A Microfluidic Cell Co-Culture Chip for the Monitoring of Interactions between Macrophages and Fibroblasts

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Construction of Fluorescent Cells
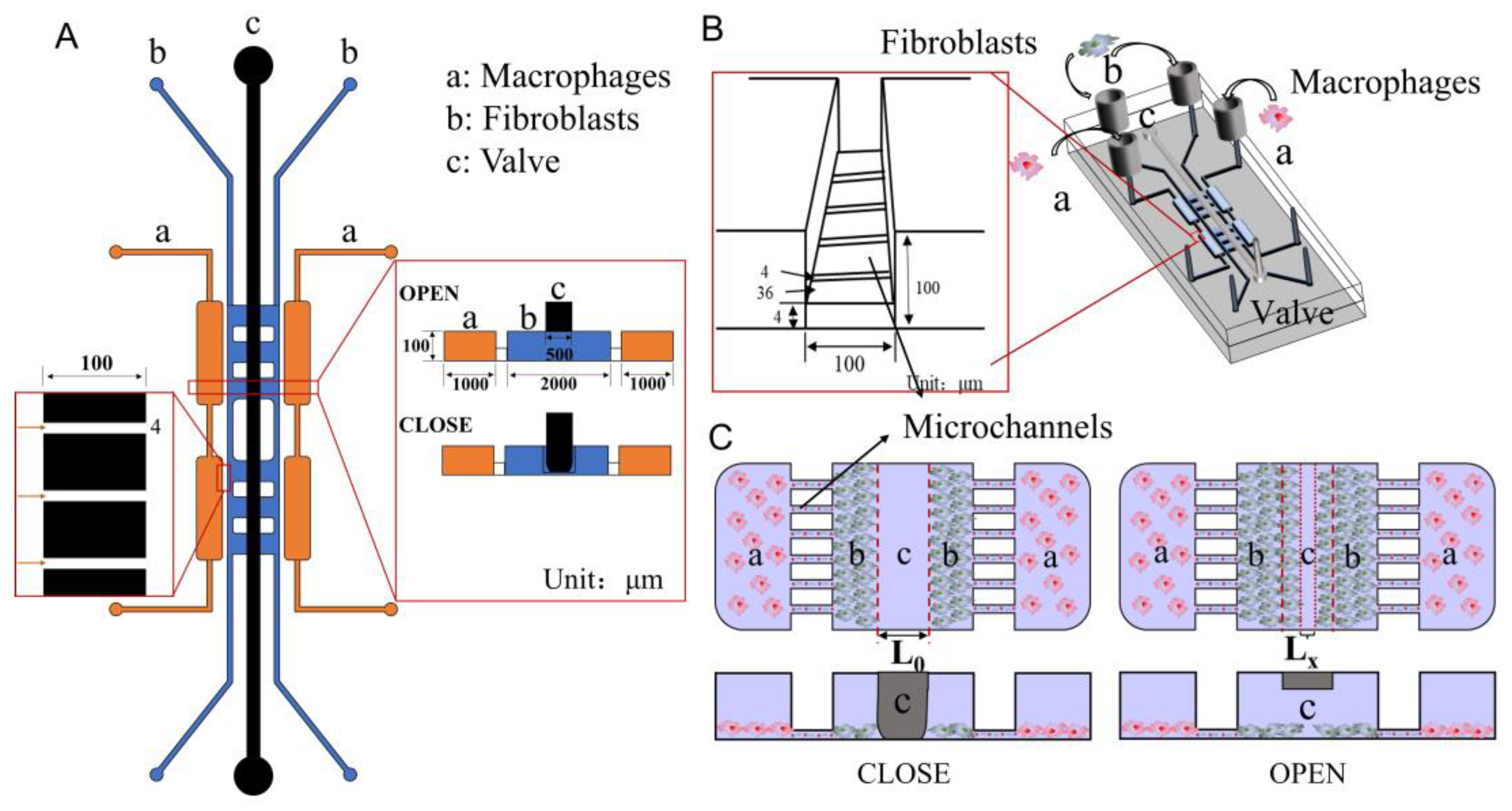
2.3. Fabrication of the Microfluidic Chip
2.4. Immunofluorescence Staining and Imaging
2.5. Scratch Assay
2.6. Statistical Analysis
3. Results and Discussion
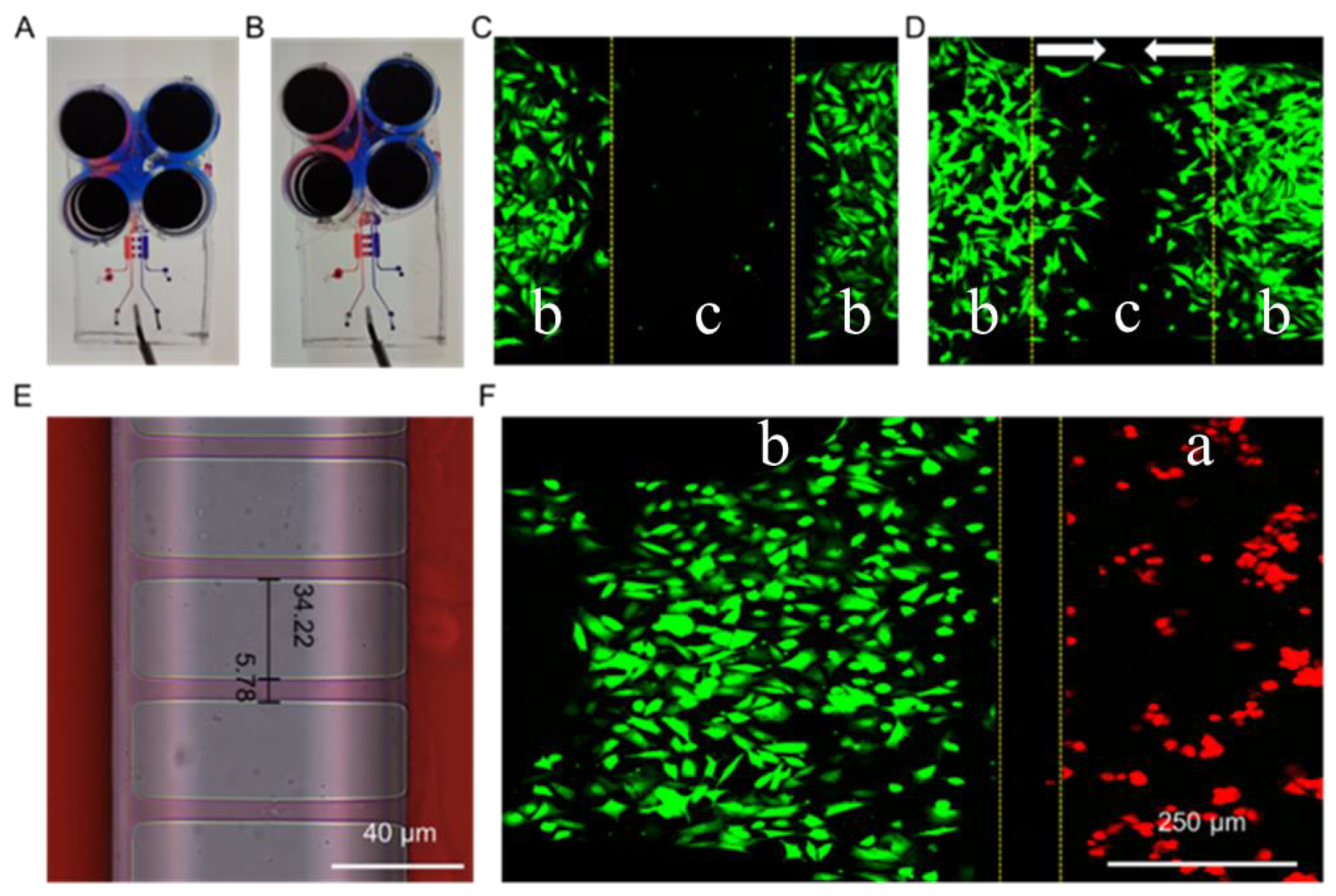
3.1. Design, Fabrication, and Characterization of the Microfluidic Chip
3.2. Culture of Macrophages and Fibroblasts in the Microfluidic Chip
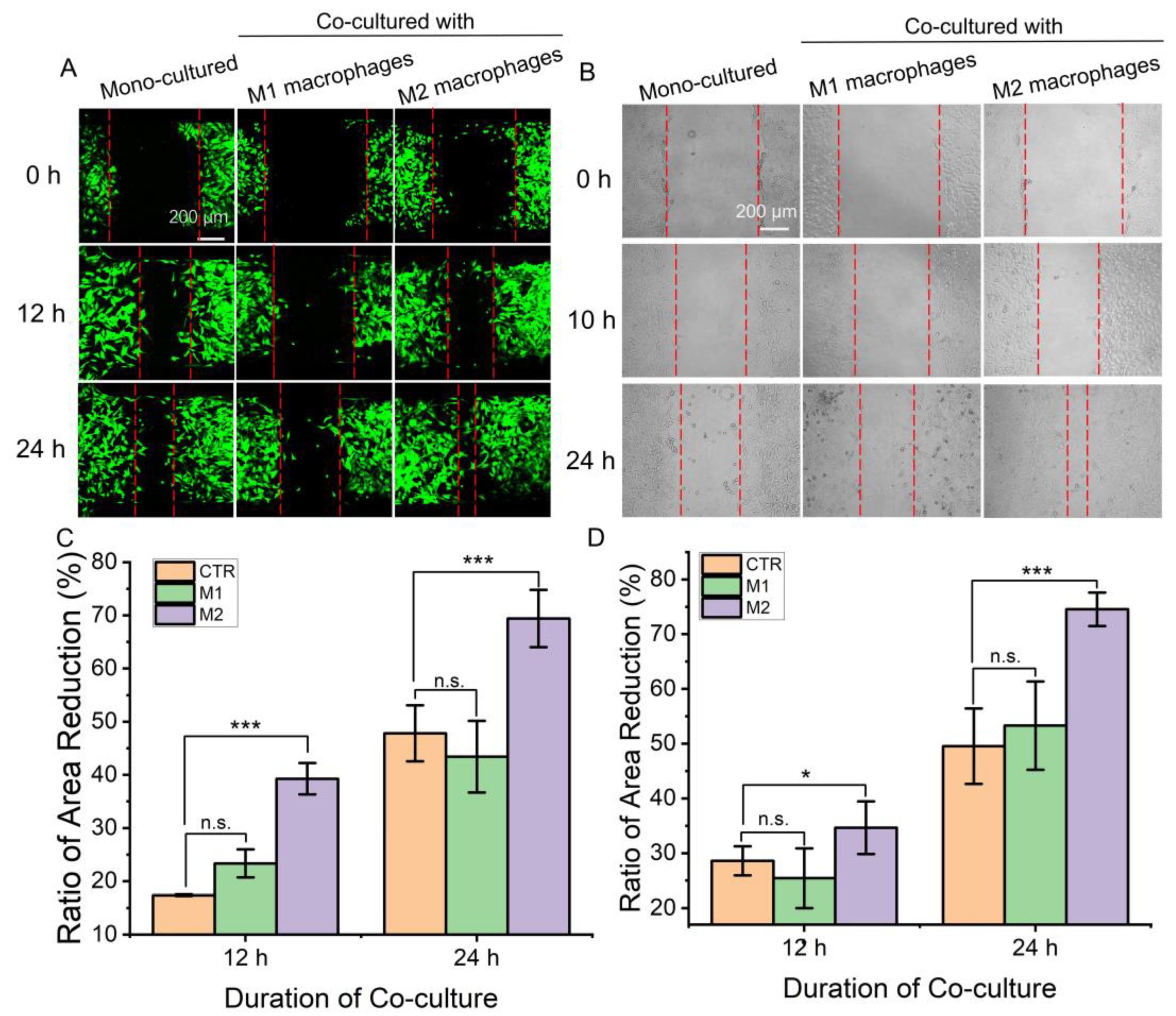
3.3. Effect of Macrophages on the Immigration of Fibroblasts
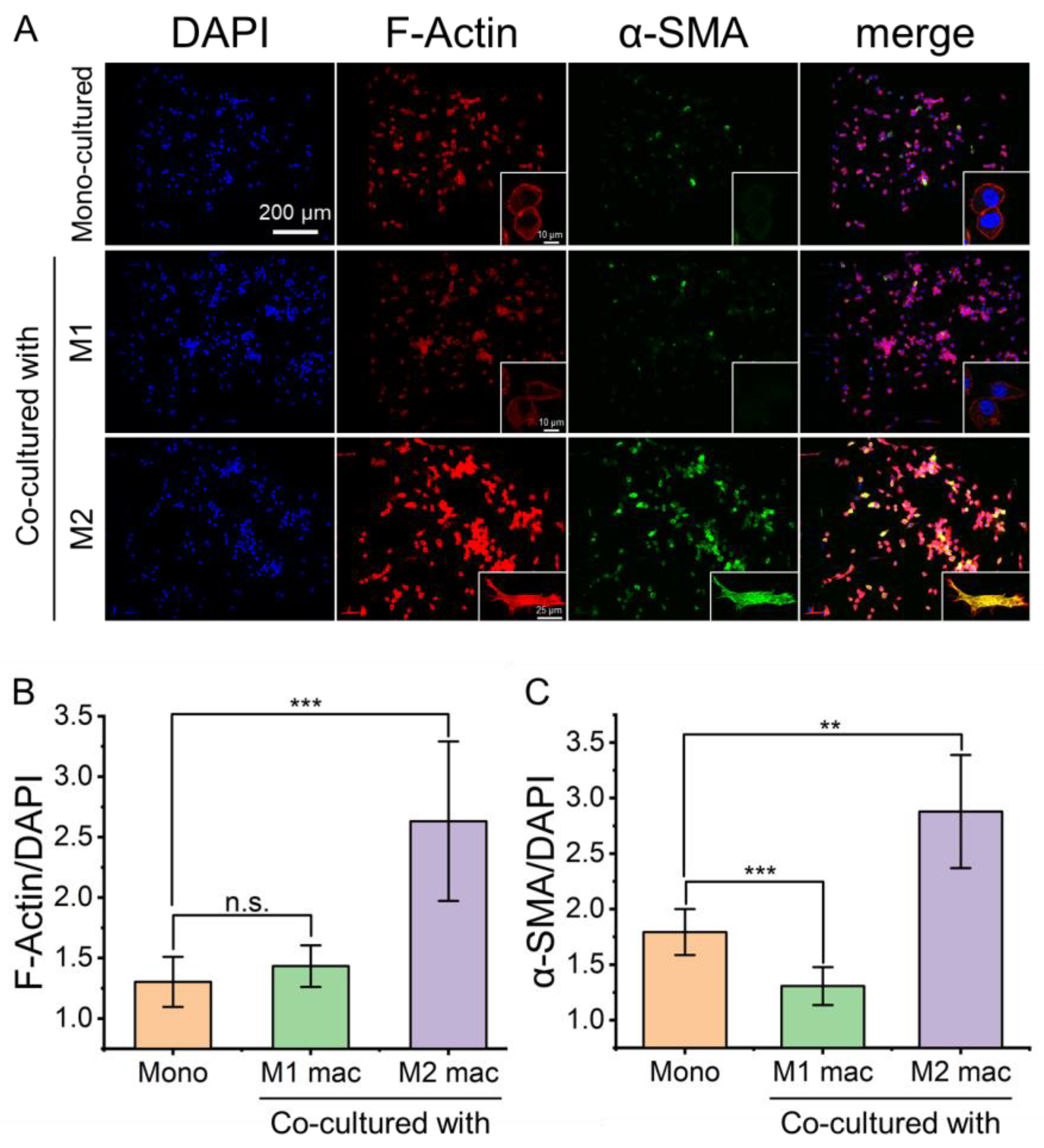
3.4. Effect of Macrophages on Activation of Fibroblasts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Sonnemann, K.J.; Bement, W.M. Wound repair: Toward understanding and integration of single-cell and multicellular wound responses. Annu. Rev. Cell Dev. Biol. 2011, 27, 237–263. [Google Scholar] [CrossRef] [PubMed]
- Song, J.W.; Nam, H.S.; Ahn, J.W.; Park, H.S.; Kang, D.O.; Kim, H.J.; Kim, Y.H.; Han, J.; Choi, J.Y.; Lee, S.Y.; et al. Macrophage targeted theranostic strategy for accurate detection and rapid stabilization of the inflamed high-risk plaque. Theranostics 2021, 11, 8874–8893. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Cánoves, P.; Serrano, A.L. Macrophages decide between regeneration and fibrosis in muscle. Trends Endocrinol. Metab. 2015, 26, 449–450. [Google Scholar] [CrossRef]
- Plikus, M.V.; Guerrero-Juarez, C.F.; Ito, M.; Li, Y.R.; Dedhia, P.H.; Zheng, Y.; Shao, M.; Gay, D.L.; Ramos, R.; Hsi, T.C.; et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 2017, 355, 748–752. [Google Scholar] [CrossRef]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef]
- Rinkevich, Y.; Walmsley, G.G.; Hu, M.S.; Maan, Z.N.; Newman, A.M.; Drukker, M.; Januszyk, M.; Krampitz, G.W.; Gurtner, G.C.; Lorenz, H.P.; et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 2015, 348, aaa2151. [Google Scholar] [CrossRef]
- Aurora, A.B.; Olson, E.N. Immune modulation of stem cells and regeneration. Cell Stem Cell 2014, 15, 14–25. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Lucas, T.; Waisman, A.; Ranjan, R.; Roes, J.; Krieg, T.; Müller, W.; Roers, A.; Eming, S.A. Differential roles of macrophages in diverse phases of skin repair. J. Immunol. 2010, 184, 3964–3977. [Google Scholar] [CrossRef] [PubMed]
- Lechner, A.J.; Driver, I.H.; Lee, J.; Conroy, C.M.; Nagle, A.; Locksley, R.M.; Rock, J.R. Recruited Monocytes and Type 2 Immunity Promote Lung Regeneration following Pneumonectomy. Cell Stem Cell 2017, 21, 120–134.e127. [Google Scholar] [CrossRef] [PubMed]
- Knipper, J.A.; Willenborg, S.; Brinckmann, J.; Bloch, W.; Maaß, T.; Wagener, R.; Krieg, T.; Sutherland, T.; Munitz, A.; Rothenberg, M.E.; et al. Interleukin-4 Receptor α Signaling in Myeloid Cells Controls Collagen Fibril Assembly in Skin Repair. Immunity 2015, 43, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.-B.; Espenel, S.; Vallard, A.; Battiston-Montagne, P.; Wozny, A.-S.; Ardail, D.; Alphonse, G.; Rancoule, C.; Rodriguez-Lafrasse, C.; Magne, N. Evaluation of the cell invasion and migration process: A comparison of the video microscope-based scratch wound assay and the boyden chamber assay. J. Vis. Exp. 2017, 129, e56337. [Google Scholar] [CrossRef]
- Kramer, N.; Walzl, A.; Unger, C.; Rosner, M.; Krupitza, G.; Hengstschläger, M.; Dolznig, H. In vitro cell migration and invasion assays. Mutat. Res./Rev. Mutat. Res. 2013, 752, 10–24. [Google Scholar] [CrossRef]
- Ascione, F.; Caserta, S.; Guido, S. The wound healing assay revisited: A transport phenomena approach. Chem. Eng. Sci. 2017, 160, 200–209. [Google Scholar] [CrossRef]
- Halldorsson, S.; Lucumi, E.; Gómez-Sjöberg, R.; Fleming, R.M.T. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens. Bioelectron. 2015, 63, 218–231. [Google Scholar] [CrossRef]
- Li, Y.; Yang, M.; Huang, Z.; Chen, X.; Maloney, M.T.; Zhu, L.; Liu, J.; Yang, Y.; Du, S.; Jiang, X.; et al. AxonQuant: A Microfluidic Chamber Culture-Coupled Algorithm That Allows High-Throughput Quantification of Axonal Damage. Neurosignals 2014, 22, 14–29. [Google Scholar] [CrossRef]
- Menon, N.V.; Chuah, Y.J.; Cao, B.; Lim, M.; Kang, Y. A microfluidic co-culture system to monitor tumor-stromal interactions on a chip. Biomicrofluidics 2014, 8, 064118. [Google Scholar] [CrossRef]
- Li, R.; Zhang, X.; Lv, X.; Geng, L.; Li, Y.; Qin, K.; Deng, Y. Microvalve controlled multi-functional microfluidic chip for divisional cell co-culture. Anal. Biochem. 2017, 539, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Brewer, B.M.; Shi, M.; Edd, J.F.; Webb, D.J.; Li, D. A microfluidic cell co-culture platform with a liquid fluorocarbon separator. Biomed. Microdevices 2014, 16, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Millet, L.J.; Gillette, M.U. Over a century of neuron culture: From the hanging drop to microfluidic devices. Yale J. Biol. Med. 2012, 85, 501–521. [Google Scholar] [PubMed]
- Lei, Y.; Tang, L.; Xie, Y.; Xianyu, Y.; Zhang, L.; Wang, P.; Hamada, Y.; Jiang, K.; Zheng, W.; Jiang, X. Gold nanoclusters-assisted delivery of NGF siRNA for effective treatment of pancreatic cancer. Nat. Commun. 2017, 8, 15130. [Google Scholar] [CrossRef]
- Rubakhin, S.S.; Romanova, E.V.; Nemes, P.; Sweedler, J.V. Profiling metabolites and peptides in single cells. Nat. Methods 2011, 8, S20–S29. [Google Scholar] [CrossRef]
- Cui, X.; Liu, Y.; Hu, D.; Qian, W.; Tin, C.; Sun, D.; Chen, W.; Lam, R.H.W. A fluorescent microbead-based microfluidic immunoassay chip for immune cell cytokine secretion quantification. Lab Chip 2018, 18, 522–531. [Google Scholar] [CrossRef]
- Uzel, S.G.; Platt, R.J.; Subramanian, V.; Pearl, T.M.; Rowlands, C.J.; Chan, V.; Boyer, L.A.; So, P.T.; Kamm, R.D. Microfluidic device for the formation of optically excitable, three-dimensional, compartmentalized motor units. Sci. Adv. 2016, 2, e1501429. [Google Scholar] [CrossRef]
- Wang, D.; Xue, M.; Chen, J.; Chen, H.; Liu, J.; Li, Q.; Xie, Y.; Hu, Y.; Ni, Y.; Zhou, Q. Macrophage-derived implantable vaccine prevents postsurgical tumor recurrence. Biomaterials 2021, 278, 121161. [Google Scholar] [CrossRef]
- Wang, D.; Chen, H.; Lei, L.; Chen, J.; Gao, J.; Liu, J.; Li, Q.; Xie, Y.; Hu, Y.; Ni, Y. Biofabricated macrophage and fibroblast membranes synergistically promote skin wound healing. Bioeng. Transl. Med. 2022, 7, e10344. [Google Scholar] [CrossRef]
- Smith, M.P.; Young, H.; Hurlstone, A.; Wellbrock, C. Differentiation of THP1 cells into macrophages for transwell co-culture assay with melanoma cells. Bio-Protocol 2015, 5, e1638. [Google Scholar] [CrossRef]
- Thorsen, T.; Maerkl, S.J.; Quake, S.R. Microfluidic large-scale integration. Science 2002, 298, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Chung, C.K. PDMS Microfabrication and Design for Microfluidics and Sustainable Energy Application: Review. Micromachines 2021, 12, 1350. [Google Scholar] [CrossRef] [PubMed]
- Potrich, C.; Pederzolli, C. Simple PDMS microdevice for biomedical applications. Talanta 2019, 193, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Sordel, T.; Kermarec-Marcel, F.; Garnier-Raveaud, S.; Glade, N.; Sauter-Starace, F.; Pudda, C.; Borella, M.; Plissonnier, M.; Chatelain, F.; Bruckert, F. Influence of glass and polymer coatings on CHO cell morphology and adhesion. Biomaterials 2007, 28, 1572–1584. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Y.; Zheng, L.; Zhan, Y.; He, L. Graphene oxide/poly-l-lysine assembled layer for adhesion and electrochemical impedance detection of leukemia K562 cancercells. Biosens. Bioelectron. 2013, 42, 112–118. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Cui, F.; Chen, H.; Yang, Y.; Li, G.; Mao, H.; Lyu, X. A Microfluidic Cell Co-Culture Chip for the Monitoring of Interactions between Macrophages and Fibroblasts. Biosensors 2023, 13, 70. https://doi.org/10.3390/bios13010070
Li P, Cui F, Chen H, Yang Y, Li G, Mao H, Lyu X. A Microfluidic Cell Co-Culture Chip for the Monitoring of Interactions between Macrophages and Fibroblasts. Biosensors. 2023; 13(1):70. https://doi.org/10.3390/bios13010070
Chicago/Turabian StyleLi, Pengcheng, Feiyun Cui, Heying Chen, Yao Yang, Gang Li, Hongju Mao, and Xiaoyan Lyu. 2023. "A Microfluidic Cell Co-Culture Chip for the Monitoring of Interactions between Macrophages and Fibroblasts" Biosensors 13, no. 1: 70. https://doi.org/10.3390/bios13010070
APA StyleLi, P., Cui, F., Chen, H., Yang, Y., Li, G., Mao, H., & Lyu, X. (2023). A Microfluidic Cell Co-Culture Chip for the Monitoring of Interactions between Macrophages and Fibroblasts. Biosensors, 13(1), 70. https://doi.org/10.3390/bios13010070