Hot-Spot-Specific Probe (HSSP) for Rapid and Accurate Detection of KRAS Mutations in Colorectal Cancer

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
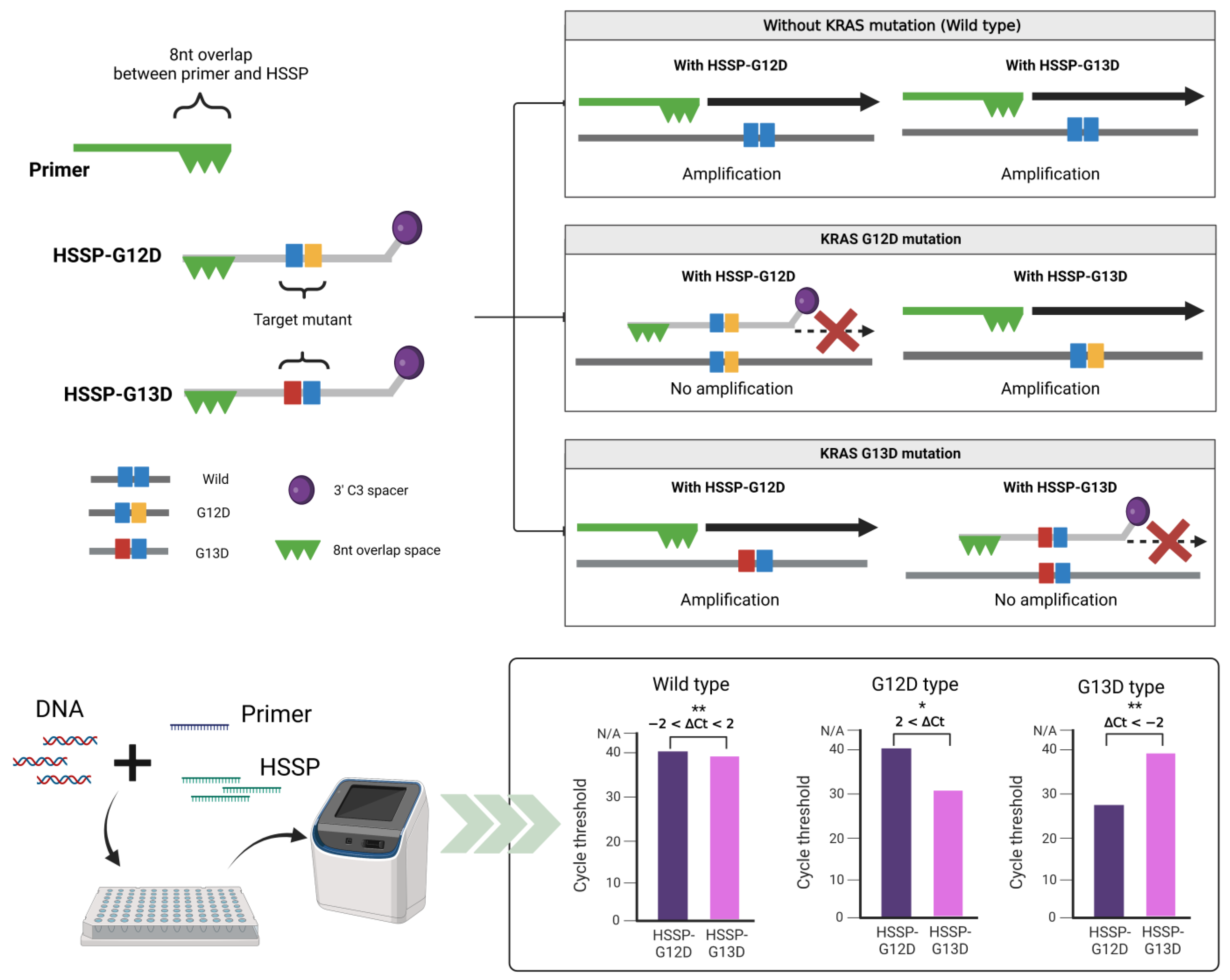
2.1. Design of Primer and HSSP
2.2. HSSP with qPCR
2.3. Cancer Cell Line
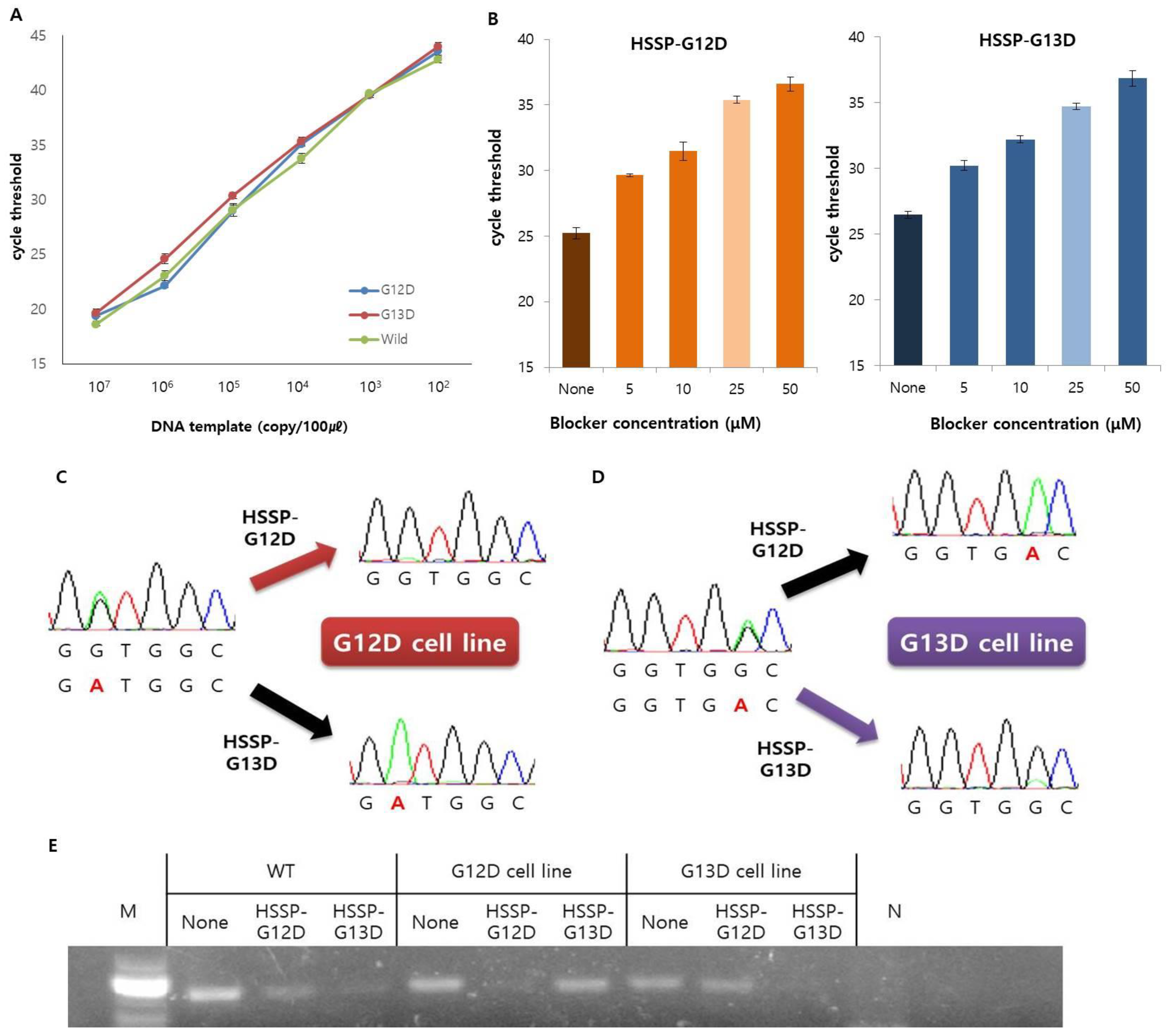
2.4. Conventional PCR and Sequencing Analysis
2.5. DNA Template Preparation
2.6. Clinical Sample Collection
3. Results
3.1. Principles of HSSP
3.2. Optimization and Application of HSSP
3.3. Detection Limit of HSSP in Mixed-Cell Populations
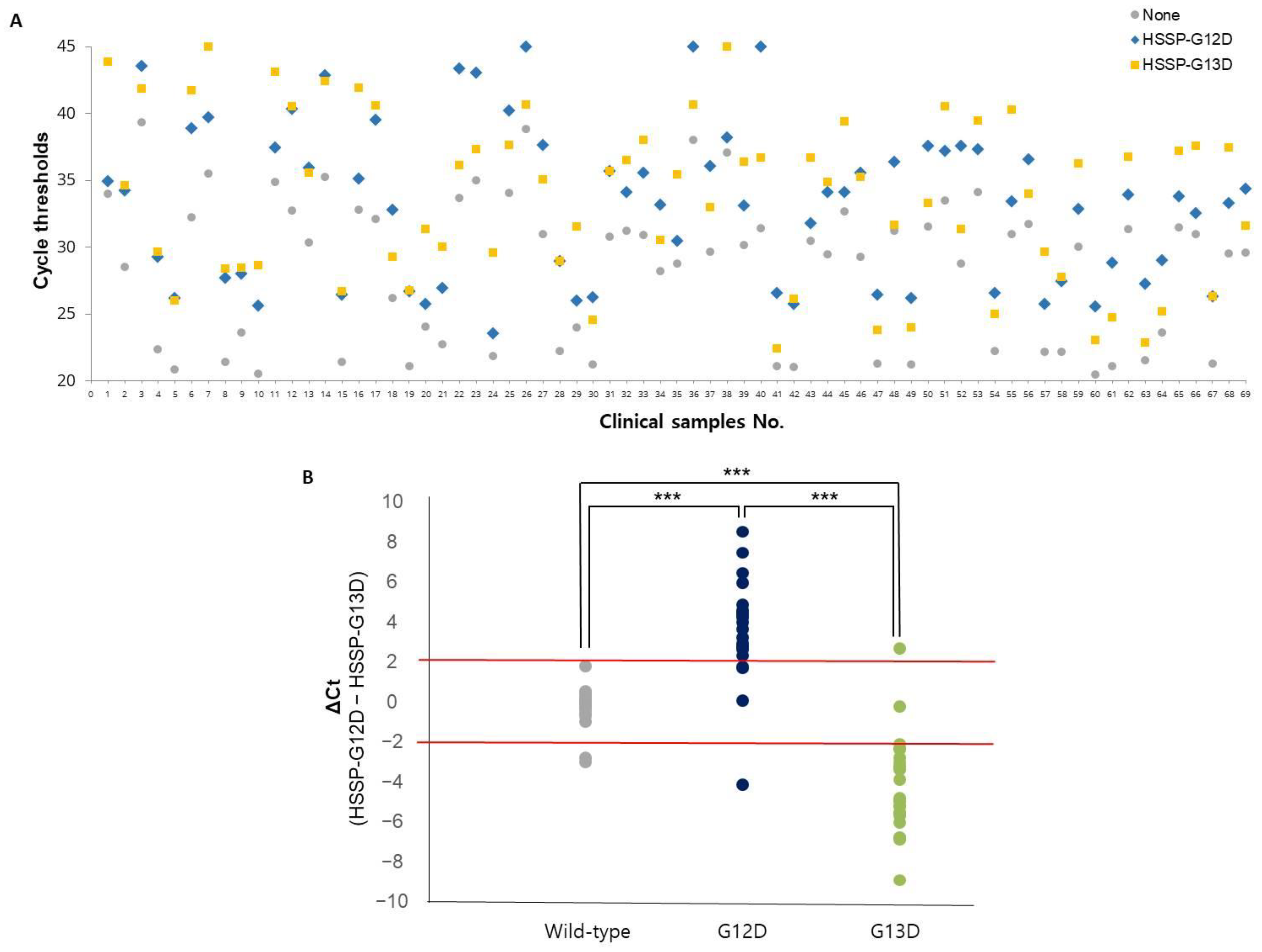
3.4. Clinical Utility of HSSP
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global Cancer Statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Cancer State Facts: Colorectal Cancer. Available online: https://seer.cancer.gov/statfacts/html/colorect.html. (accessed on 27 June 2022).
- Biller, L.H.; Schrag, D. Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA J. Am. Med. Assoc. 2021, 325, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Phipps, A.I.; Buchanan, D.D.; Makar, K.W.; Win, A.K.; Baron, J.A.; Lindor, N.M.; Potter, J.D.; Newcomb, P.A. KRAS-mutation status in relation to colorectal cancer survival: The joint impact of correlated tumour markers. Br. J. Cancer 2013, 108, 1757–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, Y.; Morikawa, T.; Liao, X.Y.; Lochhead, P.; Kuchiba, A.; Yamauchi, M.; Qian, Z.R.; Nishihara, R.; Meyerhardt, J.A.; Haigis, K.M.; et al. Specific Mutations in KRAS Codons 12 and 13, and Patient Prognosis in 1075 BRAF Wild-Type Colorectal Cancers. Clin. Cancer Res. 2012, 18, 4753–4763. [Google Scholar] [CrossRef] [Green Version]
- Timar, J.; Kashofer, K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev. 2020, 39, 1029–1038. [Google Scholar] [CrossRef]
- Laurent-Puig, P.; Pekin, D.; Normand, C.; Kotsopoulos, S.K.; Nizard, P.; Perez-Toralla, K.; Rowell, R.; Olson, J.; Srinivasan, P.; Le Corre, D.; et al. Clinical Relevance of KRAS-Mutated Subclones Detected with Picodroplet Digital PCR in Advanced Colorectal Cancer Treated with Anti-EGFR Therapy. Clin. Cancer Res. 2015, 21, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- Walther, A.; Johnstone, E.; Swanton, C.; Midgley, R.; Tomlinson, I.; Kerr, D. Genetic prognostic and predictive markers in colorectal cancer. Nat. Rev. Cancer 2009, 9, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G. Drug therapy: EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tougeron, D.; Lecomte, T.; Pages, J.C.; Villalva, C.; Collin, C.; Ferru, A.; Tourani, J.M.; Silvain, C.; Levillain, P.; Karayan-Tapon, L. Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2013, 24, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Felicioni, L.; Buscarino, M.; De Dosso, S.; Buttitta, F.; Malatesta, S.; Movilia, A.; Luoni, M.; Boldorini, R.; Alabiso, O.; et al. Increased Detection Sensitivity for KRAS Mutations Enhances the Prediction of Anti-EGFR Monoclonal Antibody Resistance in Metastatic Colorectal Cancer. Clin. Cancer Res. 2011, 17, 4901–4914. [Google Scholar] [CrossRef] [Green Version]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.R.; Chen, S.X.; Wu, Y.; Patel, A.A.; Zhang, D.Y. Multiplexed enrichment of rare DNA variants via sequence-selective and temperature-robust amplification. Nat. Biomed. Eng. 2017, 1, 714–723. [Google Scholar] [CrossRef]
- Lai, W.; Xiao, M.S.; Yang, H.H.; Li, L.; Fan, C.H.; Pei, H. Circularized blocker-displacement amplification for multiplex detection of rare DNA variants. Chem. Commun. 2020, 56, 12331–12334. [Google Scholar] [CrossRef]
- Yamin, W.; Zhong, C. Mutation detection and molecular targeted tumor therapies. STEMedicine 2020, 1, e11. [Google Scholar]
- Su, N.; Wei, K.; Zhao, N.; Wang, L.; Duan, G.J.; Ren, X.D.; Qu, X.M.; Huang, Q. Sensitive and selective detections of codon 12 and 13 KRAS mutations in a single tube using modified wild-type blocker. Clin. Chim. Acta 2019, 494, 123–131. [Google Scholar] [CrossRef]
- Dufort, S.; Richard, M.J.; de Fraipont, F. Pyrosequencing method to detect KRAS mutation in formalin-fixed and paraffin-embedded tumor tissues. Anal. Biochem. 2009, 391, 166–168. [Google Scholar] [CrossRef]
- Vivancos, A.; Aranda, E.; Benavides, M.; Elez, E.; Gomez-Espana, M.A.; Toledano, M.; Alvarez, M.; Parrado, M.R.C.; Garcia-Barberan, V.; Diaz-Rubio, E. Comparison of the Clinical Sensitivity of the Idylla Platform and the OncoBEAM RAS CRC Assay for KRAS Mutation Detection in Liquid Biopsy Samples. Sci. Rep. 2019, 9, 8976. [Google Scholar] [CrossRef] [Green Version]
- Holm, M.; Andersson, E.; Osterlund, E.; Ovissi, A.; Soveri, L.M.; Anttonen, A.K.; Kytola, S.; Aittomaki, K.; Osterlund, P.; Ristimaki, A. Detection of KRAS mutations in liquid biopsies from metastatic colorectal cancer patients using droplet digital PCR, Idylla, and next generation sequencing. PLoS ONE 2020, 15, e0239819. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.H.; Wang, S.J.; Fu, B.Q.; Wang, J. Evaluation of droplet digital PCR and next generation sequencing for characterizing DNA reference material for KRAS mutation detection. Sci. Rep.-UK 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didelot, A.; Le Corre, D.; Luscan, A.; Cazes, A.; Pallier, K.; Emile, J.F.; Laurent-Puig, P.; Blons, H. Competitive allele specific TaqMan PCR for KRAS, BRAF and EGFR mutation detection in clinical formalin fixed paraffin embedded samples. Exp. Mol. Pathol. 2012, 92, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Chubarov, A.S.; Oscorbin, I.P.; Filipenko, M.L.; Lomzov, A.A.; Pyshnyi, D.V. Allele-Specific PCR for KRAS Mutation Detection Using Phosphoryl Guanidine Modified Primers. Diagnostics 2020, 10, 872. [Google Scholar] [CrossRef]
- Vestheim, H.; Jarman, S.N. Blocking primers to enhance PCR amplification of rare sequences in mixed samples-a case study on prey DNA in Antarctic krill stomachs. Front. Zool. 2008, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Dames, S.; Margraf, R.L.; Pattison, D.C.; Wittwer, C.T.; Voelkerding, K.V. Characterization of aberrant melting peaks in unlabeled probe assays. J. Mol. Diagn. 2007, 9, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.E.; Yeom, S.S.; Koo, B.; Lee, T.Y.; Lee, J.H.; Shin, Y.; Lim, S.B. Rapid and accurate detection of KRAS mutations in colorectal cancers using the isothermal-based optical sensor for companion diagnostics. Oncotarget 2017, 8, 83860–83871. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zou, Q.; Kim, M.G.; Qiao, Z.; Nguyen, D.T.T.; Koo, B.; Lee, H.J.; Jang, Y.O.; Kim, J.K.; Shin, Y. Homobifunctional Imidoester Combined Black Phosphorus Nanosheets Used as Cofactors for Nucleic Acid Extraction. BioChip J. 2022, 16, 58–66. [Google Scholar] [CrossRef]
- Jin, C.E.; Koo, B.; Lee, E.Y.; Kim, J.Y.; Kim, S.H.; Shin, Y. Simple and label-free pathogen enrichment via homobifunctional imidoesters using a microfluidic (SLIM) system for ultrasensitive pathogen detection in various clinical specimens. Biosens. Bioelectron. 2018, 111, 66–73. [Google Scholar] [CrossRef]
- Jin, C.E.; Koo, B.; Lee, T.Y.; Han, K.; Lim, S.B.; Park, I.J.; Shin, Y. Simple and Low-Cost Sampling of Cell-Free Nucleic Acids from Blood Plasma for Rapid and Sensitive Detection of Circulating Tumor DNA. Adv. Sci. 2018, 5, 1800614. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Demuth, C.; Spindler, K.L.G.; Johansen, J.S.; Pallisgaard, N.; Nielsen, D.; Hogdall, E.; Vittrup, B.; Sorensen, B.S. Measuring KRAS Mutations in Circulating Tumor DNA by Droplet Digital PCR and Next-Generation Sequencing. Transl. Oncol. 2018, 11, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Durkee, K.H.; Moore, K.J.; Goodman, S.N.; Shuber, A.P.; Kinzler, K.W.; Vogelstein, B. Analysis of mutations in DNA isolated from plasma and stool of colorectal cancer patients. Gastroenterology 2008, 135, 489–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KRAS | HSSP with Real-Time PCR | Direct Sequencing | ||
|---|---|---|---|---|
| Sensitivity (95% CI) | Specificity (95% CI) | Sensitivity (95% CI) | Specificity (95% CI) | |
| G12D | 84% (63.92–95.46%) | 97.73% (87.98–99.94%) | 80.00% (59.30–93.17%) | 100% (91.96–100.00%) |
| G13D | 92% (73.97–99.02%) | 93.18% (81.34–98.57%) | 80.00% (59.30–93.17%) | 97.06% (87.98–99.94%) |
| WT | 89.47% (66.86–98.7%) | 92% (80.77–98.70%) | 100% (82.35–100.00%) | 82.00% (68.56–91.42%) |
| Mutation LOD (%) | Protocol Time (h) | Cost Per Sample (USD) | Refs | |
|---|---|---|---|---|
| HSSP with real-time PCR | 5–10 | 1.5 | ≤2 | This paper |
| HSSP with direct sequencing | 1–2 | 48+ | ≤6 | |
| NGS | 1 | 48+ | 1000+ | [32] |
| AS-PCR | 1–10 | 2 | ≤3 | [24] |
| Droplet digital PCR | 0.1 | 4 | 10 | [23,33] |
| OncoBEAM | 0.02 | 48+ | 24 | [34] |
| Idylla | 1 | 2.5 | 10 | [21] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.J.; Koo, B.; Jang, Y.O.; Liu, H.; Dao, T.N.T.; Lim, S.-B.; Shin, Y. Hot-Spot-Specific Probe (HSSP) for Rapid and Accurate Detection of KRAS Mutations in Colorectal Cancer. Biosensors 2022, 12, 597. https://doi.org/10.3390/bios12080597
Lee HJ, Koo B, Jang YO, Liu H, Dao TNT, Lim S-B, Shin Y. Hot-Spot-Specific Probe (HSSP) for Rapid and Accurate Detection of KRAS Mutations in Colorectal Cancer. Biosensors. 2022; 12(8):597. https://doi.org/10.3390/bios12080597
Chicago/Turabian StyleLee, Hyo Joo, Bonhan Koo, Yoon Ok Jang, Huifang Liu, Thuy Nguyen Thi Dao, Seok-Byung Lim, and Yong Shin. 2022. "Hot-Spot-Specific Probe (HSSP) for Rapid and Accurate Detection of KRAS Mutations in Colorectal Cancer" Biosensors 12, no. 8: 597. https://doi.org/10.3390/bios12080597
APA StyleLee, H. J., Koo, B., Jang, Y. O., Liu, H., Dao, T. N. T., Lim, S.-B., & Shin, Y. (2022). Hot-Spot-Specific Probe (HSSP) for Rapid and Accurate Detection of KRAS Mutations in Colorectal Cancer. Biosensors, 12(8), 597. https://doi.org/10.3390/bios12080597