2.1. Materials
The following reagents were purchased from Merck Life Science S.r.l. (Milan, Italy): methanol, acetonitrile, oxalic acid, ethanolamine, glycine, dithiothreitol (DTT), sulfanilic acid, hydrochloric acid, ammonium hydroxide, ammonium sulphate and all powders for buffer solutions.
The following microbeads were used: Dynabeads M-270 Epoxy (DynaEpoxy) and Dynabeads M-270 Streptavidin (DynaSA) from Thermo Scientific (Waltham, MA, USA); MAGAR-cN (MagarNA) from Immagina Biotechnology (Trento, Italy); PureCube NHS-Activated MagBeads (PureCubeNHS) and PureCube Maleimide-Activated MagBeads (PureCube Maleimide) from Cube Biotech (Monheim am Rhein, Germany).
All aptamers were obtained from IDT Integrated DNA Technologies (Leuven, Belgium). Aptamers are listed in
Table 1 and
Table 2.
Anti-antibiotics antibodies were obtained from Fitzgerald (Fitzgerald Industries International; Acton, MA, USA): sheep polyclonal anti-tetracycline antibody (a-TC Ab); sheep polyclonal anti-chloramphenicol antibody (a-CAF Ab); and rabbit polyclonal anti-sulfamethazine antibody (a-SMZ Ab). The fluorescent antibody Ab-A568 (rabbit anti-goat IgG (H+L), Cross-Adsorbed Secondary Antibody, AlexaFluor568 and protein G were purchased from Thermo Scientific (Waltham, MA, USA).
The following buffers were used:
Binding Buffer (BB; used for TC and SMZ): 100 mM NaCl, 2 mM MgCl2, 5 mM KCl, 1 mM CaCl2, 20 mM Tris/HCl pH 7.6
Binding Buffer Muller (BBM): 100 mM NaCl, 10 mM MgCl2, 20 mM potassium phosphate buffer pH 7.5
Binding and Washing Buffer (B&W): 1 M NaCl, 0.5 mM EDTA, 5 mM Tris/HCl pH 7.5
Coupling Buffer I (CBI): 150 mM NaCl, 100 mM sodium phosphate pH 7.4
Phosphate Buffered Saline (PBS): 138 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate pH 7.4
Binding Buffer SDM (BBSDM): 50 mM NaCl, 5 mM KCl, 5 mM MgCl2, 20 mM Tris/HCl pH 8
The Istituto Zooprofilattico Sperimentale del Piemonte, Liguria e Valle d’Aosta (IZSPLV), provided raw milk and honey, all tested for the absence of antibiotics. IZSPLV also provided pure tetracycline, sulfonamides and chloramphenicol for spiking solutions.
Microfluidic chips, with four chambers of 100 µL volume each (Rhombic Chamber Chip eP1, Fluidic Design 221, PMMA, 600 µm depth), were purchased from microfluidic ChipShop GmbH (Jena, Germany).
2.2. Interaction of Antibiotics with Their Aptamers or Antibodies
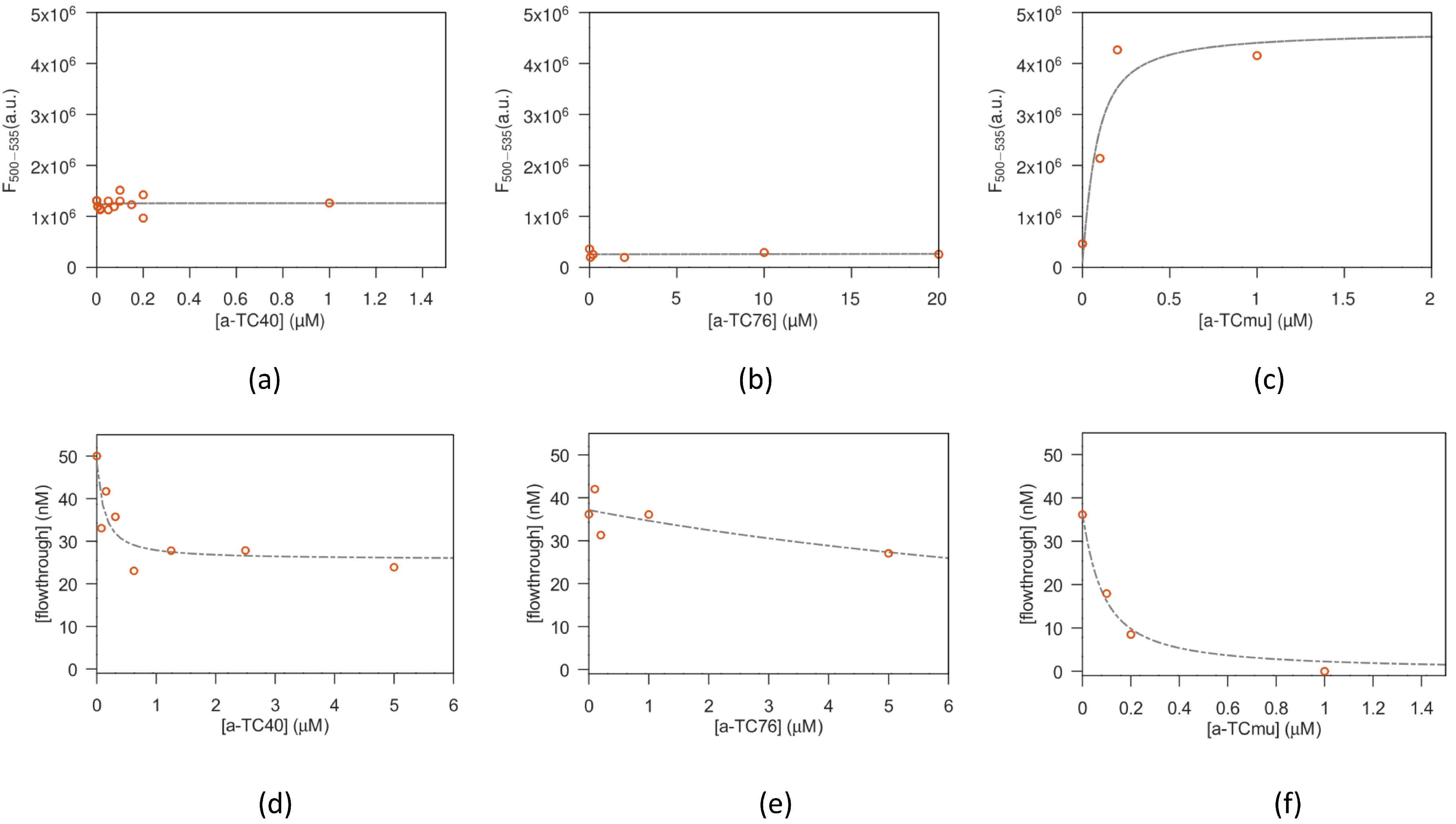
The specific recognition of aptamers and antibodies (in this section generally designed as macromolecules) with their targets was evaluated in solution, using two different methods: the effect of binding on the intrinsic fluorescence of tetracycline was exploited for testing the a-TC aptamers [
17] (
Section 2.2.1), while an equilibrium filtration method [
27,
28] was utilized for all aptamers (apart from a-TC8, which is too small for this method), and for all the antibodies (
Section 2.2.2).
2.2.1. Spectrofluorimetric Analysis
Different concentrations of TC were tested for their intrinsic fluorescence signal in order to optimize the concentration to be used as starting amount. Therefore, TC was dissolved at fixed nanomolar concentration in a total volume of 1 mL of buffer 100 mM NaCl, 2 mM MgCl2, 5 mM KCl, 1 mM CaCl2, 20 mM sodium phosphate pH 7.6 for a-TC8, a-TC40 and a-TC76 or BBM for a-TCmu and specific aptamers were added at increasing concentrations. The aptamers were titrated in such a way as to not exceed a total volume increase of 5%. The solution was stirred during each titration step and allowed to equilibrate for 5 min before data collection (longer incubation time were also tested, without significant differences in results). Fluorescence spectra were acquired for each titration point with a SPEX FluorMax spectrofluorimeter (Horiba Instruments Inc., Edison, NJ, USA) at 25 °C. An excitation wavelength of 370 nm was used to acquire the emission spectrum from 380 to 610 nm. The fluorescence emission signal from 500 to 535 nm was then integrated and plotted against the aptamer concentration.
2.2.2. Equilibrium Filtration Method
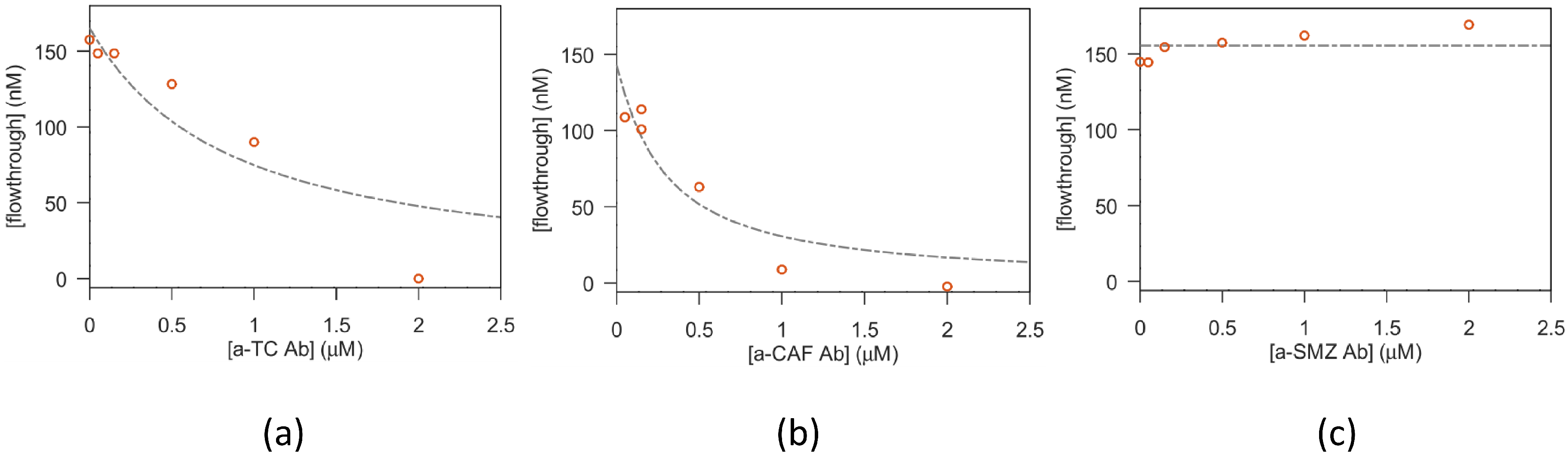
Vials with 100 µL solutions of macromolecules:antibiotic (1:1 volumes) were prepared at a fixed antibiotic concentration and increasing macromolecule concentration and left to reach equilibrium for 30 min. Afterwards, the solutions were transferred in the upper compartment of filters with a nominal cutoff of 10.000 Dalton (Microcon-10kDa Centrifugal Filter Unit with Ultracel-10 membrane, Merck Life Science S.r.l.; Milan, Italy), or 3.000 Dalton (Amicon Ultra-0.5 Centrifugal Filter Unit, Ultracel-3, Merck Life Science S.r.l.; Milan, Italy), depending on the MW of the tested aptamer. Then, the solutions were centrifuged for 8 min at 12.000× g, allowing around 50–60 µL of solution to flow to the lower filter compartment. The antibiotic concentration found in the flow through is a good estimate of the free antibiotic present in the original reaction vial. On the other hand, it is expected that the antibiotic bound to the aptamer is (mostly, see below for details) confined in the upper filter compartment.
2.2.3. Fit of the Antibiotic-Macromolecule Titration
The amount of bound antibiotic present in the solutions prepared as described above, was estimated from the saturation parameter
, i.e., defined as the moles of bound ligand (antibiotic) per mole of macromolecule (aptamer or antibody), expressed using the equation from Cantor and Schimmel Biophysical chemistry (section 15-3, [
29]). The parameters obtained from this equation were used to build two different models for TC fluorescence and equilibrium filtration experiments. The fitting of experimental data was implemented in Octave v. 6.1.0 [
30], with the optim package v.1.6.0, using TeXmacs v. 2.1.1 [
31] as the graphical front-end. The detailed description of the fitting method as well as the definition of the fitting parameters is detailed in the
Supplementary Materials file. In brief, the equilibrium constant
k is obtained in both models, while the fluorescence yields for the free (
) and bound (
) TC characterize the fit of fluorescence experiments.
Concerning the fit of equilibrium filtration experiments, the fitted parameters are (alongside k) the filter throughput losses and the aspecific antibiotic adhesion ( —for TC).
2.4. Characterization of the Functionalized Microbeads
The binding of aptamers or antibodies to the beads was monitored by binding a fluorescent aptamer or antibody to the beads and checking the resulting fluorescence via confocal microscopy. Moreover, all the solutions of aptamer/antibody used for the functionalization of the microbeads were collected and quantified with the spectrophotometer to indirectly estimate the amount of bound aptamer/antibody. When PureCubeNHS beads are functionalized, this spectrophotometric determination of the bound aptamer/antibody is impossible because during the procedure, the NHS group is released, whose absorbance maximum is close to that of aptamers and antibodies.
In addition, the XPS analysis was employed for monitoring the functionalization of beads.
2.4.1. Confocal Analysis
The microbeads functionalized with the fluorescent aptamer or antibody were resuspended in PBS, deposited on a microscope slide and covered with a coverslip. A Leica SP5-II confocal microscope (Leica Instruments, Wetzlar, Germany), equipped with a helium/neon laser (543 nm), was employed for imaging the beads, acquiring data with a 40× objective in air. Images were also acquired using the additional channel of the transmitted light photomultiplier to image all the microbeads, independently of their functionalization. Spectral data of functionalized and untreated microbeads were acquired to confirm the correspondence of the detected light with the fluorescence spectra of the used fluorophores.
2.4.2. XPS Analysis
The XPS analysis is particularly suitable for the chemical characterization of samples carrying biomolecule layers, as already shown [
33]. Samples for XPS measurements were deposited as 15 µL solutions of microbeads resuspended in pure water, deposited on 1 cm × 1 cm substrates of thermally grown silicon oxide, after improving the beads adhesion with argon plasma treatment (10.5 W, 2 mbar, 1 min). Samples were carefully dried at room temperature before their introduction in the chamber. XPS analyses were performed using a Kratos Axis Ultra
instrument equipped with a hemispherical analyzer and a monochromatic AlKα (1486.6 eV) X-ray source, in spectroscopy mode.For beads functionalized with sulfanilic acid, O 1s, C 1s, N 1s, S 2p core lines were acquired, while for beads functionalized with a-TC Ab, O 1s, C 1s, N 1s, and Si 2p core lines were measured. XPS quantification was performed using the instrument sensitivity factors and the high-resolution spectra. Charge compensation was achieved using a charge neutralizer located at the bottom of the electrostatic input lens system. The quantification, reported as a relative elemental percentage, was performed by using the integrated area of the fitted core lines and by correcting for the atomic sensitivity factors. XPS data analysis was performed using the software described in Speranza and Canteri [
34].
2.4.3. Spectrophotometric Analysis
All solutions produced during the functionalization of microbeads as well as the stock solutions added to the beads were collected and measured at the spectrophotometer (Jasco V-550) to check the amount of aptamer/antibody present. Spectra from 190 to 340 nm were acquired and the absorbance at 260 (aptamers) or 280 nm (antibodies) was used to quantify the amount of aptamers (considering A260 ssDNA = 33 µg/mL) and antibodies (considering an approximate IgG = 210,000).
2.5. Binding/Elution Test
The binding of antibiotics to their specific targets, which are bound to microbeads, was studied both in 1.5 mL vials and using fluidic microdevices. The test in vials started from the functionalized microbeads, which are incubated with the antibiotics at fixed concentrations for 1 h at room temperature using a tilting and rotation mixer. At the end of incubation, the vials were mounted on a magnet, the supernatant was collected (unbound antibiotic), and the microbeads were washed for at least three times with the suitable binding buffer. Then, different conditions of ionic strength, pH, and temperature were tested for the elution of the antibiotics. A final washing step was then performed. All fractions were collected for quantification in HPLC.
The binding test performed on the microfluidic chip was adapted from Lunelli et al. [
20] and is schematized in
Figure 1. Briefly, a microfluidic PMMA chip was mounted on the ChipGenie ®edition P device (microfluidic ChipShop, Jena, Germany; in
Figure 1b) and fluids were injected in the chip chamber inlets with two disposable syringes actuated by two syringe pumps (Legato 185, KD Scientific, Holliston, MA, USA; in
Figure 1a). The chip chambers were filled with functionalized magnetic beads at the beginning of the experiment (typically 30 µL diluted in 350 µL of suitable buffer), and the solutions containing known amounts of antibiotic were loaded in the microdevice (step in
Figure 1c). The antibiotic is captured on the beads’ surfaces, while all the unwanted materials present in the raw matrix are washed away with the buffer. The antibiotic is then released with the proper elution condition (high ionic strength solutions, extreme pH, and temperature). Finally, a washing step with buffer is performed. All fractions were collected from the outlet for the quantification via HPLC (step in
Figure 1d). Concerning the milk experiment, no preliminary treatments or dilution in the binding buffer was performed before the spiking of TC. Honey instead required a preliminary dilution of 5 times in the binding buffer.
2.6. Quantification of Antibiotic Solutions
The antibiotic present in the fractions collected during the binding/elution test (both in 1.5 mL vial and microdevice) was quantified by means of a Shimadzu (Kyoto, Japan) HPLC instrument. The instrument was equipped with the UV/Vis Detector SPD-20A, the Shimadzu prominence Communications Bus Module CBM-20A, the binary pump system Liquid Chromatograph LC- 20AB and with a degasser (Waters In-Line Degasser) to improve HPLC performance. Fractions, after centrifugation at 21,000× g for 10 min, were manually injected through a Rheodyne® model 7725i injector connected with a 50 µL loop and passed through a precolumn Security Guard by Phenomenex® before separation by means of an inverse phase column (Luna® C18 by Phenomenex®, 5 µm, 10 nm, 250 mm × 3 mm), acquiring data with the software provided with the instrument (LCSolution). The mobile phase for tetracycline was oxalic acid 0.01 M, pH 2.7 (solvent A), and the gradient was obtained with methanol and acetonitrile in a 1.5:1 ratio (solvent B). Flux was set to 0.35 mL/min. The separation method was the following: 10% solvent B for 5 min, and gradient up to 58% of solvent B in 20 min. For the quantification of sulfonamides, 0.05 M sodium acetate pH 4.8 was used as a mobile phase (solvent A) and pure acetonitrile as solvent B. The separation method was gradient from 20% of solvent B to 50% B in 23 min, and 1 min up to 70% B. For the quantification of chloramphenicol, 0.1% formic acid in water was used as solvent A and 0.1% formic acid in acetonitrile as solvent B, while the separation method was 20% of solvent B for 5 min, gradient up to 95% B in 15 min. A calibration curve was obtained for each antibiotic by injecting known amounts of antibiotic in HPLC; the fit of this curve allowed the precise quantification of the antibiotic present in each fraction.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}