tarnawski, M.; Roberti, M.J.; Koch, B.; Hiblot, J.; Johnsson, K. Engineered HaloTag Variants for Fluorescence Lifetime Multiplexing. Nat. Methods 2022, 19, 65–70. [Google Scholar] [CrossRef]
- Niehörster, T.; Löschberger, A.; Gregor, I.; Krämer, B.; Rahn, H.-J.; Patting, M.; Koberling, F.; Enderlein, J.; Sauer, M. Multi-Target Spectrally Resolved Fluorescence Lifetime Imaging Microscopy. Nat. Methods 2016, 13, 257–262. [Google Scholar] [CrossRef]
- Saimi, D.; Chen, Z. Chemical Tags and beyond: Live-cell Protein Labeling Technologies for Modern Optical Imaging. Smart Molecules 2023, 1, e20230002. [Google Scholar] [CrossRef]
- Choi, J.H.; Kim, S.; Kang, O.-Y.; Choi, S.Y.; Hyun, J.Y.; Lee, H.S.; Shin, I. Selective fluorescent labeling of cellular proteins and its biological applications. Chem. Soc. Rev. 2024, 53, 9446–9489. [Google Scholar] [CrossRef]
- Mamontova, A.V.; Shakhov, A.M.; Lukyanov, K.A.; Bogdanov, A.M. Deciphering the Role of Positions 145 and 165 in Fluorescence Lifetime Shortening in the EGFP Variants. Biomolecules 2020, 10, 1547. [Google Scholar] [CrossRef]
- Sarkisyan, K.S.; Goryashchenko, A.S.; Lidsky, P.V.; Gorbachev, D.A.; Bozhanova, N.G.; Gorokhovatsky, A.Y.; Pereverzeva, A.R.; Ryumina, A.P.; Zherdeva, V.V.; Savitsky, A.P.; et al. Green Fluorescent Protein with Anionic Tryptophan-Based Chromophore and Long Fluorescence Lifetime. Biophys. J. 2015, 109, 380–389. [Google Scholar] [CrossRef]
- Mamontova, A.V.; Solovyev, I.D.; Savitsky, A.P.; Shakhov, A.M.; Lukyanov, K.A.; Bogdanov, A.M. Bright GFP with Subnanosecond Fluorescence Lifetime. Sci. Rep. 2018, 8, 13224. [Google Scholar] [CrossRef]
- Goedhart, J.; Von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; Van Weeren, L.; Gadella, T.W.J.; Royant, A. Structure-Guided Evolution of Cyan Fluorescent Proteins towards a Quantum Yield of 93%. Nat. Commun. 2012, 3, 751. [Google Scholar] [CrossRef]
- Heo, M.; Nord, A.L.; Chamousset, D.; Van Rijn, E.; Beaumont, H.J.E.; Pedaci, F. Impact of Fluorescent Protein Fusions on the Bacterial Flagellar Motor. Sci. Rep. 2017, 7, 12583. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, C.; Chen, S.; Ye, F.; Xing, X.-H. Active Inclusion Bodies of Acid Phosphatase PhoC: Aggregation Induced by GFP Fusion and Activities Modulated by Linker Flexibility. Microb. Cell Factories 2013, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Dave, K.; Gelman, H.; Thu, C.T.H.; Guin, D.; Gruebele, M. The Effect of Fluorescent Protein Tags on Phosphoglycerate Kinase Stability Is Nonadditive. J. Phys. Chem. B 2016, 120, 2878–2885. [Google Scholar] [CrossRef]
- Aissa, H.B.; Gautier, A. Engineering Glowing Chemogenetic Hybrids for Spying on Cells. Eur. J. Org. Chem. 2020, 2020, 5637–5646. [Google Scholar] [CrossRef]
- Plamont, M.-A.; Billon-Denis, E.; Maurin, S.; Gauron, C.; Pimenta, F.M.; Specht, C.G.; Shi, J.; Quérard, J.; Pan, B.; Rossignol, J.; et al. Small Fluorescence-Activating and Absorption-Shifting Tag for Tunable Protein Imaging in Vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 497–502. [Google Scholar] [CrossRef]
- Lu, Z.; Peng, B.; Ebert, B.E.; Dumsday, G.; Vickers, C.E. Auxin-Mediated Protein Depletion for Metabolic Engineering in Terpene-Producing Yeast. Nat. Commun. 2021, 12, 1051. [Google Scholar] [CrossRef]
- Streett, H.; Charubin, K.; Papoutsakis, E.T. Anaerobic Fluorescent Reporters for Cell Identification, Microbial Cell Biology and High-Throughput Screening of Microbiota and Genomic Libraries. Curr. Opin. Biotechnol. 2021, 71, 151–163. [Google Scholar] [CrossRef]
- Flaiz, M.; Poehlein, A.; Wilhelm, W.; Mook, A.; Daniel, R.; Dürre, P.; Bengelsdorf, F.R. Refining and Illuminating Acetogenic Eubacterium Strains for Reclassification and Metabolic Engineering. Microb. Cell Factories 2024, 23, 24. [Google Scholar] [CrossRef]
- Tebo, A.G.; Pimenta, F.M.; Zoumpoulaki, M.; Kikuti, C.; Sirkia, H.; Plamont, M.-A.; Houdusse, A.; Gautier, A. Circularly Permuted Fluorogenic Proteins for the Design of Modular Biosensors. ACS Chem. Biol. 2018, 13, 2392–2397. [Google Scholar] [CrossRef]
- Tebo, A.G.; Gautier, A. A Split Fluorescent Reporter with Rapid and Reversible Complementation. Nat. Commun. 2019, 10, 2822. [Google Scholar] [CrossRef] [PubMed]
- Povarova, N.V.; Zaitseva, S.O.; Baleeva, N.S.; Smirnov, A.Y.; Myasnyanko, I.N.; Zagudaylova, M.B.; Bozhanova, N.G.; Gorbachev, D.A.; Malyshevskaya, K.K.; Gavrikov, A.S.; et al. Red-Shifted Substrates for FAST Fluorogen-Activating Protein Based on the GFP-Like Chromophores. Chem. A Eur. J. 2019, 25, 9592–9596. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Gautier, A.; Puchner, E.M. Single-Molecule Localization Microscopy with the Fluorescence-Activating and Absorption-Shifting Tag (FAST) System. ACS Chem. Biol. 2019, 14, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalapathy, M.; Belapurkar, V.; Jose, M.; Gautier, A.; Nair, D. Live Cell Super Resolution Imaging by Radial Fluctuations Using Fluorogen Binding Tags. Nanoscale 2019, 11, 3626–3632. [Google Scholar] [CrossRef]
- Tebo, A.G.; Moeyaert, B.; Thauvin, M.; Carlon-Andres, I.; Böken, D.; Volovitch, M.; Padilla-Parra, S.; Dedecker, P.; Vriz, S.; Gautier, A. Orthogonal Fluorescent Chemogenetic Reporters for Multicolor Imaging. Nat. Chem. Biol. 2021, 17, 30–38. [Google Scholar] [CrossRef]
- Myasnyanko, I.N.; Gavrikov, A.S.; Zaitseva, S.O.; Smirnov, A.Y.; Zaitseva, E.R.; Sokolov, A.I.; Malyshevskaya, K.K.; Baleeva, N.S.; Mishin, A.S.; Baranov, M.S. Color Tuning of Fluorogens for FAST Fluorogen-Activating Protein. Chem. A Eur. J. 2021, 27, 3986–3990. [Google Scholar] [CrossRef]
- Bogdanova, Y.A.; Solovyev, I.D.; Baleeva, N.S.; Myasnyanko, I.N.; Gorshkova, A.A.; Gorbachev, D.A.; Gilvanov, A.R.; Goncharuk, S.A.; Goncharuk, M.V.; Mineev, K.S.; et al. Fluorescence Lifetime Multiplexing with Fluorogen Activating Protein FAST Variants. Commun. Biol. 2024, 7, 799. [Google Scholar] [CrossRef]
- El Hajji, L.; Lam, F.; Avtodeeva, M.; Benaissa, H.; Rampon, C.; Volovitch, M.; Vriz, S.; Gautier, A. Multiplexed In Vivo Imaging with Fluorescence Lifetime-Modulating Tags. Adv. Sci. 2024, 11, e2404354. [Google Scholar] [CrossRef]
- Goncharuk, M.V.; Baleeva, N.S.; Nolde, D.E.; Gavrikov, A.S.; Mishin, A.V.; Mishin, A.S.; Sosorev, A.Y.; Arseniev, A.S.; Goncharuk, S.A.; Borshchevskiy, V.I.; et al. Structure-Based Rational Design of an Enhanced Fluorogen-Activating Protein for Fluorogens Based on GFP Chromophore. Commun. Biol. 2022, 5, 706. [Google Scholar] [CrossRef]
- Weber, E.; Engler, C.; Gruetzner, R.; Werner, S.; Marillonnet, S. A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLoS ONE 2011, 6, e16765. [Google Scholar] [CrossRef]
- Hadjantonakis, A.-K.; Papaioannou, V.E. Dynamic in Vivo Imaging and Cell Tracking Using a Histone Fluorescent Protein Fusion in Mice. BMC Biotechnol. 2004, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Natori, Y.; Sako, Y.; Kuroiwa, H.; Kuroiwa, T.; Umezawa, Y. A Minimal Peptide Sequence That Targets Fluorescent and Functional Proteins into the Mitochondrial Intermembrane Space. ACS Chem. Biol. 2007, 2, 176–186. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In Vitro Measurements | In Cellulo Measurements | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Variant | Kd, µM | ε, M−1·cm−1 | FQY, % | Brightness | τm, ns * | τi, ns * | H2B Fuse τi, ns * | IMS Fuse τi, ns * | Vimentin Fuse τi, ns * |
| FAST (original) | 0.33 ± 0.01 | 27,000 ± 410 | 26 ± 1.4 | 7000 ± 480 | 2.65 | 2.79 | 1.407 ± 0.064 | - | - |
| D65K | 0.25 ± 0.01 | 27,500 ± 410 | 26 ± 2.1 | 7100 ± 680 | 2.53 | 2.70 | 1.342 ± 0.059 | 1.254 ± 0.067 | 1.295 ± 0.043 |
| F62L | 0.55 ± 0.08 | 27,500 ± 410 | 19 ± 0.9 | 5250 ± 330 | 2.28 | 2.44 | 1.009 ± 0.054 | 1.060 ± 0.170 | 0.897 ± 0.046 |
| P68K | 0.22 ± 0.01 | 26,500 ± 400 | 18 ± 0.7 | 4750 ± 260 | 1.94 | 2.13 | 1.114 ± 0.079 | 0.978 ± 0.032 | 1.027 ± 0.051 |
| R52Y | 0.24 ± 0.02 | 29,000 ± 440 | 29 ± 1.5 | 8350 ± 560 | 2.90 | 2.90 | 1.926 ± 0.092 | 1.512 ± 0.072 | 1.591 ± 0.070 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilvanov, A.R.; Myasnyanko, I.N.; Goncharuk, S.A.; Goncharuk, M.V.; Kublitski, V.S.; Bodunova, D.V.; Sidorenko, S.V.; Maksimov, E.G.; Baranov, M.S.; Bogdanova, Y.A. Fluorescence Lifetime Multiplexing with Fluorogen-Activating FAST Protein Variants and Red-Shifted Arylidene–Imidazolone Derivative as Fluorogen. Biosensors 2025, 15, 274. https://doi.org/10.3390/bios15050274
Gilvanov AR, Myasnyanko IN, Goncharuk SA, Goncharuk MV, Kublitski VS, Bodunova DV, Sidorenko SV, Maksimov EG, Baranov MS, Bogdanova YA. Fluorescence Lifetime Multiplexing with Fluorogen-Activating FAST Protein Variants and Red-Shifted Arylidene–Imidazolone Derivative as Fluorogen. Biosensors. 2025; 15(5):274. https://doi.org/10.3390/bios15050274
Chicago/Turabian StyleGilvanov, Aidar R., Ivan N. Myasnyanko, Sergey A. Goncharuk, Marina V. Goncharuk, Vadim S. Kublitski, Daria V. Bodunova, Svetlana V. Sidorenko, Eugene G. Maksimov, Mikhail S. Baranov, and Yulia A. Bogdanova. 2025. "Fluorescence Lifetime Multiplexing with Fluorogen-Activating FAST Protein Variants and Red-Shifted Arylidene–Imidazolone Derivative as Fluorogen" Biosensors 15, no. 5: 274. https://doi.org/10.3390/bios15050274
APA StyleGilvanov, A. R., Myasnyanko, I. N., Goncharuk, S. A., Goncharuk, M. V., Kublitski, V. S., Bodunova, D. V., Sidorenko, S. V., Maksimov, E. G., Baranov, M. S., & Bogdanova, Y. A. (2025). Fluorescence Lifetime Multiplexing with Fluorogen-Activating FAST Protein Variants and Red-Shifted Arylidene–Imidazolone Derivative as Fluorogen. Biosensors, 15(5), 274. https://doi.org/10.3390/bios15050274