Threonine Phosphorylation of an Electrochemical Peptide-Based Sensor to Achieve Improved Uranyl Ion Binding Affinity

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Fabrication of U-pT-12 and U-12 Sensors on Gold Disk Electrodes
2.3. Fabrication of a U-pT-12 Sensor on a Paper Electrode
2.4. Sensor Characterization and Target Interrogation
3. Results and Discussion
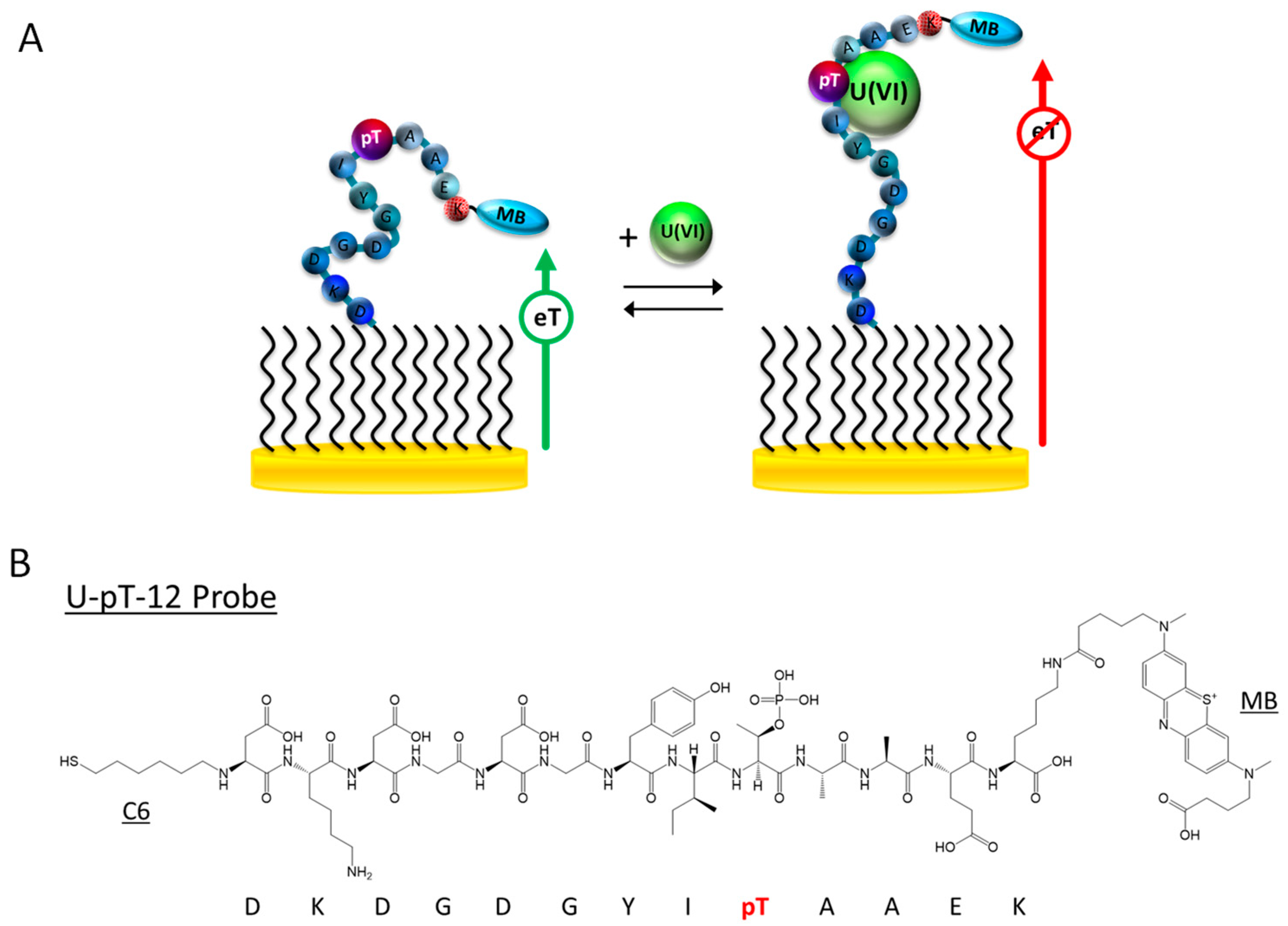
3.1. Sensor Design
3.2. Sensor Characterization
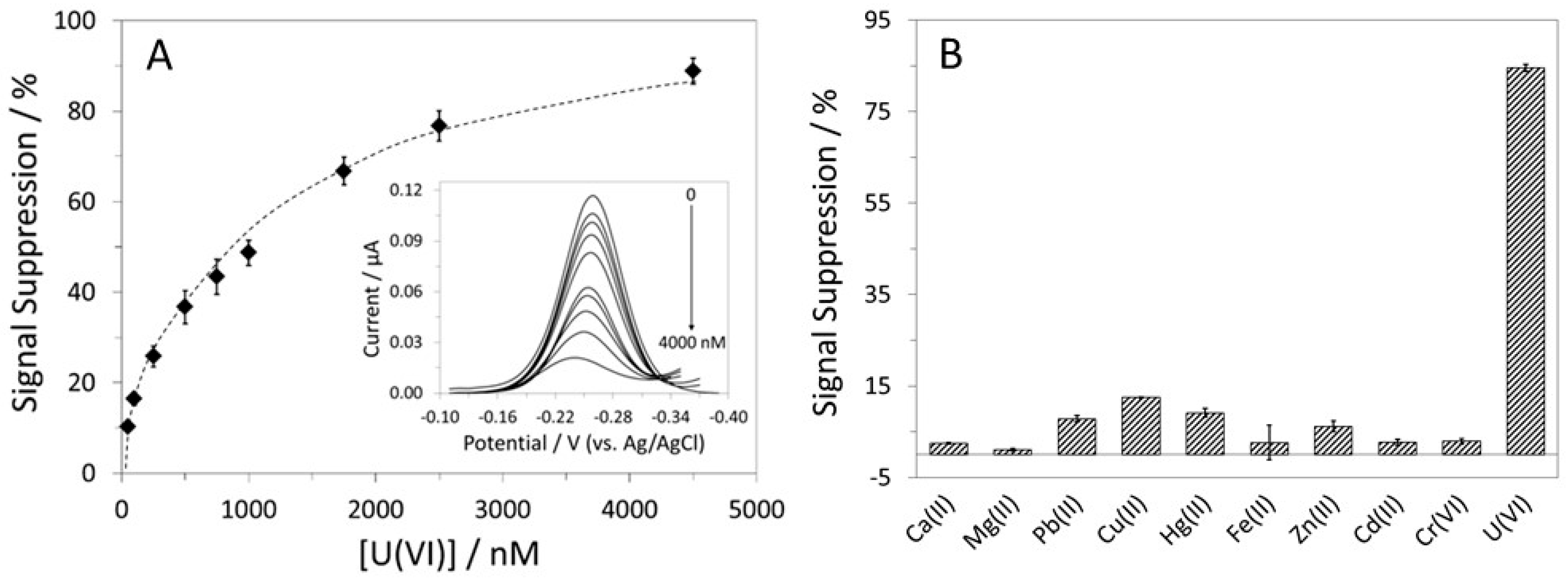
3.3. Sensor Performance: Sensitivity, Specificity, and Selectivity
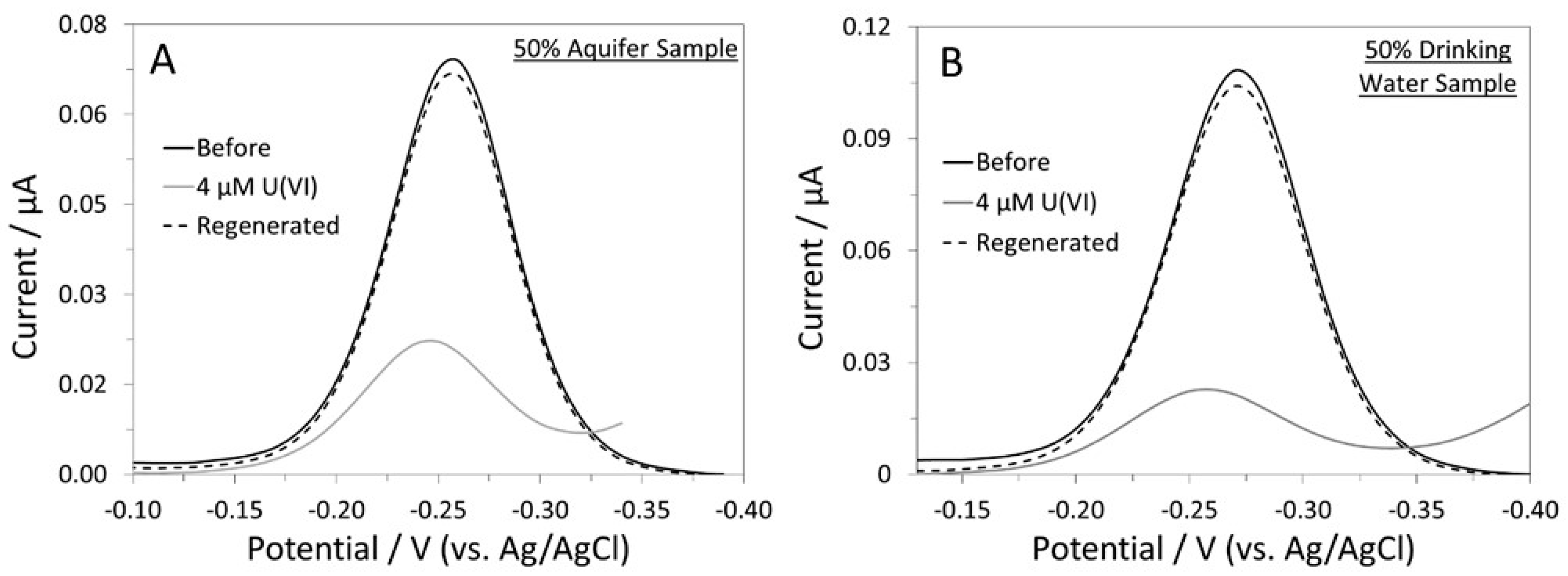
3.4. Sensor Response on a Disposable Paper Electrode
3.5. Key Analytical Properties of the U-pT-12 Sensor—A Comparison
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vicente-Vicente, L.; Quiros, Y.; Pérez-Barriocanal, F.; López-Novoa, J.M.; López-Hernández, F.J.; Morales, A.I. Nephrotoxicity of uranium: Pathophysiological, diagnostic and therapeutic perspectives. Toxicol. Sci. 2010, 118, 324–347. [Google Scholar] [CrossRef] [PubMed]
- Malard, V.; Gaillard, J.C.; Bérenguer, F.; Sage, N.; Quéméneur, E. Urine proteomic profiling of uranium nephrotoxicity. Biochim. Biophys. Acta 2009, 1794, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Bots, P.; Behrends, T. Uranium mobility in subsurface aqueous systems: The influence of redox conditions. Mineral. Mag. 2008, 72, 381–384. [Google Scholar] [CrossRef]
- Paradis, C.J.; Jagadamma, S.; Watson, D.B.; McKay, L.D.; Hazen, T.C.; Park, M.; Istok, J.D. In situ mobility of uranium in the presence of nitrate following sulfate-reducing conditions. J. Contam. Hydrol. 2016, 187, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.; Weber, K.A. Natural uranium contamination in major U.S. aquifers linked to nitrate. Environ. Sci. Technol. Lett. 2015, 2, 215–220. [Google Scholar] [CrossRef]
- Available online: https://www.epa.gov/ground-water-and-drinking-water/national-primary-drinking-water-regulations (accessed on 1 June 2021).
- Lindahl, P.; Olszewski, G.; Eriksson, M. Performance and optimisation of triple quadrupole ICP-MS for accurate measurement of uranium isotopic ratios. J. Anal. At. Spectrom. 2021, 36, 2164–2172. [Google Scholar] [CrossRef]
- Venus, M.; Puntarić, D.; Gvozdić, V.; Vidosavljević, D.; Bijelić, L.; Puntarić, A.; Puntarić, E.; Vidosavljević, M.; Matijana, J.; Jasenka, S. Determinations of uranium concentrations in soil, water, vegetables and biological samples from inhabitants of war affected areas in eastern Croatia (ICP-MS method). J. Environ. Radioact. 2019, 203, 147–153. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, G.-P.; Xiao, S.-J.; Tan, Q.-G.; Zheng, Q.-Q.; Liang, R.-P.; Qiu, J.-D. Facile construction of covalent organic framework nanozyme for colorimetric detection of uranium. Small 2021, 17, 2102944. [Google Scholar] [CrossRef]
- Wu, X.; Yin, Q.; Huang, Q.; Mao, Y.; Hu, Q.; Wang, H. Rational designing an azo colorimetric sensor with high selectivity and sensitivity for uranium environmental monitoring. Anal. Chim. Acta 2020, 1140, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; He, W.; Chao, H.; Wang, H.; Su, P.; Song, J.; Yang, Y. Insertion of hemin into metal−organic frameworks: Mimicking natural peroxidase microenvironment for the rapid ultrasensitive detection of uranium. Anal. Chem. 2022, 94, 6833–6841. [Google Scholar] [CrossRef]
- Huang, Y.-Q.; Zhang, X.; Xue, J.-H.; Liu, L.; Chen, S.-H.; Wang, Y.-S. Sensitive and selective assay of uranyl based on the aggregation induced fluorescent quenching of protamine capped gold nanoclusters. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 226, 117649. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, S.; Deng, H.; Wu, H.; Chen, J.; Liao, J. Rapid and sensitive detection of uranyl ion with citrate-stabilized silver nanoparticles by the surface-enhanced Raman scattering technique. R. Soc. Open Sci. 2018, 5, 181099. [Google Scholar] [CrossRef]
- Phan, H.T.; Geng, S.; Haes, A.J. Microporous silica membranes promote plasmonic nanoparticle stability for SERS detection of uranyl. Nanoscale 2020, 12, 23700–23708. [Google Scholar] [CrossRef]
- Akl, Z.F. Rapid electrochemical sensor for uranium(VI) assessment in aqueous media. RSC Adv. 2022, 12, 20147. [Google Scholar] [CrossRef] [PubMed]
- Jarczewska, M.; Ziόłkowski, R.; Gόrski, Ł.; Malinowska, E. Electrochemical uranyl cation biosensor with DNA oligonucleotides as receptor layer. Bioelectrochemistry 2014, 96, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Xu, H.; Chen, C.; Cao, X.; Ma, J.; Liu, Y. Determination of U(VI) by differential pulse stripping voltammetry using a polydopamine/reduced graphene oxide nanocomposite modified glassy carbon electrode. Microchem. J. 2022, 175, 107111. [Google Scholar] [CrossRef]
- Roozbahani, G.M.; Chen, X.; Zhang, Y.; Xie, R.; Ma, R.; Li, D.; Li, H.; Guan, X. Peptide-mediated nanopore detection of uranyl ions in aqueous media. ACS Sensors 2017, 2, 703–709. [Google Scholar] [CrossRef]
- Wu, Y.; Lai, R.Y. An electrochemical gold(III) sensor with high sensitivity and tunable dynamic range. Anal. Chem. 2016, 88, 2227–2233. [Google Scholar] [CrossRef]
- Guerreiro, G.V.; Zaitouna, A.J.; Lai, R.Y. Characterization of an electrochemical mercury sensor using alternating current, cyclic, square wave and differential pulse voltammetry. Anal. Chim. Acta 2014, 810, 79–85. [Google Scholar] [CrossRef]
- Wu, Y.; Lai, R.Y. A reagentless DNA-based electrochemical silver(I) sensor for real time detection of Ag(I)—The effect of probe sequence and orientation on sensor response. Biotechnol. J. 2016, 11, 788–796. [Google Scholar] [CrossRef][Green Version]
- Zhad, H.R.L.Z.; Rodriquez Torres, Y.M.; Lai, R.Y. A reagentless and reusable electrochemical aptamer-based sensor for rapid detection of Cd(II). J. Electroanal. Chem. 2017, 803, 89–94. [Google Scholar] [CrossRef]
- Chen, M.D.; Gao, Z.F.; Nian, J.J.; Hong, B.L.; Luo, Q. A sensitive and selective electrochemical biosensor for detection of mercury(II) ions based on nicking endonuclease-assisted signal amplification. Sens. Actuators B Chem. 2015, 210, 290–296. [Google Scholar] [CrossRef]
- Saidur, M.R.; Aziz, A.R.; Basirun, W.J. Recent Advances in DNA-based electrochemical biosensors for heavy metal ion detection: A review. Biosens. Bioelectron. 2017, 90, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.; Gooding, J.J. Peptide modified electrodes as electrochemical metal ion sensors. Electroanalysis 2006, 18, 1437–1448. [Google Scholar] [CrossRef]
- Gooding, J.J.; Hibbert, D.B.; Yang, Y. Electrochemical metal ion sensors. Exploiting amino acids and peptides as recognition elements. Sensors 2001, 1, 75–90. [Google Scholar] [CrossRef]
- Zhad, H.R.L.Z.; Lai, R.Y. Application of calcium-binding motif of E-cadherin for electrochemical detection of Pb(II). Anal. Chem. 2018, 90, 6519–6525. [Google Scholar] [CrossRef]
- Stellato, C.C.; Lai, R.Y. Engineering uranyl-chelating peptides from NikR for electrochemical peptide-based sensing applications. J. Electroanal. Chem. 2020, 858, 113698. [Google Scholar] [CrossRef]
- Sfragano, P.S.; Moro, G.; Polo, F.; Palchetti, I. The role of peptides in the design of electrochemical biosensors for clinical diagnostics. Biosensors 2021, 11, 246. [Google Scholar] [CrossRef]
- Wasilewski, T.; Neubauer, D.; Kamysz, W.; Gębicki, J. Recent progress in the development of peptide-based gas biosensors for environmental monitoring. CSCEE 2022, 5, 100197. [Google Scholar] [CrossRef]
- Mascini, M.; Dikici, E.; Perez-Erviti, J.A.; Deo, S.K.; Compagnone, D.; Daunert, S. A new class of sensing elements for sensors: Clamp peptides for Zika virus. Biosens. Bioelectron. 2021, 191, 113471. [Google Scholar] [CrossRef]
- Cui, M.; Ma, Y.; Wang, L.; Wang, Y.; Wang, S.; Luo, X. Antifouling sensors based on peptides for biomarker detection. Trends Anal. Chem. 2020, 127, 115903. [Google Scholar] [CrossRef]
- Pardoux, R.; Sauge-Merle, S.; Lemaire, D.; Delangle, P.; Guilloreau, L.; Adriano, J.-M.; Berthomieu, C. Modulating uranium binding affinity in engineered calmodulin EF-hand peptides: Effect of phosphorylation. PLoS ONE 2012, 7, e41922. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, C.; Starck, M.; Gathu, V.; Chenavier, Y.; Delangle, P. Engineering short peptide sequences for uranyl binding. Chem. Eur. J. 2014, 20, 16566–16573. [Google Scholar] [CrossRef]
- Sarasa, S.B.; Mahendran, R.; Muthusamy, G.; Thankappan, B.; Selta, D.R.F.; Angayarkanni, J. A brief review on the non-protein amino acid, gamma-amino butyric acid (GABA): Its production and role in microbes. Curr. Microbiol. 2019, 77, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.E.; Carter, J.M. Water-Resources Investigations Report 01–4194; U.S. Geological Survey: Reston, VA, USA, 2001.
- Korshoj, L.E.; Zaitouna, A.J.; Lai, R.Y. Methylene blue-mediated electrocatalytic detection of hexavalent chromium. Anal. Chem. 2015, 87, 2560–2564. [Google Scholar] [CrossRef]
- Patterson, K.Y.; Pehrsson, P.R.; Perry, C.R. The mineral content of tap water in United States households. J. Food Compost. Anal. 2013, 31, 46–50. [Google Scholar] [CrossRef]
- Yang, W.; Gerasimov, J.Y.; Lai, R.Y. Folding-based electrochemical DNA sensor fabricated on a gold-plated screen-printed carbon electrode. Chem. Commun. 2009, 20, 2902–2904. [Google Scholar] [CrossRef]
- Lai, R.Y.; Walker, B.; Stormberg, K.; Zaitouna, A.J.; Yang, W. Electrochemical techniques for characterization of stem-loop probe and linear probe-based DNA sensors. Methods 2013, 64, 267–275. [Google Scholar] [CrossRef]
- Yang, W.; Lai, R.Y. Comparison of the stem-loop and linear probe-based electrochemical DNA sensors by alternating current voltammetry and cyclic voltammetry. Langmuir 2011, 27, 14669–14677. [Google Scholar] [CrossRef]
- Vetter, S.W.; Leclerc, E. Novel aspects of calmodulin target recognition and activation. Eur. J. Biochem. 2003, 270, 404–414. [Google Scholar] [CrossRef]
- Halling, D.B.; Liebeskind, B.J.; Hall, A.M.; Aldrich, R.W. Conserved properties of individual Ca2+-binding sites in calmodulin. Proc. Natl. Acad. Sci. USA 2016, 113, E1216–E1225. [Google Scholar] [CrossRef]
- Finn, B.E.; Forsén, S. The evolving model of calmodulin structure, function and activation. Structure 1995, 3, 7–11. [Google Scholar] [CrossRef]
- Ikura, M.; Hiraoki, T.; Hikichi, K.; Mikuni, T.; Yazawa, M.; Yagi, K. Nuclear magnetic resonance studies on calmodulin: Calcium-induced conformational change. Biochemistry 1983, 22, 2573–2579. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.Y. Calmodulin and its activation by cadmium ions. Ann. N. Y. Acad. Sci. 1988, 522, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Gerasimov, J.Y.; Lai, R.Y. Design and characterization of an electrochemical peptide-based sensor fabricated via “click” chemistry. Chem. Commun. 2011, 47, 8688–8690. [Google Scholar] [CrossRef] [PubMed]
- McQuistan, A.; Zaitouna, A.J.; Echeverria, E.; Lai, R.Y. Use of thiolated oligonucleotides as anti-fouling diluents in electrochemical peptide-based sensors. Chem. Commun. 2014, 50, 4690–4692. [Google Scholar] [CrossRef]
- Gerasimov, J.Y.; Lai, R.Y. An electrochemical peptide-based biosensing platform for HIV detection. Chem. Commun. 2010, 46, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Zaitouna, A.J.; Joyce, J.; Cerny, R.L.; Dussault, P.H.; Lai, R.Y. Comparison of mannose, ethylene glycol and methoxy-terminated diluents on specificity and selectivity of electrochemical peptide-based sensors. Anal. Chem. 2015, 87, 6966–6973. [Google Scholar] [CrossRef]
- Pyo, M.; Jeong, S.-H. pH dependence of electrochemical behaviors of methylene blue on self-assembled monolayers. Bull. Korean Chem. Soc. 1998, 19, 122–124. [Google Scholar]
- Zhao, F.; Zeng, B.; Pang, D. Voltammetric study of methylene blue at thiol SAMs-modified gold electrodes. Electroanalysis 2003, 15, 1060–1066. [Google Scholar] [CrossRef]
- Sumner, J.J.; Creager, S.E. Redox kinetics in monolayers on electrodes: electron transfer is sluggish for ferrocene groups buried within the monolayer interior. J. Phys. Chem. B 2001, 105, 8739–8745. [Google Scholar] [CrossRef]
- Creager, S.E.; Wooster, T.T. A new way of using AC voltammetry to study redox kinetics in electroactive monolayers. Anal. Chem. 1998, 70, 4257–4263. [Google Scholar] [CrossRef]
- Zhao, S.; Yang, W.; Lai, R.Y. A folding-based electrochemical aptasensor for detection of vascular endothelial growth factor in human whole blood. Biosens. Bioelectron. 2011, 26, 2442–2447. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Lai, R.Y.; Heeger, A.J.; Plaxco, K.W.; Sumner, J.J. Effect of molecular crowding on the response of an electrochemical DNA sensor. Langmuir 2007, 23, 6827–6834. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Sensor/Material | Detection Method | LOD (nM) | Dynamic Range or Linear Dynamic Range (µM) | Assay Time (min) | Ref. |
|---|---|---|---|---|---|
| Covalent organic framework nanozyme | UV-Vis (Colorimetric) | 50 | 0.18–75 | 10 | [9] |
| Coordination ligand W1H | UV-Vis (Colorimetric) | 9.33 | 0–16 | 30 | [10] |
| Hemin-modified metal- organic frameworks | UV-Vis (Colorimetric) | 79 | 0.25–40 | 3 | [11] |
| Protamine capped gold nanoclusters | Fluorescence | 6.1 | 0.0204–9.74 | 35 | [12] |
| Citrate-stabilized silver nanoparticles | SERS | 60 | 0.2–5 | NR | [13] |
| Plasmonic nanoparticle | SERS | 110 | 0–13.6 | 30 | [14] |
| Schiff base ionophore | Electrochemical (Potentiometric) | 390 | 1–100,000 | 0.15 | [15] |
| DNA-modified electrode | Electrochemical (SWV) | 30 | 0.1–1 | 60 | [16] |
| Polydopamine/rGO -modified electrode | Electrochemical (DPSV) | 50 | 0.1–50 | 15 | [17] |
| E-PB U-pT-12 sensor | Electrochemical (ACV) | 50 | 0.05–4.5 | ~40 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, C.C.; Lai, R.Y. Threonine Phosphorylation of an Electrochemical Peptide-Based Sensor to Achieve Improved Uranyl Ion Binding Affinity. Biosensors 2022, 12, 961. https://doi.org/10.3390/bios12110961
Thompson CC, Lai RY. Threonine Phosphorylation of an Electrochemical Peptide-Based Sensor to Achieve Improved Uranyl Ion Binding Affinity. Biosensors. 2022; 12(11):961. https://doi.org/10.3390/bios12110961
Chicago/Turabian StyleThompson, Channing C., and Rebecca Y. Lai. 2022. "Threonine Phosphorylation of an Electrochemical Peptide-Based Sensor to Achieve Improved Uranyl Ion Binding Affinity" Biosensors 12, no. 11: 961. https://doi.org/10.3390/bios12110961
APA StyleThompson, C. C., & Lai, R. Y. (2022). Threonine Phosphorylation of an Electrochemical Peptide-Based Sensor to Achieve Improved Uranyl Ion Binding Affinity. Biosensors, 12(11), 961. https://doi.org/10.3390/bios12110961