Melanoma Mediated Disruption of Brain Endothelial Barrier Integrity Is Not Prevented by the Inhibition of Matrix Metalloproteinases and Proteases

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.1.1. Human Brain Endothelial Cells (hCMVECs)
2.1.2. Malignant Melanoma Cells (NZM)
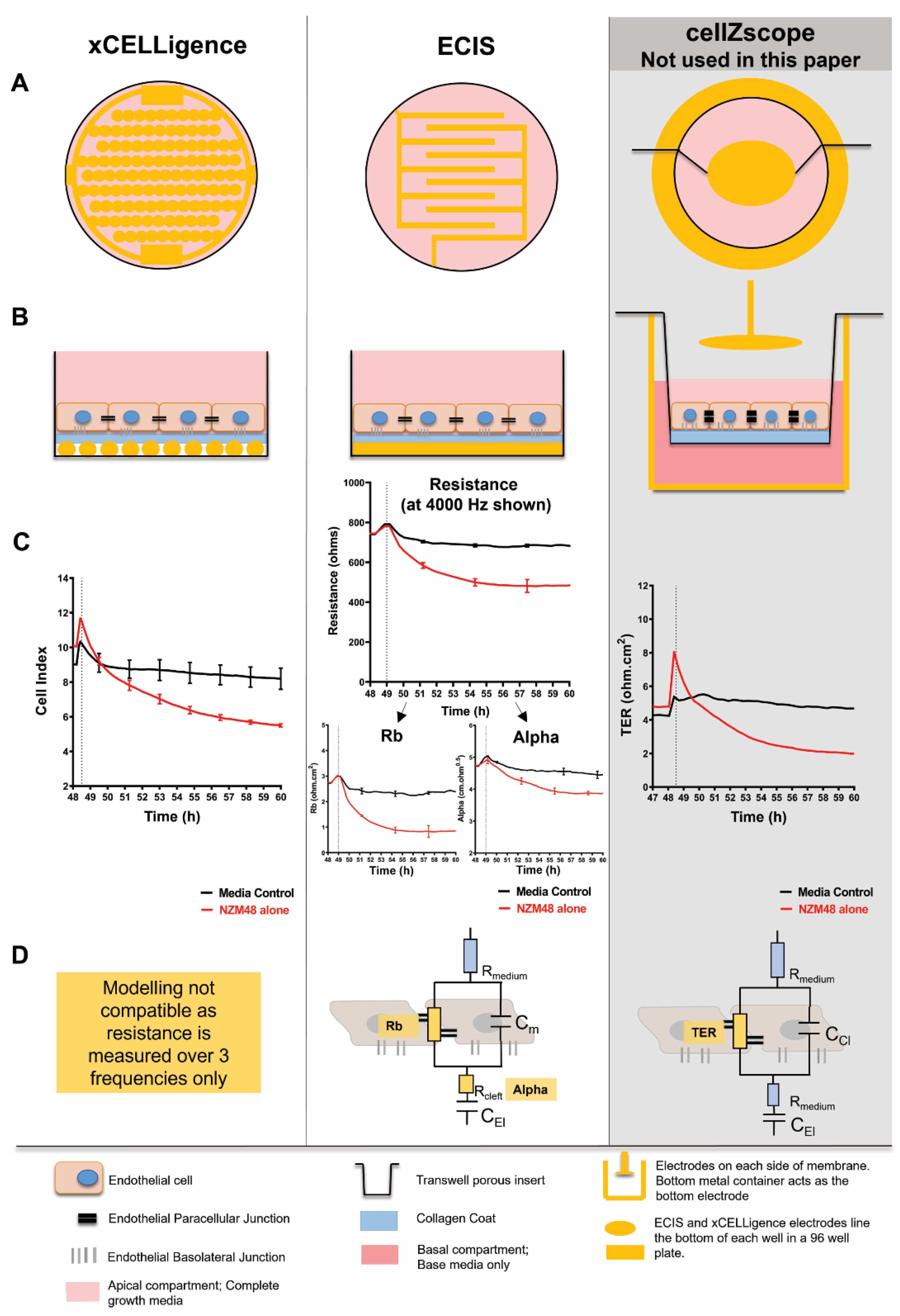
2.2. Biosensors
2.2.1. xCELLigence Theory and Setup
2.2.2. Electric Cell-Substrate Impedance Sensing (ECIS) Setup

2.3. Protease Based Treatment of Brain Endothelial Cells
Protease Based Treatment of Melanoma Addition to Brain Endothelial Cells
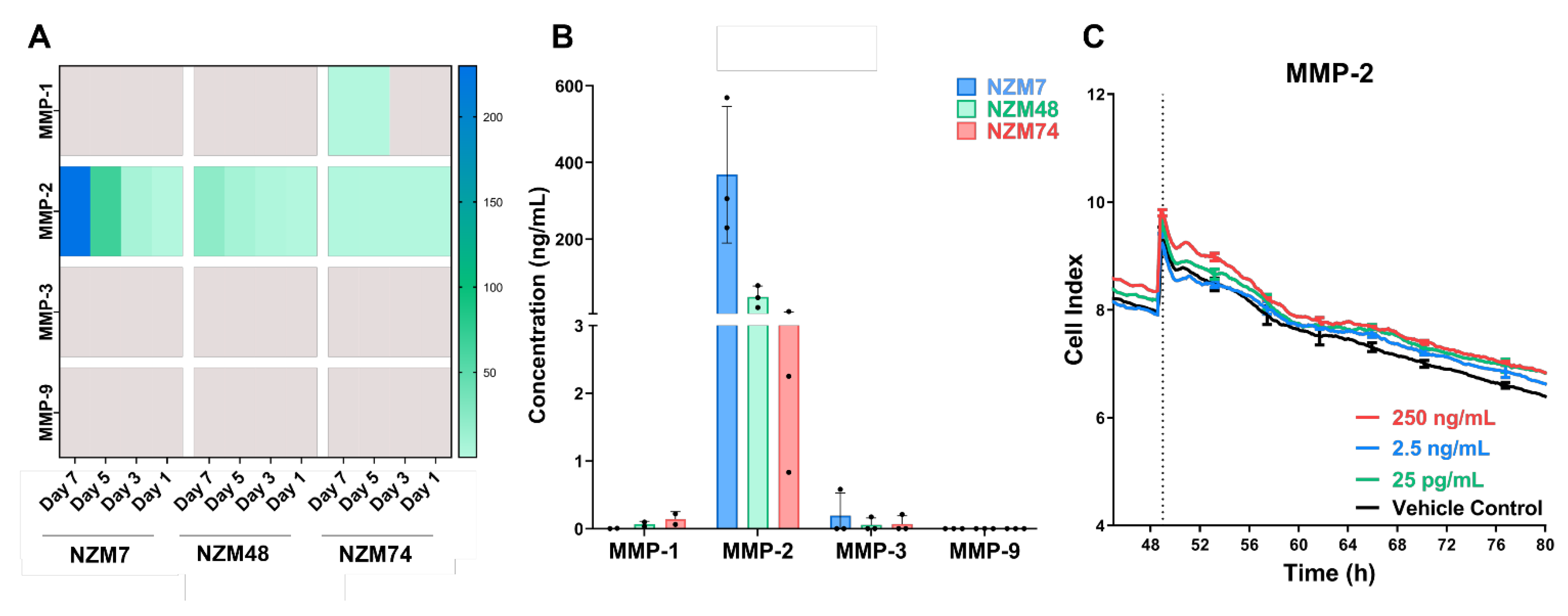
2.4. Melanoma Protease Detection
2.4.1. Luminex Immunoassay
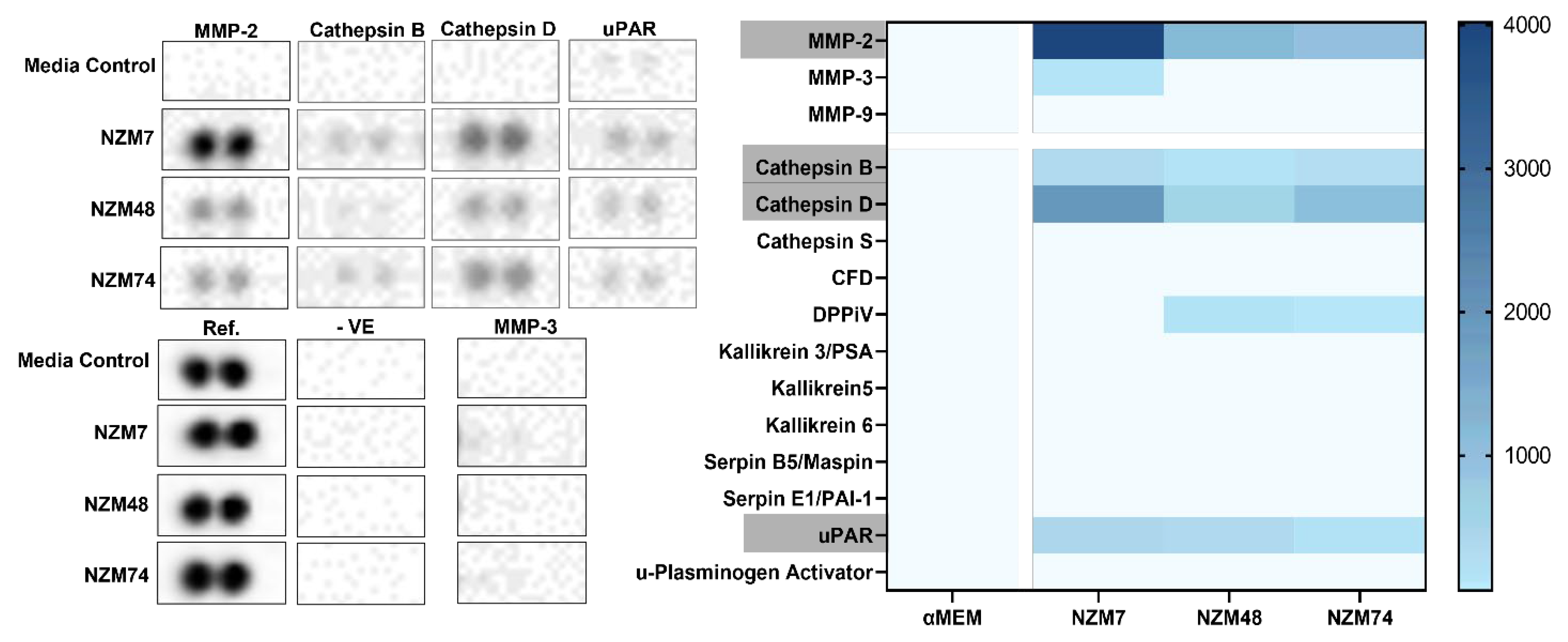
2.4.2. Screening Proteome Profiler Arrays
2.5. Biosensor Statistics
3. Results
3.1. MMP-2 Is Expressed in Melanoma Conditioned Media but Does Not Disrupt the Brain Endothelial Barrier
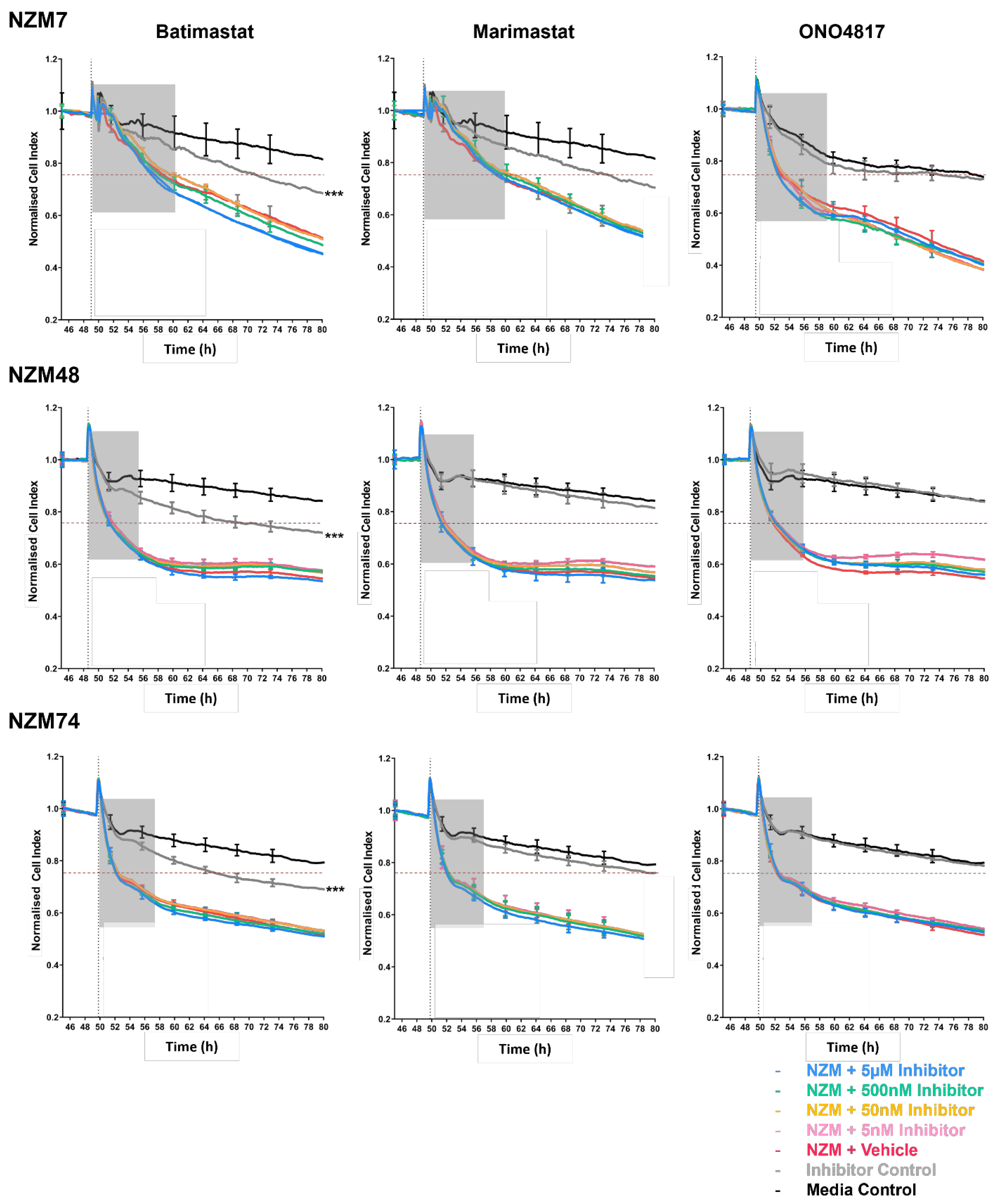
3.2. Blocking Melanoma Cells with MMP Inhibitors Does Not Prevent Melanoma Mediated Brain Endothelial Disruption
3.3. Cathepsin D, Cathepsin B and uPAR Is Expressed in Melanoma Conditioned Media, as seen with MMP-2
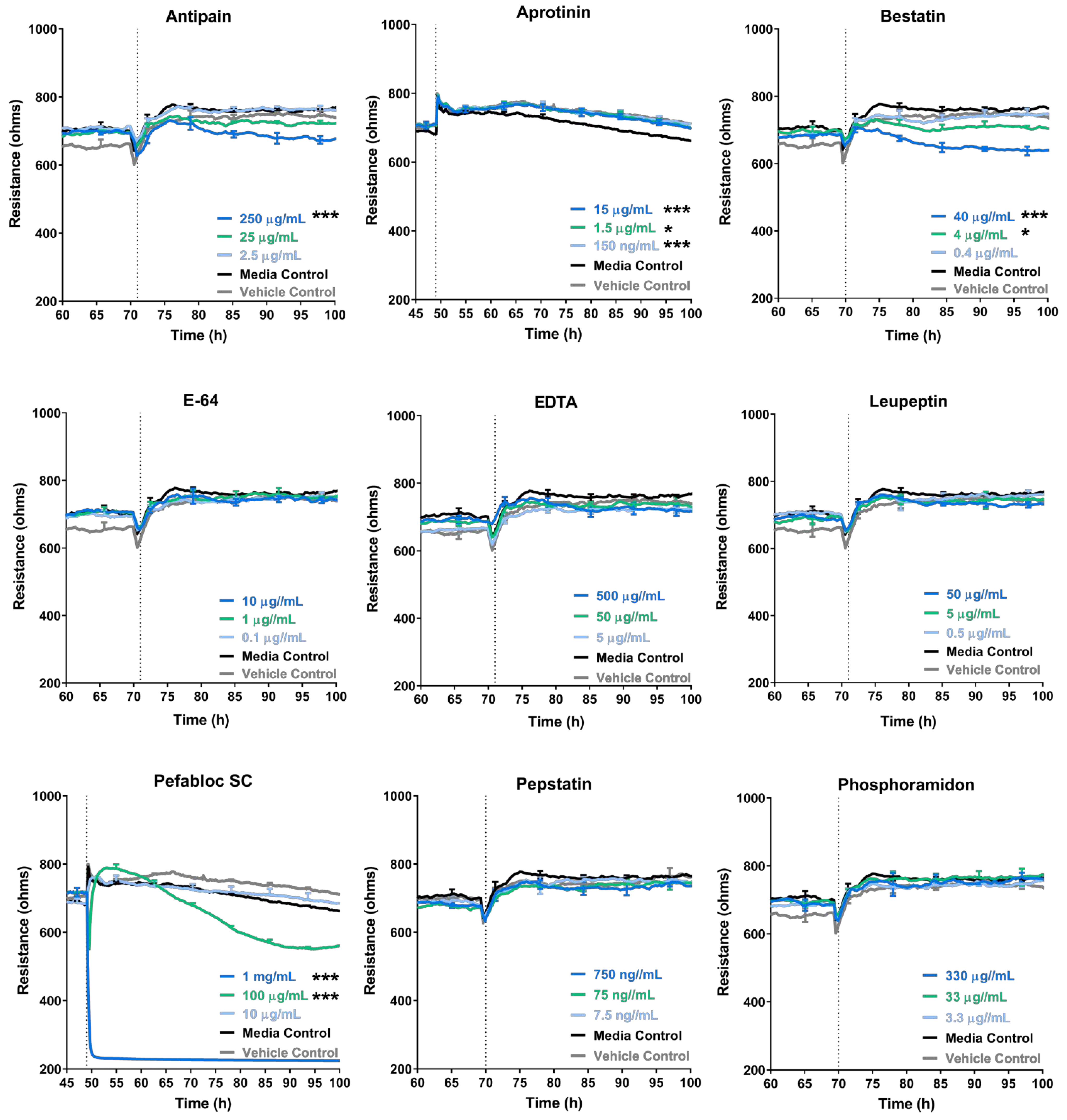
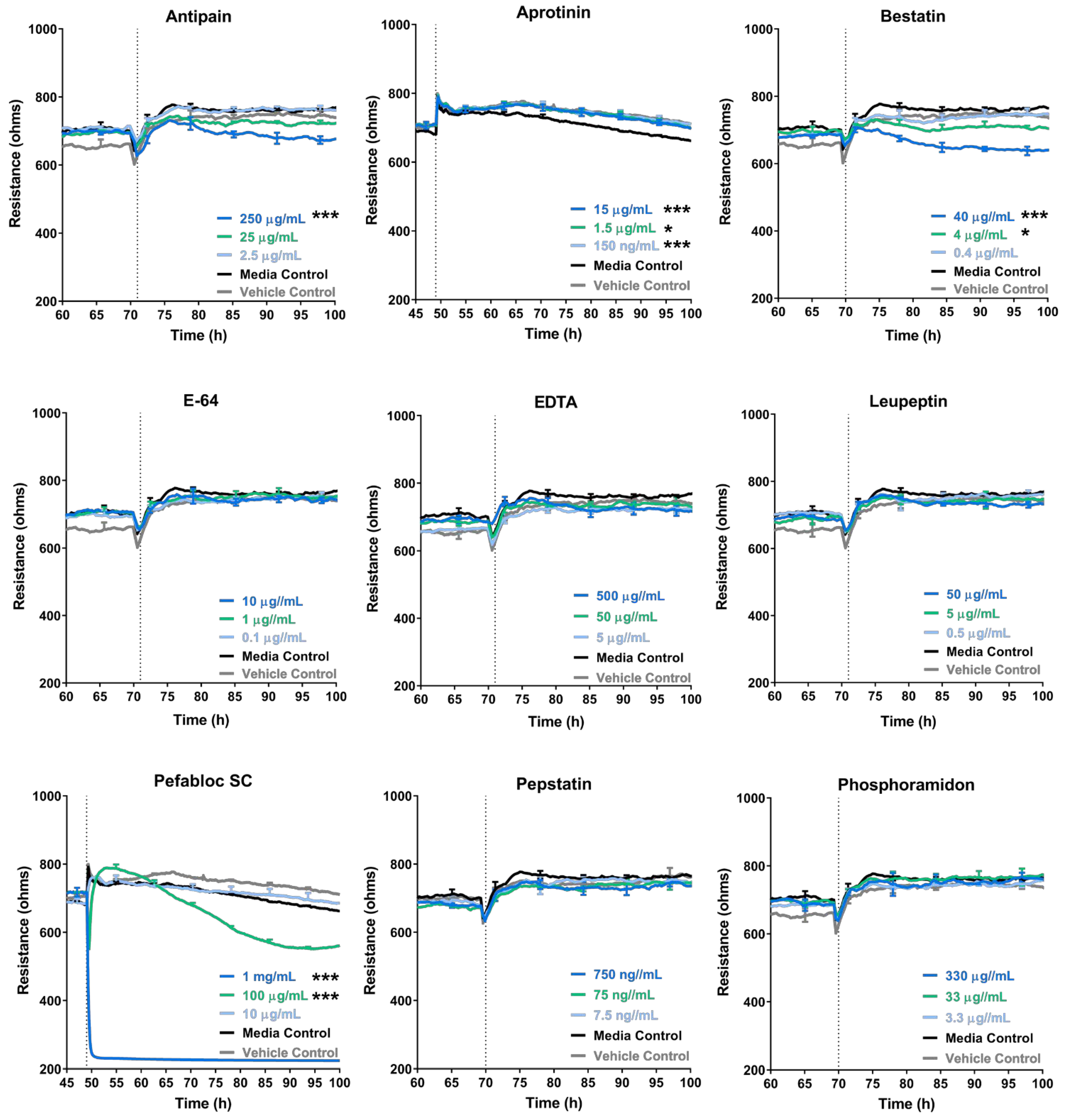
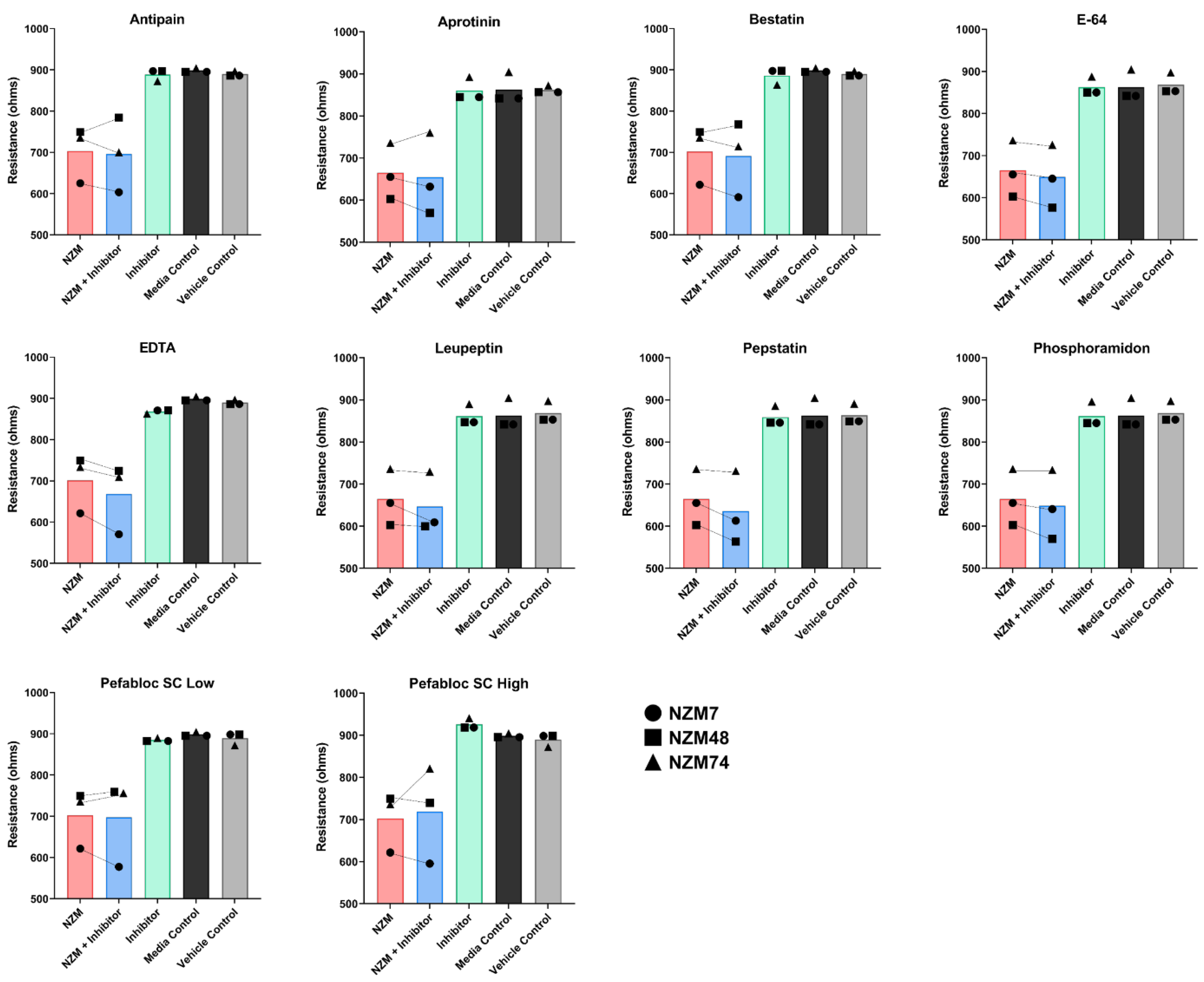
3.4. Some Protease Inhibitors Such as Pefabloc SC Disrupt the Endothelial Barrier at High Concentrations
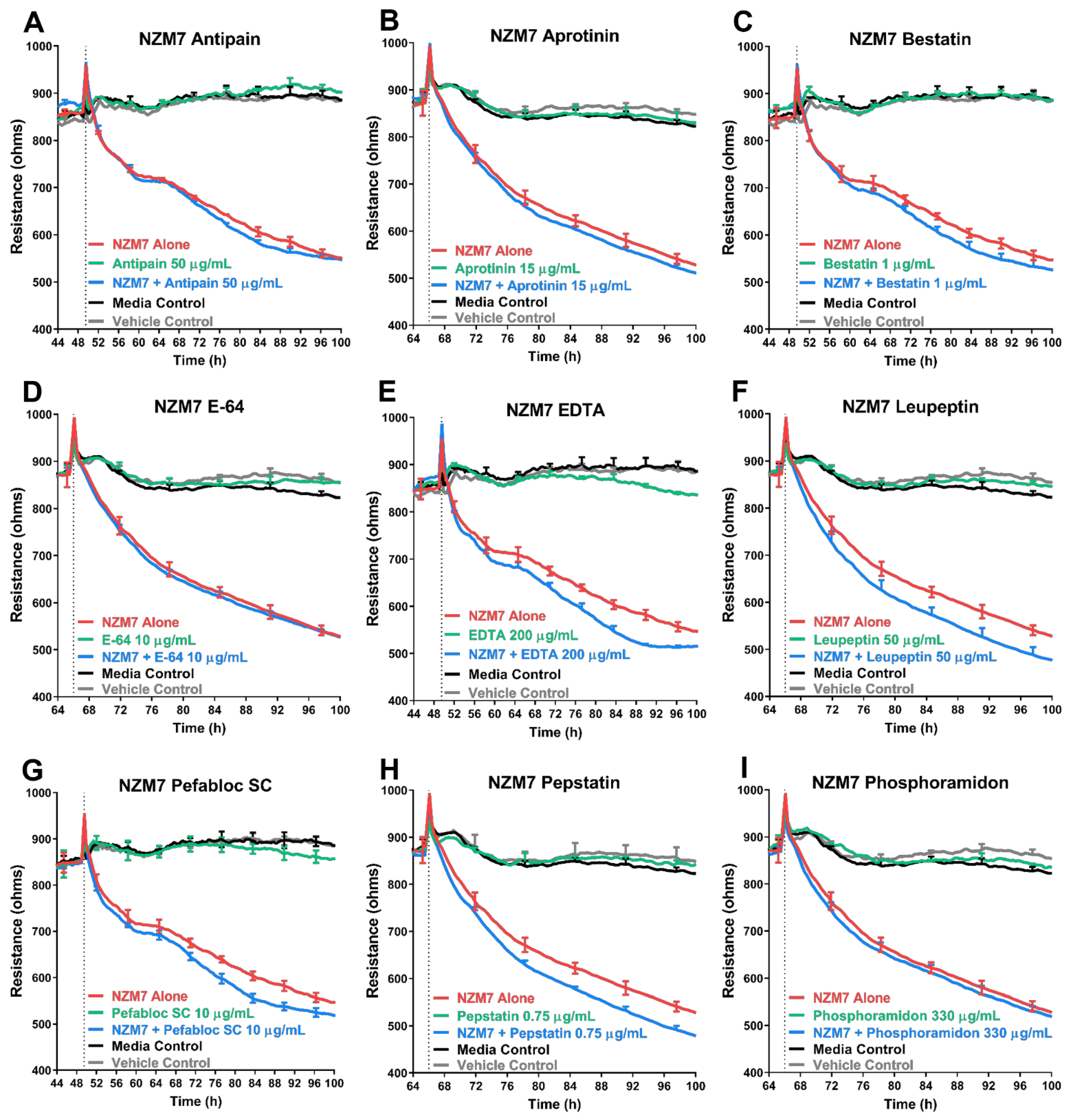
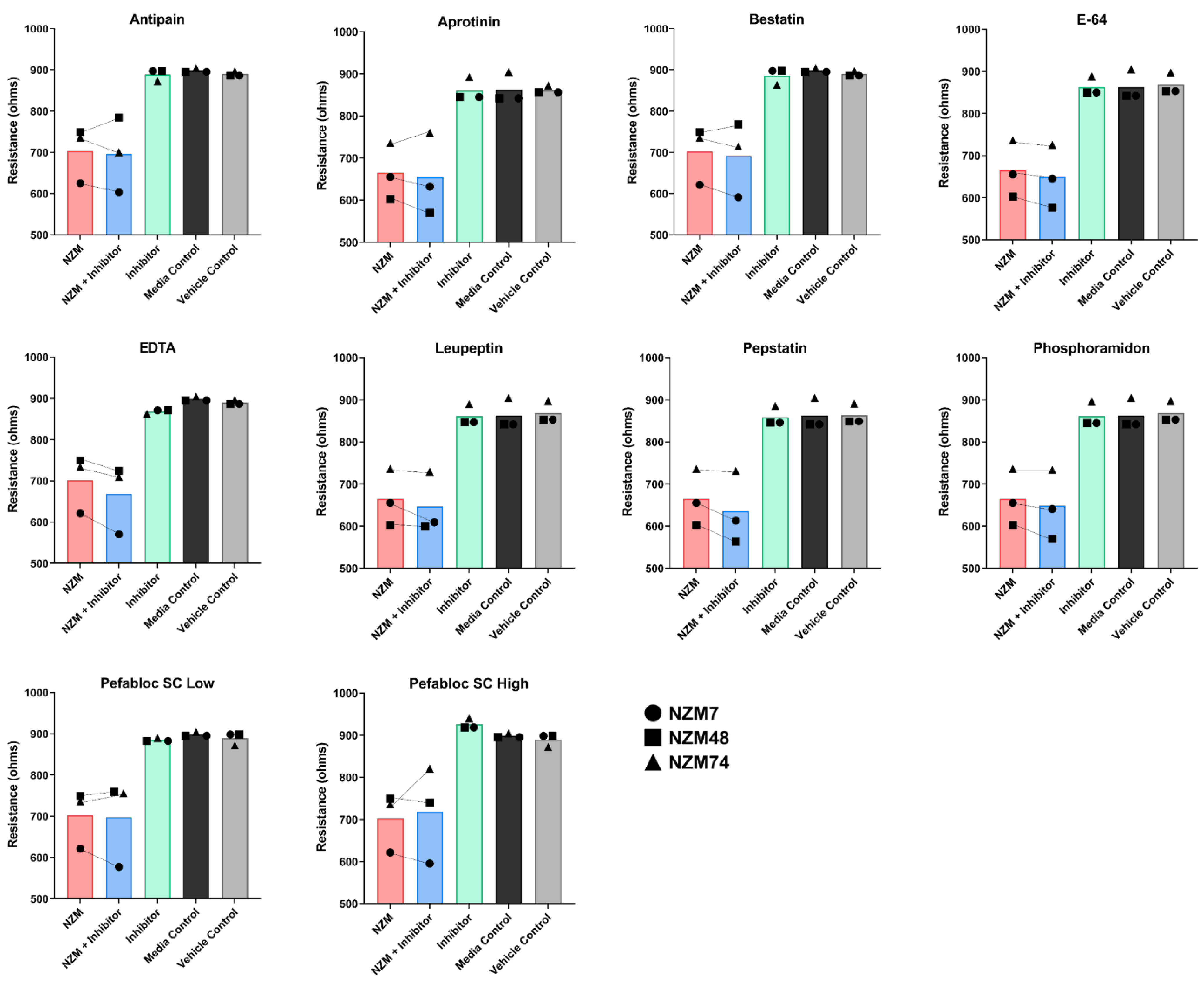
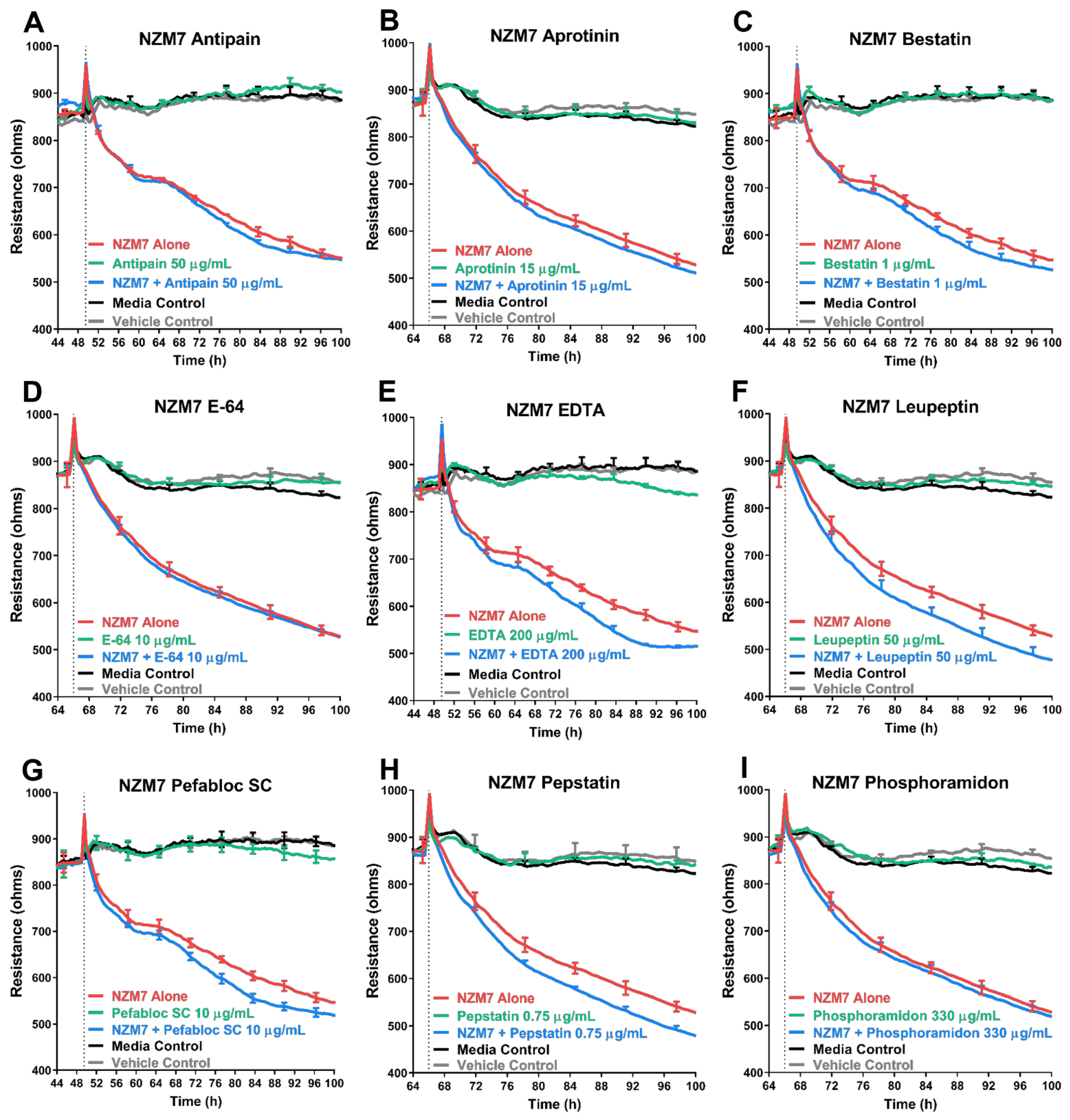
3.5. Treatment of Melanoma Cells with a Range of Proteases Inhibitors Does Not Prevent Melanoma Mediated Brain Endothelial Disruption
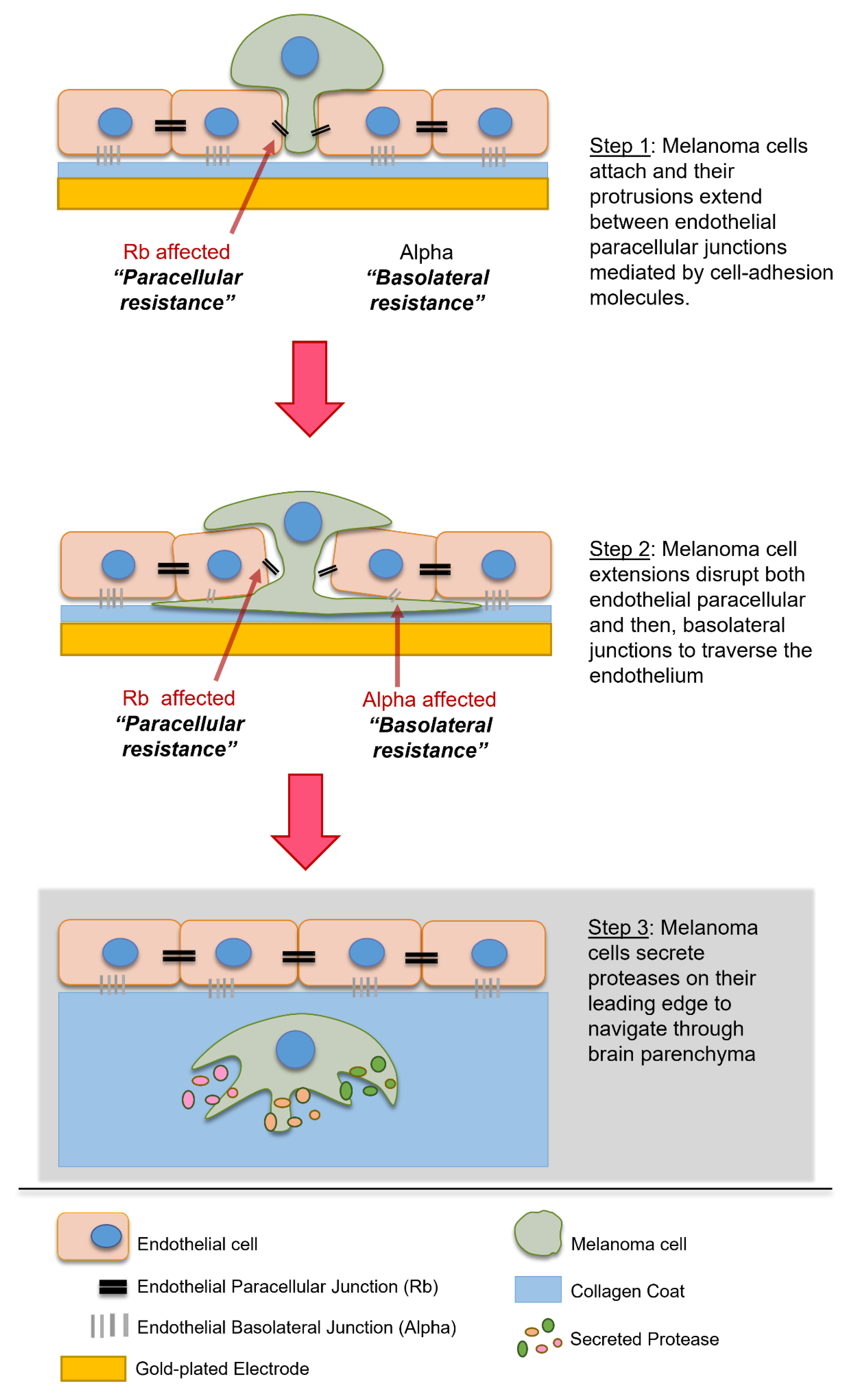
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Company | Catalogue Number |
|---|---|---|
| Collagen I—rat tail | Gibco | A1048301 |
| M199 | Gibco | 11150-067 |
| FBS | Sigma-Aldrich St. Louis, MO, USA | 12203C-500ML |
| Hydrocortison | Sigma-Aldrich | H0888 |
| hFGF | PeproTech | PTAF10018B50 |
| hEGF | PeproTech | PTAF10015100 |
| Heparin | Sigma-Aldrich | H-3393 |
| GlutaMAX | Gibco | 305050-061 |
| dibutyryl-cAMP | Sigma-Aldrich | D0627 |
| aMEM | Gibco | 12561072 |
| Insulin-Transferrin-Sodium Selenite | Sigma-Aldrich | 11074547001 |
| TrypLE™ Express Enzyme | Gibco | 12604021 |
| PBS 1× | Gibco | 10010-023 |
| Material | Assay | Company | Catalogue No. | Standard Curve (pg/mL) |
|---|---|---|---|---|
| MMP-2 | Exogenous Protein Treatment | PeproTech | 420-02 | - |
| Human MMP-1 | Luminex | R&D Systems | LXSAH-4 | 39.59–9620 |
| Human MMP-2 | Luminex | R&D Systems | LXSAHM-6 | 334.94–81,390 |
| Human MMP-3 | Luminex | R&D Systems | LXSAHM-6 | 75.68–18,390 |
| Human MMP-9 | Luminex | R&D Systems | LXSAHM-6 | 115.72–28,120 |
| Human XL Cytokine Array Kit | Proteome Profiler | R&D Systems | ARY022B | - |
| Human XL Oncology Array Kit | Proteome Profiler | R&D Systems | ARY026 | - |
| Batimastat | MMP Inhibition | R&D | RDS2961 | - |
| Marimastat | MMP Inhibition | R&D | RDS2931 | - |
| ONO4817 | MMP Inhibition | R&D | RDS2628 | - |
| Pefabloc SC | Protease Inhibition | Sigma-Aldrich | 11206893001 | - |
| Antipain dihydrochloride | Protease Inhibition | Sigma-Aldrich | ||
| Bestatin | Protease Inhibition | Sigma-Aldrich | ||
| E-64 | Protease Inhibition | Sigma-Aldrich | ||
| Leupeptin | Protease Inhibition | Sigma-Aldrich | ||
| Pepstatin | Protease Inhibition | Sigma-Aldrich | ||
| Phosphoramidon | Protease Inhibition | Sigma-Aldrich | ||
| EDTA, disodium salt | Protease Inhibition | Sigma-Aldrich | ||
| Aprotinin | Protease Inhibition | Sigma-Aldrich |
References
- Sawaya, R.; Bindal, R.K.; Lang, F.F.; Suki, D. 45—Metastatic brain tumors. In Brain Tumors, 3rd ed.; Kaye, A.H., Laws, E.R., Eds.; W.B. Saunders: Edinburgh, UK, 2012; pp. 864–892. [Google Scholar]
- Abbott, N.J.; Friedman, A. Overview and introduction: The blood-brain barrier in health and disease. Epilepsia 2012, 53 (Suppl. S6), 1–6. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Leibold, A.T.; Monaco, G.N.; Dey, M. The role of the immune system in brain metastasis. Curr. Neurobiol. 2019, 10, 33–48. [Google Scholar] [PubMed]
- Anchan, A.; Kalogirou-Baldwin, P.; Johnson, R.; Kho, D.T.; Joseph, W.; Hucklesby, J.; Finlay, G.J.; O’Carroll, S.J.; E Angel, C.; Graham, E.S. Real-Time Measurement of Melanoma Cell-Mediated Human Brain Endothelial Barrier Disruption Using Electric Cell-Substrate Impedance Sensing Technology. Biosensors 2019, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Anchan, A.; Martin, O.; Hucklesby, J.J.W.; Finlay, G.; Johnson, R.H.; Robilliard, L.D.; O’Carroll, S.J.; Angel, C.E.; Graham, E.S. Analysis of Melanoma Secretome for Factors That Directly Disrupt the Barrier Integrity of Brain Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 8193. [Google Scholar] [CrossRef]
- Oda, K. New families of carboxyl peptidases: Serine-carboxyl peptidases and glutamic peptidases. J. Biochem. 2012, 151, 13–25. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2013, 42, D503–D509. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.B.; Westphal, J.R.; Van Muijen, G.N.; Ruiter, D.J. Matrix metalloproteinases in human melanoma. J. Investig. Dermatol. 2000, 115, 337–344. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Stetler-Stevenson, W.G. Matrix metalloproteinases and metastasis. Cancer Chemother. Pharmacol. 1999, 43, S42–S51. [Google Scholar] [CrossRef]
- Lattanzi, S.; Di Napoli, M.; Ricci, S.; Divani, A.A. Matrix Metalloproteinases in Acute Intracerebral Hemorrhage. Neurotherapeutics 2020, 17, 484–496. [Google Scholar] [CrossRef]
- Stetler-Stevenson, W.G.; Aznavoorian, S.; Liotta, L.A. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu. Rev. Cell Biol. 1993, 9, 541–573. [Google Scholar] [CrossRef] [PubMed]
- Woessner, J.F., Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991, 5, 2145–2154. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, I. Matrix metalloproteinases in tumor invasion and metastasis. Semin. Cancer Biol. 2000, 10, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Raffo, D.; Pontiggia, O.; Simian, M. Role of MMPs in metastatic dissemination: Implications for therapeutic advances. Curr. Pharm. Biotechnol. 2011, 12, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar] [CrossRef]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef]
- Verma, R.P.; Hansch, C. Matrix metalloproteinases (MMPs): Chemical-biological functions and (Q)SARs. Bioorg. Med. Chem. 2007, 15, 2223–2268. [Google Scholar] [CrossRef]
- Roomi, M.W.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Modulation of MMP-2 and -9 secretion by cytokines, inducers and inhibitors in human melanoma A-2058 cells. Oncol. Rep. 2017, 37, 3681–3687. [Google Scholar] [CrossRef]
- Sato, H.; Takino, T.; Kinoshita, T.; Imai, K.; Okada, Y.; Stetler Stevenson, W.G.; Seiki, M. Cell surface binding and activation of gelatinase A induced by expression of membrane-type-1-matrix metalloproteinase (MT1-MMP). FEBS Lett. 1996, 385, 238–240. [Google Scholar] [CrossRef]
- Sato, H.; Takino, T.; Okada, Y.; Cao, J.; Shinagawa, A.; Yamamoto, E.; Seiki, M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 1994, 370, 61–65. [Google Scholar] [CrossRef]
- Brooks, P.C.; Stromblad, S.; Sanders, L.C.; von Schalscha, T.L.; Aimes, R.T.; Stetler-Stevenson, W.G.; Quigley, J.P.; Cheresh, D.A. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 1996, 85, 683–693. [Google Scholar] [CrossRef]
- Nakahara, H.; Howard, L.; Thompson, E.W.; Sato, H.; Seiki, M.; Yeh, Y.; Chen, W.-T. Transmembrane/cytoplasmic domain-mediated membrane type 1-matrix metalloprotease docking to invadopodia is required for cell invasion. Proc. Natl. Acad. Sci. USA 1997, 94, 7959–7964. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Feng, X.; Zhan, Y.; Wang, R.; Zheng, S.; Liu, W.; Zeng, X. Matrix metalloproteinase-2 Promotes αvβ3 Integrin-Mediated Adhesion and Migration of Human Melanoma Cells by Cleaving Fibronectin. PLoS ONE 2012, 7, e41591. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A.; Martinka, M.; Li, G. MMP2 expression is a prognostic marker for primary melanoma patients. Cell Oncol. 2012, 35, 207–216. [Google Scholar] [CrossRef]
- Hofmann, U.B.; Westphal, J.R.; Waas, E.T.; Zendman, A.J.W.; Cornelissen, I.M.H.A.; Ruiter, D.J.; Van Muijen, G.N.P. Matrix metalloproteinases in human melanoma cell lines and xenografts: Increased expression of activated matrix metalloproteinase-2 (MMP-2) correlates with melanoma progression. Br. J. Cancer 1999, 81, 774–782. [Google Scholar] [CrossRef]
- Blackburn, J.S.; Liu, I.; Coon, C.I.; Brinckerhoff, C.E. A matrix metalloproteinase-1/protease activated receptor-1 signaling axis promotes melanoma invasion and metastasis. Oncogene 2009, 28, 4237–4248. [Google Scholar] [CrossRef]
- Blackburn, J.S.; Rhodes, C.H.; Coon, C.I.; Brinckerhoff, C.E. RNA interference inhibition of matrix metalloproteinase-1 prevents melanoma metastasis by reducing tumor collagenase activity and angiogenesis. Cancer Res. 2007, 67, 10849–10858. [Google Scholar] [CrossRef]
- Nikkola, J.; Vihinen, P.; Vlaykova, T.; Hahka-Kemppinen, M.; Kahari, V.M.; Pyrhonen, S. High expression levels of collagenase-1 and stromelysin-1 correlate with shorter disease-free survival in human metastatic melanoma. Int. J. Cancer 2002, 97, 432–438. [Google Scholar] [CrossRef]
- Witty, J.P.; Lempka, T.; Coffey, R.J.; Jr Matrisian, L.M. Decreased tumor formation in 7,12-dimethylbenzanthracene-treated stromelysin-1 transgenic mice is associated with alterations in mammary epithelial cell apoptosis. Cancer Res. 1995, 55, 1401–1406. [Google Scholar]
- Zhang, Q.; Zheng, M.; Betancourt, C.E.; Liu, L.; Sitikov, A.; Sladojevic, N.; Zhao, Q.; Zhang, J.H.; Liao, J.K.; Wu, R. Increase in Blood-Brain Barrier (BBB) Permeability Is Regulated by MMP3 via the ERK Signaling Pathway. Oxid. Med. Cell Longev. 2021, 2021, 6655122. [Google Scholar] [CrossRef]
- Salemi, R.; Falzone, L.; Madonna, G.; Polesel, J.; Cina, D.; Mallardo, D.; Ascierto, P.A.; Libra, M.; Candido, S. MMP-9 as a Candidate Marker of Response to BRAF Inhibitors in Melanoma Patients with BRAF(V600E) Mutation Detected in Circulating-Free DNA. Front. Pharmacol. 2018, 9, 856. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, Y.; Bai, J.; Xue, Y.; Peng, Q. MMP1 and MMP9 are potential prognostic biomarkers and targets for uveal melanoma. BMC Cancer. 2021, 21, 1068. [Google Scholar] [CrossRef] [PubMed]
- Schultz, R.M.; Silberman, S.; Persky, B.; Bajkowski, A.S.; Carmichael, D.F. Inhibition by human recombinant tissue inhibitor of metalloproteinases of human amnion invasion and lung colonization by murine B16-F10 melanoma cells. Cancer Res. 1988, 48, 5539–5545. [Google Scholar] [PubMed]
- Fazakas, C.; Wilhelm, I.; Nagyőszi, P.; Farkas, A.E.; Haskó, J.; Molnar, J.; Bauer, H.; Bauer, H.-C.; Ayaydin, F.; Dung, N.T.K.; et al. Transmigration of melanoma cells through the blood-brain barrier: Role of endothelial tight junctions and melanoma-released serine proteases. PLoS ONE 2011, 6, e20758. [Google Scholar] [CrossRef] [PubMed]
- Krenzer, S.; Peterziel, H.; Mauch, C.; Blaber, S.I.; Blaber, M.; Angel, P.; Hess, J. Expression and function of the kallikrein-related peptidase 6 in the human melanoma microenvironment. J. Investig. Dermatol. 2011, 131, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Matarrese, P.; Ascione, B.; Ciarlo, L.; Vona, R.; Leonetti, C.; Scarsella, M.; Mileo, A.M.; Catricalà, C.; Paggi, M.G.; Malorni, W. Cathepsin B inhibition interferes with metastatic potential of human melanoma: An in vitro and in vivo study. Mol. Cancer. 2010, 9, 207. [Google Scholar] [CrossRef]
- Podhajcer, O.L.; Bover, L.; Bravo, A.I.; Ledda, M.F.; Kairiyama, C.; Calb, I.; Guerra, L.; Capony, F.; Mordoh, J. Expression of Cathepsin D in Primary and Metastatic Human Melanoma and Dysplastic Nevi. J. Investig. Dermatol. 1995, 104, 340–344. [Google Scholar] [CrossRef]
- Quintanilla-Dieck, M.J.; Codriansky, K.; Keady, M.; Bhawan, J.; Rünger, T.M. Cathepsin K in melanoma invasion. J. Investig. Dermatol. 2008, 128, 2281–2288. [Google Scholar] [CrossRef]
- Yang, Z.; Cox JLCathepsin, L. Increases invasion and migration of B16 melanoma. Cancer Cell Int. 2007, 7, 8. [Google Scholar] [CrossRef]
- Bartenjev, I.; Rudolf, Z.; Stabuc, B.; Vrhovec, I.; Perkovic, T.; Kansky, A. Cathepsin D expression in early cutaneous malignant melanoma. Int. J. Dermatol. 2000, 39, 599–602. [Google Scholar] [CrossRef]
- Eding, C.B.; Domert, J.; Wäster, P.; Jerhammar, F.; Rosdahl, I.; Öllinger, K. Melanoma growth and progression after ultraviolet a irradiation: Impact of lysosomal exocytosis and cathepsin proteases. Acta Derm. Venereol. 2015, 95, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Bajkowski, A.S.; Steiner, D.F.; Chan, S.J.; Frankfater, A. Expression of Five Cathepsins in Murine Melanomas of Varying Metastatic Potential and Normal Tissues. Cancer Res. 1989, 49, 4870–4875. [Google Scholar] [PubMed]
- Yin, M.; Soikkeli, J.; Jahkola, T.; Virolainen, S.; Saksela, O.; Hölttä, E. TGF-β signaling, activated stromal fibroblasts, and cysteine cathepsins B and L drive the invasive growth of human melanoma cells. Am. J. Pathol. 2012, 181, 2202–2216. [Google Scholar] [CrossRef]
- Robilliard, L.D.; Kho, D.T.; Johnson, R.H.; Anchan, A.; O’Carroll, S.J.; Graham, E.S. The Importance of Multifrequency Impedance Sensing of Endothelial Barrier Formation Using ECIS Technology for the Generation of a Strong and Durable Paracellular Barrier. Biosensors 2018, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Kho, D.; Johnson, R.H.; Robilliard, L.; Du Mez, E.; McIntosh, J.; O’Carroll, S.J.; Angel, C.; Graham, E.S. ECIS technology reveals that monocytes isolated by CD14+ve selection mediate greater loss of BBB integrity than untouched monocytes, which occurs to a greater extent with IL-1β activated endothelium in comparison to TNFα. PLoS ONE 2017, 12, e0180267. [Google Scholar] [CrossRef] [PubMed]
- Kho, D.; MacDonald, C.; Johnson, R.; Unsworth, C.P.; O’Carroll, S.J.; Du Mez, E.; Angel, C.E.; Graham, E.S. Application of xCELLigence RTCA Biosensor Technology for Revealing the Profile and Window of Drug Responsiveness in Real Time. Biosensors 2015, 5, 199–222. [Google Scholar] [CrossRef] [PubMed]
- Hucklesby, J.J.W.; Anchan, A.; O’Carroll, S.J.; Unsworth, C.P.; Graham, E.S.; Angel, C.E. Comparison of Leading Biosensor Technologies to Detect Changes in Human Endothelial Barrier Properties in Response to Pro-Inflammatory TNFα and IL1β in Real-Time. Biosensors 2021, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, S.J.; Kho, D.T.; Wiltshire, R.; Nelson, V.; Rotimi, O.; Johnson, R.; Angel, C.E.; Graham, E.S. Pro-inflammatory TNFalpha and IL-1beta differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J. Neuroinflammation 2015, 12, 131. [Google Scholar] [CrossRef]
- Benson, K.; Cramer, S.; Galla, H.-J. Impedance-based cell monitoring: Barrier properties and beyond. Fluids Barriers CNS 2013, 10, 5. [Google Scholar] [CrossRef]
- Cacopardo, L.; Costa, J.; Giusti, S.; Buoncompagni, L.; Meucci, S.; Corti, A.; Mattei, G.; Ahluwalia, A. Real-time cellular impedance monitoring and imaging of biological barriers in a dual-flow membrane bioreactor. Biosens. Bioelectron. 2019, 140, 111340. [Google Scholar] [CrossRef]
- Kulczar, C.; Lubin, K.E.; Lefebvre, S.; Miller, D.W.; Knipp, G.T. Development of a direct contact astrocyte-human cerebral microvessel endothelial cells blood-brain barrier coculture model. J. Pharm. Pharmacol. 2017, 69, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Eigenmann, D.E.; Xue, G.; Kim, K.S.; Moses, A.V.; Hamburger, M.; Oufir, M. Comparative study of four immortalized human brain capillary endothelial cell lines, hCMEC/D3, hBMEC, TY10, and BB19, and optimization of culture conditions, for an in vitro blood–brain barrier model for drug permeability studies. Fluids Barriers CNS 2013, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Tiruppathi, C.; Malik, A.B.; Del Vecchio, P.J.; Keese, C.R.; Giaever, I. Electrical method for detection of endothelial cell shape change in real time: Assessment of endothelial barrier function. Proc. Natl. Acad. Sci. USA 1992, 89, 7919–7923. [Google Scholar] [CrossRef] [PubMed]
- Giaever, I.; Keese, C.R. Micromotion of mammalian cells measured electrically. Proc. Natl. Acad. Sci. USA 1991, 88, 7896. [Google Scholar] [CrossRef] [PubMed]
- Burkert, K.; Moodley, K.; Angel, C.E.; Brooks, A.; Graham, E.S. Detailed analysis of inflammatory and neuromodulatory cytokine secretion from human NT2 astrocytes using multiplex bead array. Neurochem. Int. 2012, 60, 573–580. [Google Scholar] [CrossRef]
- Nikkola, J.; Vihinen, P.; Vuoristo, M.S.; Kellokumpu-Lehtinen, P.; Kahari, V.M.; Pyrhonen, S. High serum levels of matrix metalloproteinase-9 and matrix metalloproteinase-1 are associated with rapid progression in patients with metastatic melanoma. Clin. Cancer Res. 2005, 11, 5158–5166. [Google Scholar] [CrossRef]
- Redondo, P.; Lloret, P.; Idoate, M.; Inoges, S. Expression and serum levels of MMP-2 and MMP-9 during human melanoma progression. Clin Exp Dermatol. 2005, 30, 541–545. [Google Scholar] [CrossRef]
- Cotignola, J.; Reva, B.; Mitra, N.; Ishill, N.; Chuai, S.; Patel, A.; Shah, S.; Vanderbeek, G.; Coit, D.; Klaus, B.; et al. Matrix Metalloproteinase-9 (MMP-9) polymorphisms in patients with cutaneous malignant melanoma. BMC Med. Genet. 2007, 8, 10. [Google Scholar] [CrossRef]
- Cotignola, J.; Roy, P.; Patel, A.; Ishill, N.; Shah, S.; Houghton, A.; Coit, D.; Halpern, A.; Busam, K.; Berwick, M.; et al. Functional polymorphisms in the promoter regions of MMP2 and MMP3 are not associated with melanoma progression. J. Negat. Results Biomed. 2007, 6, 9. [Google Scholar] [CrossRef]
- Scorilas, A.; Karameris, A.; Arnogiannaki, N.; Ardavanis, A.; Bassilopoulos, P.; Trangas, T.; Talieri, M. Overexpression of matrix-metalloproteinase-9 in human breast cancer: A potential favourable indicator in node-negative patients. Br. J. Cancer. 2001, 84, 1488–1496. [Google Scholar] [CrossRef]
- Reuning, U.; Sperl, S.; Kopitz, C.; Kessler, H.; Kruger, A.; Schmitt, M.; Magdolen, V. Urokinase-type plasminogen activator (uPA) and its receptor (uPAR): Development of antagonists of uPA/uPAR interaction and their effects in vitro and in vivo. Curr. Pharm. Des. 2003, 9, 1529–1543. [Google Scholar] [CrossRef] [PubMed]
- Sidenius, N.; Sier, C.F.M.; Blasi, F. Shedding and cleavage of the urokinase receptor (uPAR): Identification and characterisation of uPAR fragments in vitro and in vivo. FEBS Lett. 2000, 475, 52–56. [Google Scholar] [CrossRef]
- Höyhtyä, M.; Hujanen, E.; Turpeenniemi-Hujanen, T.; Thorgeirsson, U.; Liotta, L.A.; Tryggvason, K. Modulation of type-iv collagenase activity and invasive behavior of metastatic human melanoma (A2058) cells in vitro by monoclonal antibodies to type-iv collagenase. Int. J. Cancer 1990, 46, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Voura, E.B.; English, J.L.; Yu, H.Y.; Ho, A.T.; Subarsky, P.; Hill, R.P.; Hojilla, C.V.; Khokha, R. Proteolysis during tumor cell extravasation in vitro: Metalloproteinase involvement across tumor cell types. PLoS ONE 2013, 8, e78413. [Google Scholar] [CrossRef] [PubMed]
- Wylie, S.; Macdonald, I.C.; Varghese, H.J.; Schmidt, E.E.; Morris, V.L.; Groom, A.C.; Chambers, A.F. The matrix metalloproteinase inhibitor batimastat inhibits angiogenesis in liver metastases of B16F1 melanoma cells. Clin. Exp. Metastasis 1999, 17, 111–117. [Google Scholar] [CrossRef]
- Amer, M.H.; Al-Sarraf, M.; Baker, L.H.; Vaitkevicius, V.K. Malignant melanoma and central nervous system metastases. Incidence, diagnosis, treatment and survival. Cancer 1978, 42, 660–668. [Google Scholar] [CrossRef]
- Chason, J.L.; Walker, F.B.; Landers, J.W. Metastatic carcinoma in the central nervous system and dorsal root ganglia. Prospect. Study Cancer 1963, 16, 781–787. [Google Scholar]
- Madajewicz, S.; Karakousis, C.; West, C.R.; Caracandas, J.; Avellanosa, A.M. Malignant melanoma brain metastases. Review of Roswell Park Memorial Institute experience. Cancer 1984, 53, 2550–2552. [Google Scholar] [CrossRef]
- Abate-Daga, D.; Ramello, M.C.; Smalley, I.; Forsyth, P.A.; Smalley, K.S.M. The biology and therapeutic management of melanoma brain metastases. Biochem. Pharmacol. 2018, 153, 35–45. [Google Scholar] [CrossRef]
- Kotecha, R.; Miller, J.A.; Venur, V.A.; Mohammadi, A.M.; Chao, S.T.; Suh, J.H.; Barnett, G.H.; Murphy, E.S.; Funchain, P.; Yu, J.S.; et al. Melanoma brain metastasis: The impact of stereotactic radiosurgery, BRAF mutational status, and targeted and/or immune-based therapies on treatment outcome. J. Neurosurg. 2018, 129, 50–59. [Google Scholar] [CrossRef]
- Sloot, S.; Chen, Y.A.; Zhao, X.; Weber, J.L.; Benedict, J.J.; Mule, J.J.; Smalley, K.S.; Weber, J.S.; Zager, J.S.; Forsyth, P.A.; et al. Improved survival of patients with melanoma brain metastases in the era of targeted BRAF and immune checkpoint therapies. Cancer 2018, 124, 297–305. [Google Scholar] [CrossRef] [PubMed]
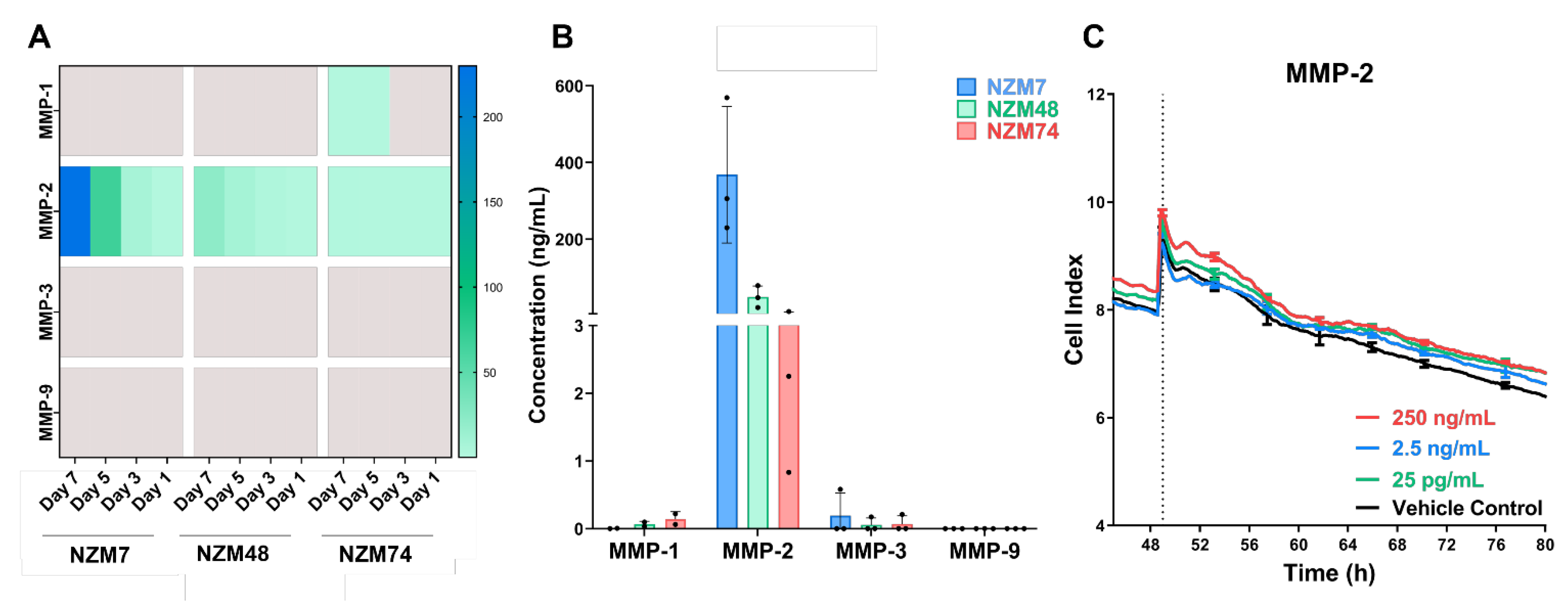

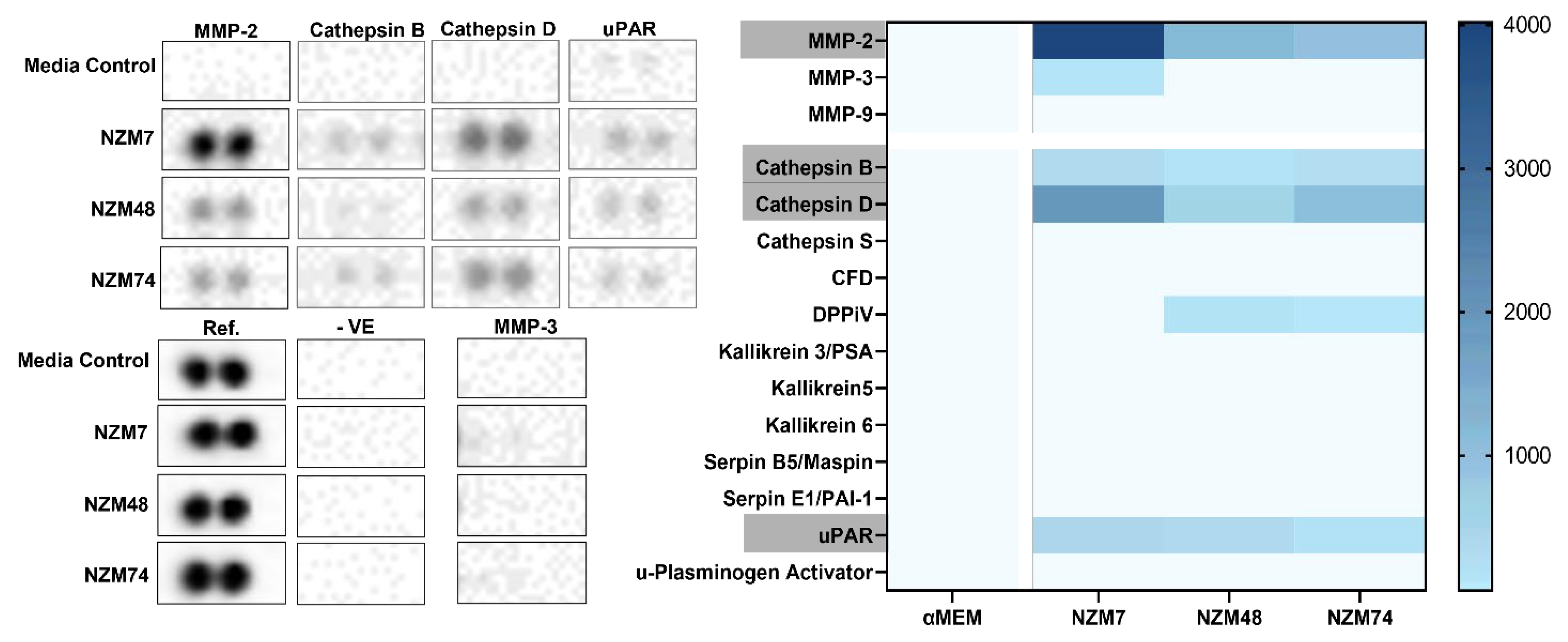

| Melanoma Line | Research Resource ID |
|---|---|
| NZM7 | CVCL_D843 |
| NZM48 | CVCL_S423 |
| NZM74 | CVCL_0D38 |
| Assay | Inhibitor | Starting Concentration | Concentration at Melanoma Treatment | Final Concentration |
|---|---|---|---|---|
| xCELLigence | Batimastat | 20 μM–20 nM | 10 μM–10 nM | 5 μM–5 nM |
| Marimastat | 20 μM–20 nM | 10 μM–10 nM | 5 μM–5 nM | |
| ONO4817 | 20 μM–20 nM | 10 μM–10 nM | 5 μM–5 nM | |
| ECIS | Antipain dihydrochloride | 200 µg/mL | 100 µg/mL | 50 µg/mL |
| Aprotinin | 60 µg/mL | 30 µg/mL | 15 µg/mL | |
| Bestatin | 4 µg/mL | 2 µg/mL | 1 µg/mL | |
| E-64 | 40 µg/mL | 20 µg/mL | 10 µg/mL | |
| EDTA | 800 µg/mL | 400 µg/mL | 200 µg/mL | |
| Leupeptin | 200 µg/mL | 100 µg/mL | 50 µg/mL | |
| Pefabloc SC | 40 µg/mL | 20 µg/mL | 10 µg/mL | |
| Pepstatin | 3 µg/mL | 1.5 µg/mL | 0.75 µg/mL | |
| Phosphoramidon | 1320 µg/mL | 660 µg/mL | 330 µg/mL |
| Batimastat | IC50: nM | Marimastat | IC50: nM | ONO4817 | Ki (IC50: nM for MMP-1) |
|---|---|---|---|---|---|
| MMP-1 | 3 | MMP-9 | 3 | MMP-12 | 0.45 |
| MMP-2 * | 4 * | MMP-1 | 5 | MMP-2 * | 0.73 * |
| MMP-9 | 4 | MMP-2 * | 6 * | MMP-8 | 1.1 |
| MMP-7 | 6 | MMP-14 | 9 | MMP-13 | 1.1 |
| MMP-3 | 20 | MMP-7 | 13 | MMP-9 | 2.1 |
| MMP-3 | 42 | ||||
| MMP-7 | 2500 | ||||
| MMP-1 | 1600 |
| Assessed Proteases | Mean Pixel Intensity | |||||
|---|---|---|---|---|---|---|
| Protease Name | Protease Type | αMEM | NZM7 | NZM48 | NZM74 | Present |
| Cathepsin B | Cysteine Protease | 320.814 | 128.485 | 247.203 | Yes | |
| Cathepsin D | Aspartic Protease | 35.95 | 1908.8495 | 556.6415 | 1020.4705 | Yes |
| Cathepsin S | Cysteine Protease | 54.571 | 88.5355 | 64.742 | 95.6315 | No |
| Complement Factor D | Serine Protease | No | ||||
| DPPiV * | Serine Protease | 147.778 | 61.0355 | Yes * | ||
| Kallikrein 5 | Serine Protease | No | ||||
| Kallikrein 3/PSA | Serine Protease | No | ||||
| Kallikrein 6 | Serine Protease | No | ||||
| uPAR | Involved in Plasmin Activation | 99.339 | 475.228 | 423.4705 | 255.607 | Yes |
| uPA | Plasmin Activator | No | ||||
| Serpin B5/Maspin | Endopeptidase Regulator | No | ||||
| Serpin E1/PAI-1 | Serine Protease Inhibitor (e.g., uPA) | 53.121 | 30.3285 | No | ||
| Assessed Protease Inhibitors | Recommended Concentration | |||
|---|---|---|---|---|
| Inhibitor | Specificity | Concentration Tested (µg/mL) | Concentration Used (µg/mL) | Toxic to Barrier |
| Antipain dihydrochloride | Broad-spectrum | 250 | 50 | Yes |
| Aprotinin | Serine Protease | 15 | 15 | No |
| Bestatin | Amino Peptidase | 40 | 1 | Yes |
| E-64 | Cysteine Protease | 10 | 10 | No |
| EDTA | Metallo Protease | 500 | 200 | Yes |
| Leupeptin | Serine, Cysteine Protease | 50 | 50 | No |
| Pefabloc SC | Serine Protease | 1000 | 10 | Yes |
| Pepstatin | Aspartic Protease | 0.75 | 0.75 | No |
| Phosphoramidon | Metallo-endopeptidase | 330 | 330 | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anchan, A.; Finlay, G.; Angel, C.E.; Hucklesby, J.J.W.; Graham, E.S. Melanoma Mediated Disruption of Brain Endothelial Barrier Integrity Is Not Prevented by the Inhibition of Matrix Metalloproteinases and Proteases. Biosensors 2022, 12, 660. https://doi.org/10.3390/bios12080660
Anchan A, Finlay G, Angel CE, Hucklesby JJW, Graham ES. Melanoma Mediated Disruption of Brain Endothelial Barrier Integrity Is Not Prevented by the Inhibition of Matrix Metalloproteinases and Proteases. Biosensors. 2022; 12(8):660. https://doi.org/10.3390/bios12080660
Chicago/Turabian StyleAnchan, Akshata, Graeme Finlay, Catherine E. Angel, James J. W. Hucklesby, and E. Scott Graham. 2022. "Melanoma Mediated Disruption of Brain Endothelial Barrier Integrity Is Not Prevented by the Inhibition of Matrix Metalloproteinases and Proteases" Biosensors 12, no. 8: 660. https://doi.org/10.3390/bios12080660
APA StyleAnchan, A., Finlay, G., Angel, C. E., Hucklesby, J. J. W., & Graham, E. S. (2022). Melanoma Mediated Disruption of Brain Endothelial Barrier Integrity Is Not Prevented by the Inhibition of Matrix Metalloproteinases and Proteases. Biosensors, 12(8), 660. https://doi.org/10.3390/bios12080660