1. Introduction
Histamine receptors belong to the aminergic family of Gprotein-coupled receptors (GPCRs), which includes the serotonin, muscarinic, acetylcholine, adrenergic, and dopamine receptors. Histamine receptors are involved in a variety of cellular processes, from allergies and gastric acid secretion to neurotransmission and immunomodulation [
1]. There are four histamine receptor subtypes. Histamine receptor 1, HR
H1, is involved in allergic responses and has been extensively characterized both structurally [
2,
3] and experimentally [
4]. Histamine receptor 2, HR
H2, is involved in gastric acid secretion and gastroesophageal reflux diseases (GERD) [
5]. Histamine receptor 3, HR
H3, is expressed in the central nervous system and participates in neuronal histamine turnover as well as the modulation of the release of other neurotransmitters, such as dopamine and serotonin [
6]. Histamine receptor 4, HR
H4, is expressed in peripheral immune cells and is involved in immunomodulation [
7].
Over the counter HR
H2 blockers, such as cimetidine (Tagamet
®) and famotidine (Pepcid
®), are used to reduce gastric acid secretion and treat peptic ulcers and acid reflux. The chemical structure of some of these blockers, such as ranitidine (Zantac
®), contains a tertiary amine that decomposes into the human carcinogen N-nitrosodimethylamine (NDMA) [
8], which has led to their recall from the market [
9]. Recently, the 8-hydroxyquinoline (8HQ)-containing compounds, chlorquinaldol and chloroxine, have been shown to block HR
H2 activity in mammalian cells [
10]. Importantly, 8HQ-based blockers lack the tertiary ammine that decomposes into NDMA, making them a promising starting point for next-generation HR
H2 therapeutics.
Interestingly, 8HQ-based blockers lack the positively charged amine commonly present in HRH2 blockers. Limited mutagenesis studies of the HRH2 binding site hamper our ability to understand the mode of action of 8HQ-based blockers. Here, we leverage an HRH2-based sensor in yeast to evaluate the role of key residues in the HRH2 active site on histamine and 8HQ-based blocker binding. First, using molecular docking, we identify the HRH2 residues involved in the binding of therapeutically relevant histamine tautomers and the 8HQ-based blockers. Then, we computationally and experimentally perform an alanine scanning of the HRH2 binding site and find that only HRH2:D186A and HRH2:T190A have residual activity in the presence of histamine. Finally, we determine that 8HQ-based blockers can still inactivate HRH2:D186A by swapping their interaction from D98 to Y250 in the case of chlorquinaldol and D186 to Y182 in the case of chloroxine. The internal stabilization of chlorinated 8HQ-based blockers, where the proton in the hydroxy group interacts with the nitrogen lone pair, is pivotal for interactions with tyrosine. Taken together, this work expands our understanding of histamine and 8HQ-blocker binding to HRH2, and provides evidence that GPCR-based sensors in yeast have utility in elucidating the mode of action of novel ligands for GPCRs.
2. Materials and Methods
Materials. Luciferase expression was assayed using the NanoGlo
® Luciferase Assay System (Promega N1120). Histamine dihydrochloride (Sigma H7250), chlorquinaldol (Selleck S4192), and chloroxine (Selleck S1839) were purchased from the specified vendors. The HR
H2 point mutants (HR
H2: D98A, HR
H2: Y182A, HR
H2: D186A, HR
H2: T190A, HR
H2: Y250A, and HR
H2: F254A) were codon optimized for
Saccharomyces cerevisiae and commercially synthesized. Tables of plasmids (
Supplementary Table S1), strains (
Supplementary Table S2), and primers (
Supplementary Table S3), as well as HR
H2 mutant sequences can be found in the
supplementary materials.
Docking of 8HQ-based blockers to HRH2 wild type and mutants. Histamine (ZINC388081), chlorquinaldol (ZINC119403), and chloroxine (ZINC1131) [
11] were docked to the AlphaFold structure of HR
H2 (Alphafold: P25021) [
12,
13]. Computational alanine scanning of HR
H2 was conducted using the PyMOL mutagenesis wizard, with P25021 as the template. Hydrogens were introduced to the ligands using CACTUS (
https://cactus.nci.nih.gov/translate/ (accessed on 27 February 2023). The HR
H2 binding site was defined by D98, D186, and T190 [
14]. Ligands were docked using AutoDock 4.2.6. Results were visualized in AutoDockTools 1.5.7 [
15] and PyMOL.
HRH2 mutant construction. HR
H2:D98A, HR
H2:Y182A, HR
H2:D186A, HR
H2:T190A, HR
H2:Y250A, and HR
H2:F254A were cloned into pESC-HIS3-P
TEF-P
ADH [
16] between
BamHI/
SacII via Gibson assembly to generate pESC-HIS3-P
TEF-HR
H2_D98A (pPM43), pESC-HIS3-P
TEF-HR
H2_Y182A (pPM50), pESC-HIS3-P
TEF-HR
H2_D186A (pPM52), pESC-HIS3-P
TEF-HR
H2_T190A (pPM53), pESC-HIS3-P
TEF-HR
H2_Y250A (pPM54), and pESC-HIS3-P
TEF-HR
H2_F254A (pPM49), respectively. Constructs were sequence-verified using primers LT62/LT63.
HRH2 mutant-based yeast sensors. PPY140 (
S. cerevisiae W303 Δfar1, Δste2, Δsst2) [
16] was co-transformed with pRS415-Leu2-P
FIG1-NanoLuc [
17] and either pPM43, pPM50, pPM52, pPM53, pPM54, or pPM49 to generate PPY2370, PPY2393, PPY2390, PPY2391, PPY2394, and PPY2392, respectively. To generate the no-receptor control, PPY140 was co-transformed with pRS415-Leu2-P
FIG1-NanoLuc and pESC-HIS3-P
TEF-P
ADH (PPY1809).
Histamine sensing. An overnight culture of PPY2370, PPY2393, PPY2390, PPY2391, PPY2394, or PPY2392 was used to inoculate 50 mL of synthetic complete medium with 2% glucose lacking histidine and leucine (SD(HL−)) to an OD600 = 0.06. After 18 h at 15 °C (150 rpm), the cultures were centrifuged (3500 rpm, 10 min), and resuspended in SD(HL−) to an OD600 = 1. In a white, flat-bottomed, 96-well plate, 190 μL pH = 7 SD (HL−), 8 μL of cells, and 2 μL of histamine (final concentration 10−2–104 μM), or DMSO as a control were added. After chemical incubation (2.5 h, 30 °C, 250 rpm), 20 μL of 1:100 mixture of NanoLuc substrate to NanoLuc buffer were added, and the reaction was incubated for 30 min (30 °C, 250 rpm). Luminescence was read in a Biotek Synergy 2 using the default settings.
HRH2 blocker sensing. The histamine sensing protocol was followed except as described. In a white, flat-bottomed, 96-well plate, 188 µL pH = 7 SD (HL−), 8 µL of cells, 2 µL of histamine (1 mM final concentration), and 2 µL chlorquinaldol or chloroxine (final concentration 10−3–10 μM) were added. For the no chemical control, no histamine or blocker was added, only 4 µL of DMSO. The no-receptor control strain was tested under the same conditions as the HRH2 mutant sensor strains.
3. Results and Discussion
Molecular docking of HRH2 to histamine. The topological view of HR
H2 highlighting its active site is seen in
Figure 1A. Early on, it was proposed that histamine’s positively charged amine interacts with D98, and that the imidazole ring tautomerizes to interact with both D186 and T190 [
18]. Experimental studies in mammalian cells confirmed the necessity for D98, as the activation of HR
H2:D98A with histamine resulted in no cAMP accumulation [
14]. Positions 190 and 186 do not seem as critical to histamine binding, as the activation of HR
H2:D186A and HR
H2:T190A with histamine resulted in 50% and 17% of wild-type HR
H2 activation, respectively [
14].
To understand the binding mode of histamine to HR
H2, we docked histamine to the AlphaFold structure of HR
H2. Notably, although the Cryo-EM structure of HR
H2 has been recently elucidated (PDB: 7UL3) [
19], it is in the inactive state. Additionally, although the 3 Å resolution provides a good overall view of the receptor, the sidechain location is not precise.
At pH 7.4, histamine is protonated and present in four tautomer states. The trans tautomers are the predominant form, with the gauche tautomers representing 25% of the population [
20]. As shown in
Figure 1B, the histamine trans tautomer 1 (TT1) makes two electrostatic interactions with D98 via the charged amine and with Y250 via the protonated nitrogen in the imidazole ring. In contrast, histamine trans tautomer 2 (TT2) interacts with D186 and T190 via the charged amine and, similarly to histamine TT1, with Y250 via the protonated nitrogen in the imidazole ring. Both histamine gauche tautomers (GTs) interact with T190 and D186, with GT2 additionally interacting with Y250 via the protonated nitrogen in the imidazole ring. Taken together, between all tautomers, histamine interacts with both sides of the binding pocket, the one described by D98/Y250 and the one described by D186/T190. Within the major tautomers, histamine TT1 engages D98 in transmembrane 2 and Y250 in transmembrane 6 (TM6) while histamine TT2 engages D186/T190 in transmembrane 5 and Y250 in TM6.
Molecular docking of 8HQ-based blockers to HRH2. Generally, HR
H2 blockers carry a positively charged amine that interacts with D98 and are long enough to interact with both ends of the HR
H2 binding site. Such is the case for famotidine, whose sulfone end interacts with D98 and Y250 and guanidine group interacts with T190 and D186 [
19] (
Figure 1C). As shown in
Figure 1D, 8HQ-based blockers interact with only one side of the HR
H2 binding pocket. Chlorquinaldol interacts with D98 and Y250 via the hydroxyl group of the hydroxyquinoline ring (
Figure 1D). Chloroxine interacts with D186 with the same moiety (
Figure 1E).
Virtual alanine scanning of the HRH2 binding site and its effect on histamine binding. We computationally determined residues within 5 Å of histamine—D98 (D
3×32), Y182 (Y
5×39), D186 (D
5×3), T190 (T
5×461), Y250 (Y
6×51), and F254 (F
6×55)—and mutated them to alanine. As shown in
Figure 2, the mutation of these residues to alanine significantly changes the interaction between the different histamine tautomers and HR
H2.
None of the histamine tautomers interact with any of the HRH2 mutants in a similar fashion to histamine TT1 interacting with wild-type HRH2. Specifically, we do not see the histamine tautomers using two electrostatic interactions via the protonated amine to engage D98 and using the protonated amine in the imidazole ring to interact with Y250. We do observe that in HRH2:D186A all histamine tautomers engage D98 via two electrostatic interactions using the charged amine. The only other instance where D98 is engaged via the charged amine is in HRH2:T190A with histamine GT2. In five HRH2 mutants, D98 is engaged via a single electrostatic interaction with the protonated nitrogen in the imidazole ring, specifically histamine TT2 with HRH2:T190A, HRH2:F254A, and HRH2:Y182A and histamine GT2 with HRH2:F254A and HRH2:Y182A. Of note, in no HRH2 mutant are histamine tautomers able to interact with Y250 via the protonated nitrogen in the imidazole ring. Taken together, the engagement of D98 is possible in some of the single-point mutants, specifically HRH2:D186A > HRH2:T190A > HRH2:F254A = HRH2:Y182A. In none of the HRH2 mutants do the histamine tautomers engage Y250 via the protonated amine, as seen in histamine TT1 with wild-type HRH2.
Experimental alanine scanning of the HRH2 active site and its impact on histamine-driven activation. We constructed the six HR
H2 alanine mutants—HR
H2:D98A, HR
H2:D186A, HR
H2:T190A, HR
H2:F254A, HR
H2:Y182A, HR
H2:Y250A—and expressed them in a previously developed GPCR-based sensor strain that links GPCR activation to cell luminescence (
Figure 3A) [
17].
As shown in
Figure 3B, all mutants, except for HR
H2:D186A and HR
H2:T190A, lost the ability to be activated by histamine. These results are consistent with previous experimental work in mammalian cells [
14]. Specifically, HR
H2:D186A retained 50% of HR
H2 wild-type activity in the presence of histamine. This is consistent with the computationally predicted engagement of all histamine tautomers with D98 with the protonated amine (
Figure 2). The 50% reduction in signal after activation could be attributed to the lack of proper engagement of Y250 via the protonated nitrogen in the imidazole ring. HR
H2:T190A resulted in an 83% decrease in histamine activation in the yeast system. This result is consistent with the computationally predicted engagement of D98 by histamine GT2 via the protonated amine and by histamine TT2 via the protonated imidazole ring (
Figure 2). The additional reduction in signal observed in HR
H2:T190A vs. HR
H2:D186A could be attributed to the lack of proper engagement of Y250 and the reduced number of histamine tautomers that could engage D98 via double electrostatic interactions with the protonated amine. Of note, in mammalian cells, HR
H2:T190A results in a 50% reduction in cAMP accumulation when compared to wild-type HR
H2 [
14]. The difference could be attributed to the signaling strength of HR
H2 in mammalian versus yeast cells.
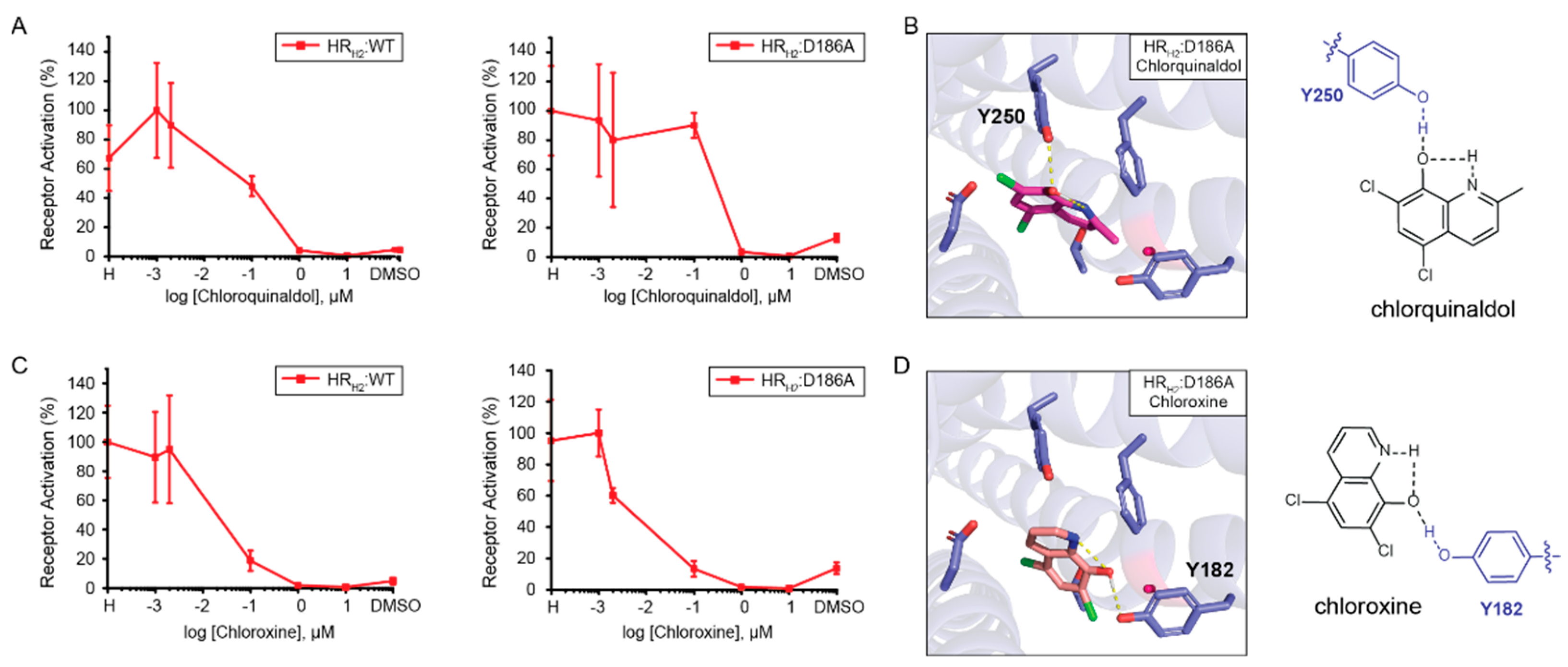
Assessing the role of position D186 in 8HQ-based blocker binding. Given that HR
H2:D186A results in a measurable increase in signal after activation in the presence of histamine, we assessed the role of position D186 in the binding of 8HQ-based blockers. As shown in
Figure 4A, both chlorquinaldol and chloroxine block the signal after the activation of wild-type HR
H2 and HR
H2:D186A in the presence of histamine in a similar fashion. Docking of chlorquinaldol to HR
H2:D186A shows chlorquinaldol intramolecular stabilization and interaction with Y250 (
Figure 4B). In chlorquinaldol, the proton in the hydroxy group is highly acidic due to the two electron withdrawing chlorines on the phenyl ring. This makes the proton likely to interact with the nitrogen lone pair in the pyridine ring. The oxygen in the hydroxy group is also able to interact with the proton of the hydroxyl group of Y250. Docking of chloroxine to HR
H2:D186A reveals a similar strategy, with chloroxine being internally stabilized and interacting with Y182 (
Figure 4D). In conclusion, the intramolecular stabilization of 8HQ-based blockers is pivotal in enabling its interaction with the tyrosine residues in HR
H2:D186A. Importantly, 8HQ-based blockers do not need to interact with D98 to inactivate the receptor. Chlorquinaldol interaction with Y250 is sufficient to inactivate HR
H2:D186A, and chloroxine interaction with D186 is exchanged for an interaction with Y182 in order to inactivate the receptor.
4. Conclusions
GPCRs are targeted by 30% of FDA-approved drugs [
21]. GPCR-based sensors in yeast that link GPCR activation to reporter gene transcription can be used to gain valuable insight into the structure–activity relationship of human GPCRs. In this work, we use a HR
H2-based sensor to elucidate the extent to which residues D98, D186, F254, Y182, and Y250 are necessary for HR
H2 activation with histamine. We find residual activation in HR
H2:D186A and HR
H2:T190A, which docking studies suggest is due to the engagement of D98 via the protonated amine by some histamine tautomers. Importantly, the 50% reduction in signal activation seen in HR
H2:D186A can be attributed to a lack of engagement of Y250. With respect to 8HQ-based blockers, we find that they engage only one end of the HR
H2 binding site, either the end described by D98/Y250 or T190/D186. In blocking HR
H2:D186A, intramolecular stabilization of the 8HQ-based blockers aids in the interaction with Y250 (chlorquinaldol) or Y182 (chloroxine). The results described in this work should aid in our understanding of the mode of action of 8HQ-based blockers and pave the way for the development of improved HR
H2 blockers. More generally, this work demonstrates that GPCR-based sensors in yeast are valuable in illuminating the mode of action of novel GPCR ligands.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}



