Advances in the Development of Phage-Based Probes for Detection of Bio-Species

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Fluorescent Signal Generated by Genetically Engineered Phages
3. Formation of Visible Monoclonal Plaques
4. Signals from Nanomaterials Conjugated with Phages
5. Phage-Based PCR for the Detection of Target of Interest
6. Techniques for Editing Phage Genome
6.1. Homologous Recombination Technique for Editing of Phage Genome
6.2. CRISPR-Cas System for Editing of Phage Genome
7. Challenges and Outlook of Phage-Based Biological Detection
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McGrath, S.; Sinderen, D.V. Bacteriophage: Genetics and Molecular Biology; Caister Academic Press: Norfolk, UK, 2007. [Google Scholar]
- De Paepe, M.; Leclerc, M.; Tinsley, C.R.; Petit, M.A. Bacteriophages: An underestimated role in human and animal health? Front. Cell. Infect. Microbiol. 2014, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef]
- Koonin, E.V. The wonder world of microbial viruses. Expert Rev. Anti-Infect. Ther. 2010, 8, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Keen, E.C. A century of phage research: Bacteriophages and the shaping of modern biology. Bioessays 2015, 37, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Sunderland, K.S.; Yang, M.; Mao, C. Phage-enabled nanomedicine: From probes to therapeutics in precision medicine. Angew. Chem. Int. Ed. 2017, 56, 1964–1992. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Moon, J.S.; Choi, E.J.; Jeong, N.N.; Sohn, J.R.; Han, D.W.; Oh, J.W. Research Progress of M13 Bacteriophage-Based Biosensors. Nanomaterials 2019, 9, 1448. [Google Scholar] [CrossRef]
- Gamkrelidze, M.; Dąbrowska, K. T4 bacteriophage as a phage display platform. Arch. Microbiol. 2014, 196, 473–479. [Google Scholar] [CrossRef]
- Yue, H.; Li, Y.; Yang, M.; Mao, C. T7 Phage as an Emerging Nanobiomaterial with Genetically Tunable Target Specificity. Adv. Sci. 2021, e2103645. [Google Scholar] [CrossRef]
- Trinh, J.T.; Alkahtani, M.H.; Rampersaud, I.; Rampersaud, A.; Scully, M.; Young, R.F.; Hemmer, P.; Zeng, L. Fluorescent nanodiamond-bacteriophage conjugates maintain host specificity. Biotechnol. Bioeng. 2018, 115, 1427–1436. [Google Scholar] [CrossRef]
- Kehoe, J.W.; Kay, B.K. Filamentous Phage Display in the New Millennium. Chem. Rev. 2005, 105, 4056–4072. [Google Scholar] [CrossRef] [PubMed]
- Malys, N.; Chang, D.Y.; Baumann, R.G.; Xie, D.; Black, L.W. A Bipartite Bacteriophage T4 SOC and HOC Randomized Peptide Display Library: Detection and Analysis of Phage T4 Terminase (gp17) and Late σ Factor (gp55) Interaction. J. Mol. Biol. 2002, 319, 289–304. [Google Scholar] [CrossRef]
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef]
- Loessner, M.J.; Rees, C.E.D.; Stewart, G.S.A.B.; Scherer, S. Construction of luciferase reporter bacteriophage A511::luxAB for rapid and sensitive detection of viable Listeria cells. Appl. Environ. Microbiol. 1996, 62, 1133–1140. [Google Scholar] [CrossRef]
- Jaye, D.L.; Geigerman, C.M.; Fuller, R.E.; Akyildiz, A.; Parkos, C.A. Direct fluorochrome labeling of phage display library clones for studying binding specificities: Applications in flow cytometry and fluorescence microscopy. J. Immunol. Methods 2004, 295, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Slootweg, E.J.; Keller, H.J.H.G.; Hink, M.A.; Borst, J.W.; Bakker, J.; Schots, A. Fluorescent T7 display phages obtained by translational frameshift. Nucleic Acids Res. 2006, 34, 137. [Google Scholar] [CrossRef]
- Tanji, Y.; Furukawa, C.; Na, S.H.; Hijikata, T.; Miyanaga, K.; Unno, H. Escherichia coli detection by GFP-labeled lysozyme-inactivated T4 bacterio-phage. J. Biotechnol. 2004, 114, 11–20. [Google Scholar] [CrossRef]
- Bakhshinejad, B. Bacteriophages and their applications in the diagnosis and treatment of hepatitis B virus infection. World J. Gastroenterol. 2014, 20, 11671. [Google Scholar] [CrossRef]
- Schofield, D.; Sharp, N.J.; Westwater, C. Phage-based platforms for the clinical detection of human bacterial pathogens. Bacteriophage 2012, 2, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Warner, C.M.; Jin, H.E.; Barnes, E.; Poda, A.R.; Perkins, E.J.; Lee, S.W. Production of tunable nanomaterials using hierarchically assembled bacteriophages. Nat. Protoc. 2017, 12, 1999–2013. [Google Scholar] [CrossRef] [PubMed]
- Hasmoni, S.S.; Yusoff, K.; Tan, W.S. Detection and precipitation of hepatitis B core antigen using a fusion bacteriophage. J. Gen. Appl. Microbiol. 2005, 51, 125–131. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, S.; Qi, J.; de Quilettes, D.W.; Huang, M.; Lin, C.W.; Bardhan, N.M.; Dang, X.; Bulović, V.; Belcher, A.M. M13 Virus-Based Framework for High Fluorescence Enhancement. Small 2019, 15, e1901233. [Google Scholar] [CrossRef] [PubMed]
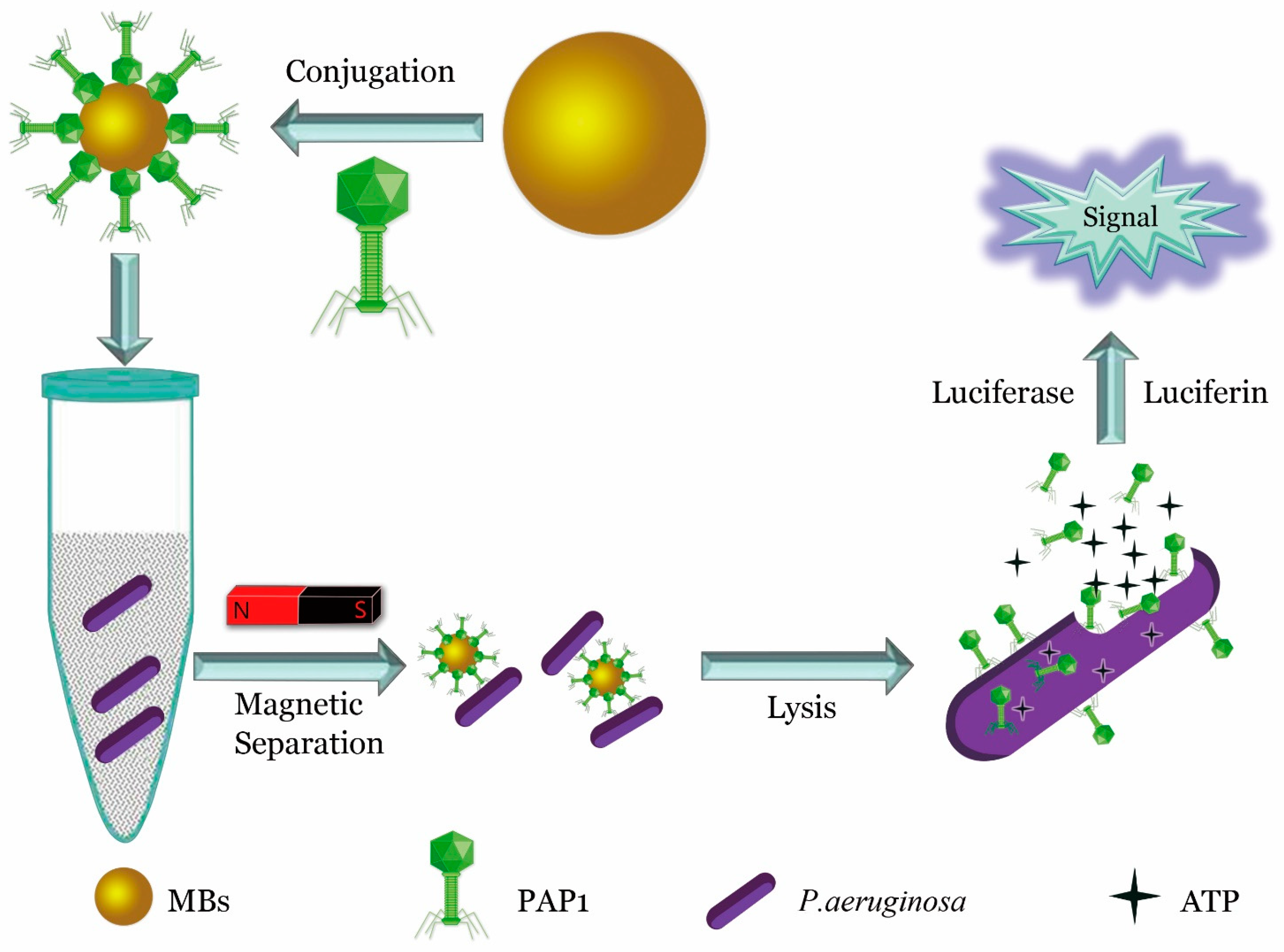
- He, Y.; Wang, M.; Fan, E.; Ouyang, H.; Yue, H.; Su, X.; Liao, G.; Wang, L.; Lu, S.; Fu, Z. Highly Specific Bacteriophage-Affinity Strategy for Rapid Separation and Sensitive Detection of Viable Pseudomonas aeruginosa. Anal. Chem. 2017, 89, 1916–1921. [Google Scholar] [CrossRef] [PubMed]
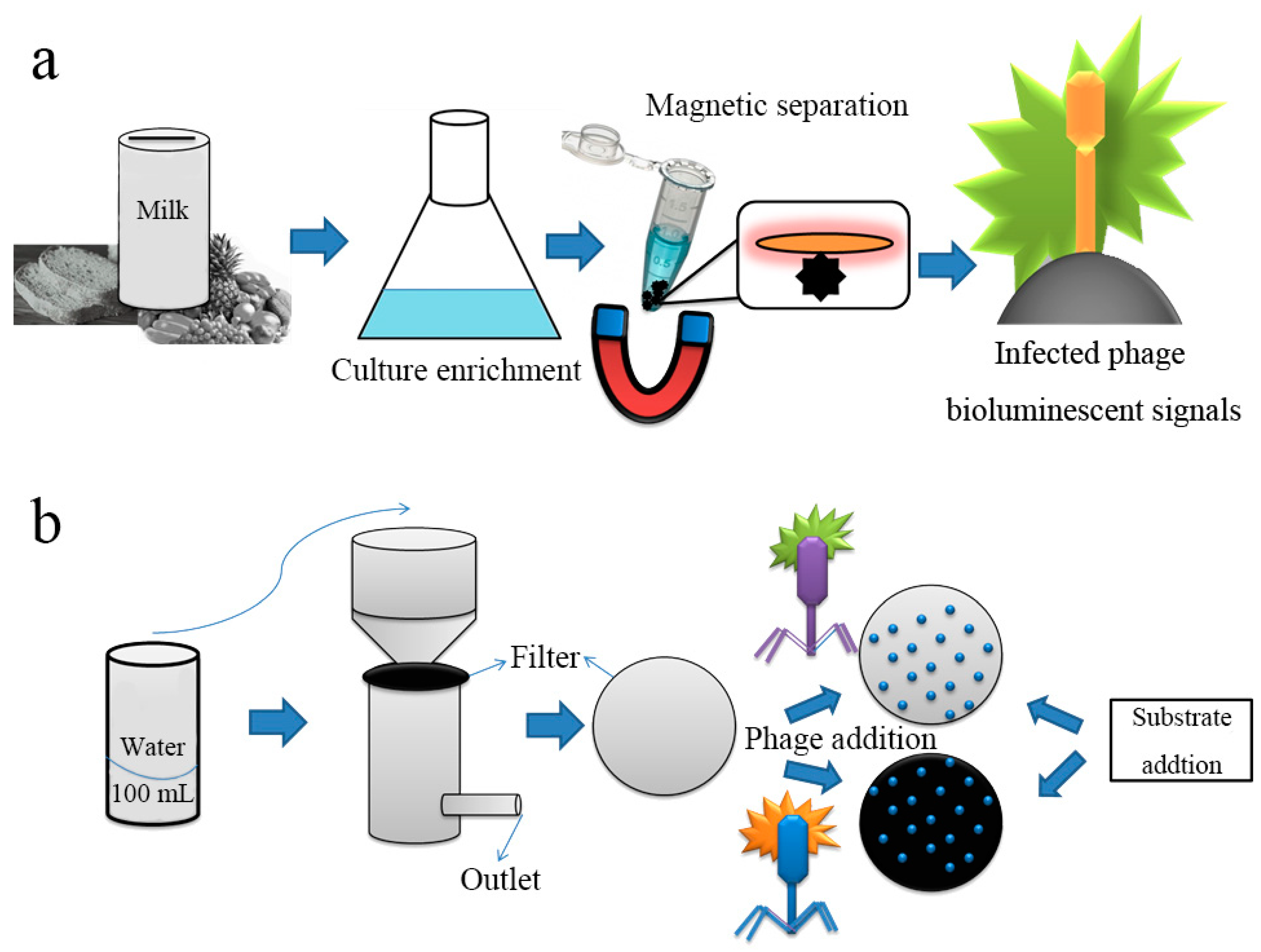
- Kretzer, J.W.; Schmelcher, M.; Loessner, M.J. Ultrasensitive and Fast Diagnostics of Viable Listeria Cells by CBD Magnetic Separation Combined with A511::luxAB Detection. Viruses 2018, 10, 626. [Google Scholar] [CrossRef] [PubMed]
- Hinkley, T.C.; Singh, S.; Garing, S.; Le Ny, A.M.; Nichols, K.P.; Peters, J.E.; Talbert, J.N.; Nugen, S.R. A phage-based assay for the rapid, quantitative, and single CFU visualization of E. coli (ECOR #13) in drinking water. Sci. Rep. 2018, 8, 14630. [Google Scholar] [PubMed]
- Chen, A.; Wang, D.; Nugen, S.R.; Chen, J. An Engineered Reporter Phage for the Fluorometric Detection of Escherichia coli in Ground Beef. Microorganisms 2021, 9, 436. [Google Scholar] [CrossRef]
- Chanishvili, N. Phage therapy—History from Twort and d’Herelle through Soviet experience to current approaches. Adv. Virus Res. 2012, 83, 3–40. [Google Scholar] [PubMed]
- Kafatos, G.; Andrews, N.; Gillespie, I.A.; Charlett, A.; Adak, G.K.; De Pinna, E.; Threlfall, E.J. Impact of reduced numbers of isolates phage-typed on the detection of Salmonella outbreaks. Epidemiol. Infect. 2009, 137, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Mi, X.; He, F.; Xiang, M.; Lian, Y.; Yi, S. Novel phage amplified multichannel series piezoelectric quartz crystal sensor for rapid and sensitive detection of Mycobacterium tuberculosis. Anal. Chem. 2012, 84, 939–946. [Google Scholar] [CrossRef]
- Goodridge, L.; Chen, J.; Griffiths, M. Development and characterization of a fluorescent-bacteriophage assay for detection of Escherichia coli O157:H7. Appl. Environ. Microbiol. 1999, 65, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Smartt, A.E.; Xu, T.; Jegier, P.; Carswell, J.J.; Blount, S.A.; Sayler, G.S.; Ripp, S. Pathogen detection using engineered bacteriophages. Anal. Bioanal. Chem. 2012, 402, 3127–3146. [Google Scholar] [CrossRef] [PubMed]
- Tawil, N.; Sacher, E.; Mandeville, R.; Mandeville, R.; Meunier, M. Bacteriophages: Biosensing tools for multi-drug resistant pathogens. Analyst 2014, 139, 1224–1236. [Google Scholar] [CrossRef]
- Van Der Merwe, R.G.; Van Helden, P.D.; Warren, R.M.; Sampson, S.L.; van Pittius, N.G. Phage-based detection of bacterial pathogens. Analyst 2014, 139, 2617–2626. [Google Scholar] [CrossRef]
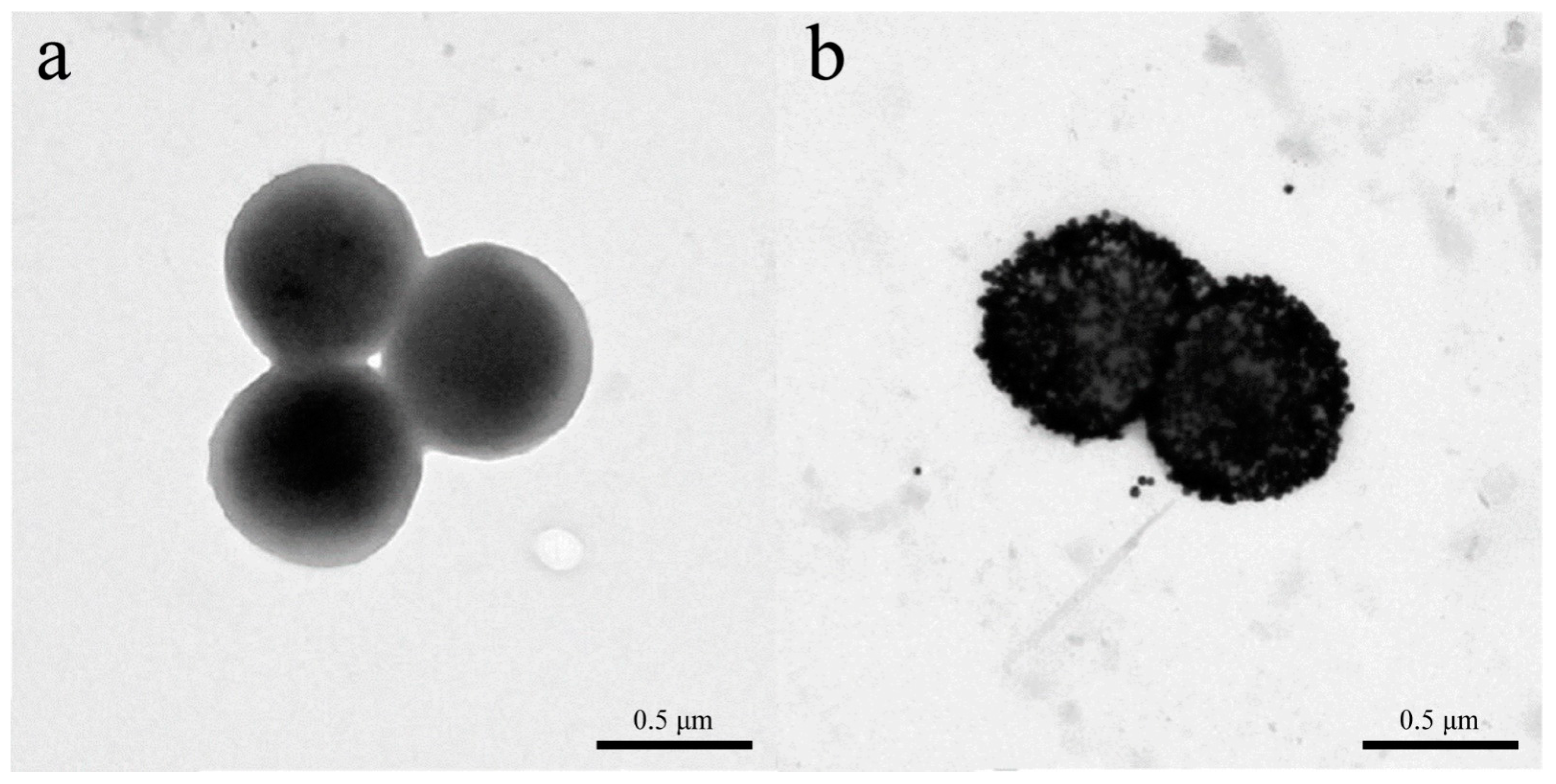
- Zhou, X.; Cao, P.; Zhu, Y.; Lu, W.; Gu, N.; Mao, C. Phage-mediated counting by the naked eye of miRNA molecules at attomolar concentrations in a Petri dish. Nat. Mater. 2015, 14, 1058–1064. [Google Scholar] [CrossRef]
- Peng, H.; Chen, I.A. Rapid Colorimetric Detection of Bacterial Species through the Capture of Gold Nanoparticles by Chimeric Phages. ACS Nano 2019, 13, 1244–1252. [Google Scholar] [CrossRef]
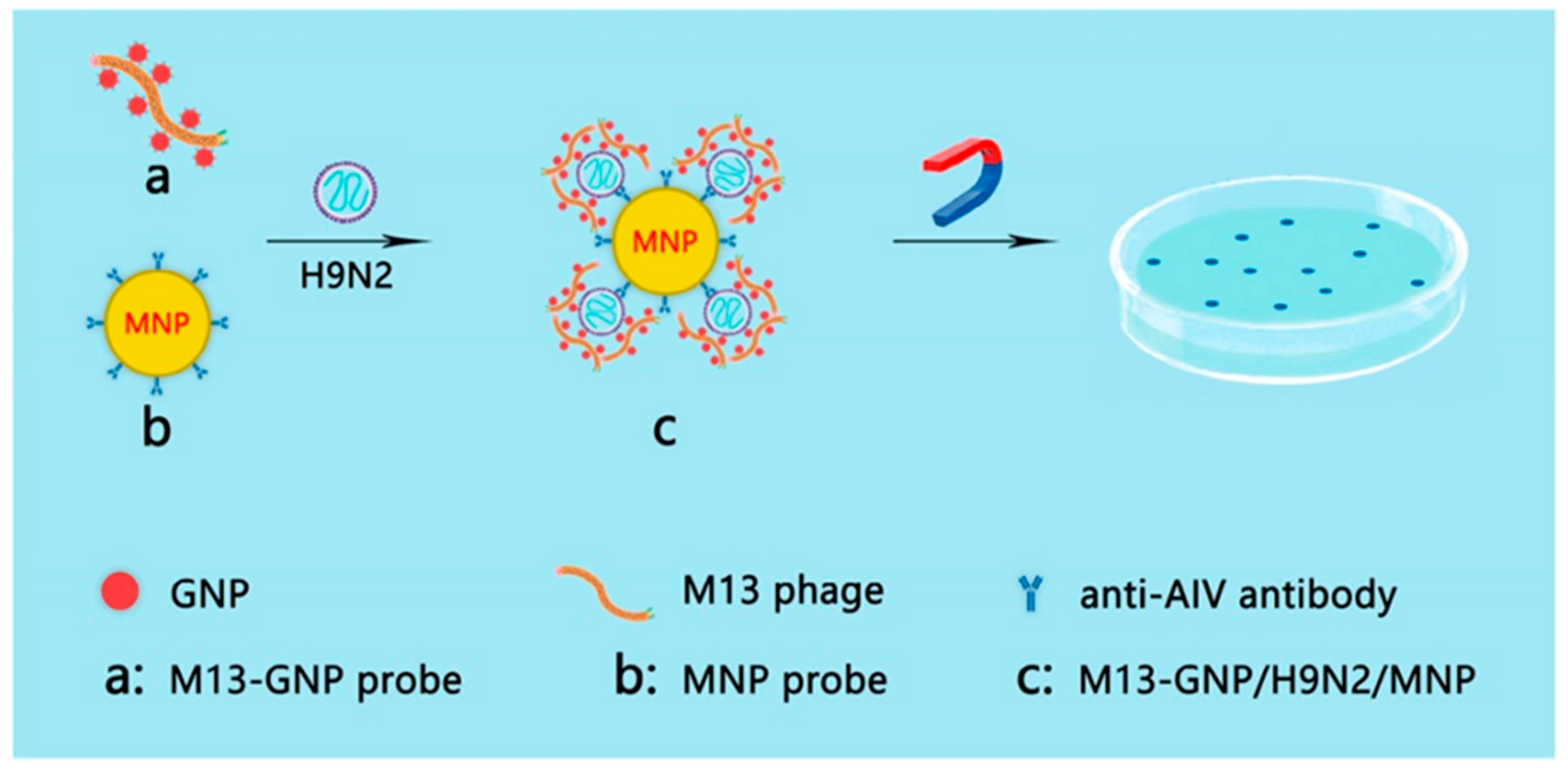
- Xu, H.; Shen, J.; Yang, C.T.; Thierry, B.; Zhu, Y.; Mao, C.B.; Zhou, X. Naked-eye counting of pathogenic viruses by phage-gold nanobiomaterials as probes. Mater. Today Adv. 2021, 10, 100122. [Google Scholar] [CrossRef]
- Paczesny, J.; Bielec, K. Application of bacteriophages in nanotechnology. Nanomaterials 2020, 10, 1944. [Google Scholar] [CrossRef]
- Chi, X.; Huang, D.; Zhao, Z.; Zhou, Z.; Yin, Z.; Gao, J. Nanoprobes for in vitro diagnostics of cancer and infectious diseases. Biomaterials 2012, 33, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; McKinstry, M.; Hwang, J.; Oppenheim, A.B.; Fekete, R.A.; Giulian, G.; Merril, C.; Nagashima, K.; Adhya, S. High-sensitivity bacterial detection using biotin-tagged phage and quantum-dot nanocomplexes. Proc. Natl. Acad. Sci. USA 2006, 103, 4841–4845. [Google Scholar] [CrossRef]
- Wang, F.; Horikawa, S.; Hu, J.; Wikle, H.C.; Chen, I.H.; Du, S.; Liu, Y.; Chin, B.A. Detection of Salmonella typhimurium on Spinach Using Phage-Based Magnetoelastic Biosensors. Sensors 2017, 17, 386. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ju, Z.; Cao, B.; Gao, X.; Zhu, Y.; Qiu, P.; Xu, H.; Pan, P.; Bao, H.; Wang, L.; et al. Ultrasensitive rapid detection of human serum antibody biomarkers by biomarker-capturing viral nanofibers. ACS Nano 2015, 9, 4475–4483. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Han, L.; Wang, F.; Petrenko, V.A.; Liu, A. Gold nanoprobe functionalized with specific fusion protein selection from phage display and its application in rapid, selective and sensitive colorimetric biosensing of Staphylococcus aureus. Biosens. Bioelectron. 2016, 82, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, T.; Zhang, X.; Chen, M.; Wang, J. In situ growth of gold nanoparticles on Hg2+-binding M13 phages for mercury sensing. Nanoscale 2017, 9, 16728–16734. [Google Scholar] [CrossRef]
- Xie, J.; Zheng, Y.; Ying, J. Highly selective and ultrasensitive detection of Hg(2+) based on fluorescence quenching of Au nanoclusters by Hg(2+)-Au(+) interactions. Chem. Commun. 2010, 46, 961–963. [Google Scholar] [CrossRef]
- De Plano, L.M.; Scibilia, S.; Rizzo, M.G.; Crea, S.; Franco, D.; Mezzasalma, A.M.; Guglielmino, S.P. One-step production of phage–silicon nanoparticles by PLAL as fluorescent nanoprobes for cell identification. Appl. Phys. 2018, 124, 222. [Google Scholar] [CrossRef]
- Lai, J.Y.; Inoue, N.; Oo, C.W.; Kawasaki, H.; Lim, T.S. One-step synthesis of M13 phage-based nanoparticles and their fluorescence properties. RSC Adv. 2021, 11, 1367–1375. [Google Scholar] [CrossRef]
- Chen, J.; Huang, Y.; Zhu, C.; Li, Q.; Wu, Y.; Liu, Q.; Cheng, Q. Early detection of Alzheimer’s disease by peptides from phage display screening. Brain Res. 2019, 1721, 146306. [Google Scholar] [CrossRef]
- Peng, H.; Borg, R.E.; Nguyen, A.; Chen, I.A.; Nguyen, A.; Chen, I.A. Chimeric Phage Nanoparticles for Rapid Characterization of Bacterial Pathogens: Detection in Complex Biological Samples and Determination of Antibiotic Sensitivity. ACS Sens. 2020, 5, 1491–1499. [Google Scholar] [CrossRef]
- Ceppi, L.; Bardhan, N.M.; Na, Y.; Siegel, A.; Rajan, N.; Fruscio, R.; Del Carmen, M.G.; Belcher, A.M.; Birrer, M.J. Real-time single-walled carbon nanotube-based fluorescence imaging improves survival after debulking surgery in an ovarian cancer model. ACS Nano 2019, 13, 5356–5365. [Google Scholar] [CrossRef]
- Fiskin, E.; Lareau, C.A.; Ludwig, L.S.; Eraslan, G.; Liu, F.; Ring, A.M.; Xavier, R.J.; Regev, A. Single-cell profiling of proteins and chromatin accessibility using PHAGE-ATAC. Nat. Biotechnol. 2021, 1–8. [Google Scholar] [CrossRef]
- Rusling, J.F.; Kumar, C.V.; Gutkind, J.S.; Patel, V. Measurement of biomarker proteins for point-of-care early detection and monitoring of cancer. Analyst 2010, 135, 2496–2511. [Google Scholar] [CrossRef]
- Vandenberg, O.; Martiny, D.; Rochas, O.; van Belkum, A.; Kozlakidis, Z. Considerations for diagnostic COVID-19 tests. Nat. Rev. Microbiol. 2021, 19, 171–183. [Google Scholar] [CrossRef]
- Shen, M.; Li, N.; Lu, Y.; Cheng, J.; Xu, Y. An enhanced centrifugation-assisted lateral flow immunoassay for the point-of-care detection of protein biomarkers. Lab Chip 2020, 20, 2626–2634. [Google Scholar] [CrossRef]
- Assumpção, A.L.; da Silva, R.C. Immuno-PCR in cancer and non-cancer related diseases: A review. Vet. Q. 2016, 36, 63–70. [Google Scholar] [CrossRef]
- Sano, T.; Smith, C.L.; Cantor, C.R. Immuno-PCR: Very sensitive antigen detection by means of specific antibody-DNA conjugates. Science 1992, 258, 120–122. [Google Scholar] [CrossRef]
- Rizzo, M.G.; Carnazza, S.; De Plano, L.M.; Franco, D.; Guglielmino, S. Rapid detection of bacterial pathogens in blood through engineered phages-beads and integrated real-time pcr into microchip. Sens. Actuators Chem. 2021, 329, 129227. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Adler, M.; Pignataro, B.; Lenhert, S.; Gao, S.; Chi, L.; Fuchs, H.; Blohm, D. Self-assembly of DNA-streptavidin nanostructures and their use as reagents in immuno-PCR. Nucleic Acids Res. 1999, 27, 4553–4561. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Wacker, R.; Adler, M. Hapten-Functionalized DNA-Streptavidin Nanocircles as Supramolecular Reagents in a Competitive Immuno-PCR Assay. Angew. Chem. Int. Ed. 2001, 40, 3169–3172. [Google Scholar] [CrossRef]
- Clement, J.P. Hantavirus. Antivir. Res. 2003, 57, 121–127. [Google Scholar] [CrossRef]
- Pan, T.; Chang, B.; Wong, P.; Li, C.; Li, R.; Kang, S.C.; Robinson, J.D.; Thompsett, A.R.; Tein, P.; Yin, S.; et al. An aggregation-specific enzyme-linked immunosorbent assay: Detection of conformational differences between recombinant PrP protein dimers and PrP(Sc) aggregates. J. Virol. 2005, 79, 12355–12364. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wuertzer, C.A.; Sullivan, M.A.; Qiu, X.; Federoff, H.J. CNS delivery of vectored prion-specific single-chain antibodies delays disease onset. Mol. Ther. 2008, 16, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.C.; Zhou, Y.F.; Zhang, X.E.; Zhang, Z.P.; Qiao, Y.M.; Bi, L.J.; Wen, J.K.; Liang, M.F.; Zhang, J.B. Phage display mediated immuno-PCR. Nucleic Acids Res. 2006, 34, e62. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, Q.; Xu, Y.; Liu, X.; Shu, M.; Tu, Z.; Li, Y.; Wang, W.; Cao, D. Anti-idiotypic VHH phage display-mediated immuno-PCR for ultrasensitive determination of mycotoxin zearalenone in cereals. Talanta 2016, 147, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, Y.; Huang, Q.; Yi, C.; Xiao, T.; Li, Q. Natural phage nanoparticle-mediated real-time immuno-PCR for ultrasensitive detection of protein marker. Chem. Commun. 2013, 49, 3778. [Google Scholar] [CrossRef]
- Hou, J.; Shen, J.; Zhao, N.; Yang, C.T.; Thierry, B.; Zhou, X.; Zhu, J.; Mao, C. Detection of a single circulating tumor cell using a genetically engineered antibody-like phage nanofiber probe. Mater. Today Adv 2021, 12, 100168. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, Q.; Wu, W.; Yan, T.; Tang, X.; Zhang, W.; Yu, L.; Li, P. Anti-idiotypic nanobody-phage display-mediated real-time immuno-PCR for sensitive, simultaneous and quantitative detection of total aflatoxins and zearalenone in grains. Food Chem. 2019, 297, 124912. [Google Scholar] [CrossRef]
- Nzuma, R.M.; Liu, F.; Grant, I.R. Generation and characterization of a novel recombinant scFv antibody specific for Campylobacter jejuni. Appl. Microbiol. Biotechnol. 2018, 102, 4873–4885. [Google Scholar] [CrossRef]
- Garrido-Maestu, A.; Fuciños, P.; Azinheiro, S.; Carvalho, C.; Carvalho, J.; Prado, M. Specific detection of viable Salmonella Enteritidis by phage amplification combined with qPCR (PAA-qPCR) in spiked chicken meat samples. Food Control 2019, 99, 79–83. [Google Scholar] [CrossRef]
- Luo, J.; Jiang, M.; Xiong, J.; Li, J.; Wei, H.; Yu, J. Rapid Ultrasensitive Diagnosis of Pneumonia Caused by Acinetobacter Baumannii Using a Combination of Enrichment and Phage-Based qPCR Assay; Research Square: Durham, NC, USA, 2020. [Google Scholar]
- Karam, J.D. Molecular Biology of Bacteriophage T4 Washington; American Society for Microbiology: Washington, DC, USA, 1994. [Google Scholar]
- Oda, M.; Morita, M.; Unno, H.; Tanji, Y. Rapid detection of Escherichia coli O157:H7 by using green fluorescent protein-labeled PP01 bacteriophage. Appl. Environ. Microbiol. 2004, 70, 527–534. [Google Scholar] [CrossRef]
- Namura, M.; Hijikata, T.; Miyanaga, K.; Tanji, Y. Detection of Escherichia coli with fluorescent labeled phages that have a broad host range to E. coli in sewage water. Biotechnol. Prog. 2008, 24, 481–486. [Google Scholar] [CrossRef]
- Sarkis, G.J.; Jacobs, W.R., Jr.; Hatfull, G.F. L5 luciferase reporter mycobacteriophages: A sensitive tool for the detection and assay of live mycobacteria. Mol. Microbiol. 1995, 15, 1055–1067. [Google Scholar] [CrossRef]
- Rao, V.B.; Mitchell, M.S. The N-terminal ATPase site in the large terminase protein gp17 is critically required for DNA packaging in bacteriophage T4. J. Mol. Biol. 2001, 314, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.J.; Piuri, M.; Swigonova, Z.; Balachandran, A.; Oldfield, L.M.; Van Kessel, J.C.; Hatfull, G.F. BRED: A simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS ONE 2008, 3, e3957. [Google Scholar] [CrossRef] [PubMed]
- Thomason, L.C.; Oppenheim, A.B.; Court, D.L. Modifying bacteriophage lambda with recombineering. Methods Mol. Biol. 2009, 501, 239–251. [Google Scholar] [PubMed]
- Marinelli, L.J.; Hatfull, G.F.; Piuri, M. Recombineering: A powerful tool for modification of bacteriophage genomes. Bacteriophage 2012, 2, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.C. Phage recombinases and their applications. Adv. Virus Res. 2012, 83, 367–414. [Google Scholar] [PubMed]
- Nafissi, N.; Slavcev, R. Bacteriophage recombination systems and biotechnical applications. Appl. Microbiol. Biotechnol. 2014, 98, 2841–2851. [Google Scholar] [CrossRef]
- Erickson, S.; Paulson, J.; Brown, M.; Hahn, W.; Gil, J.; Barron-Montenegro, R.; Moreno-Switt, A.I.; Eisenberg, M.; & Nguyen, M.M. Isolation and engineering of a Listeria grayi bacteriophage. Sci. Rep. 2021, 11, 18947. [Google Scholar] [CrossRef]
- Masuda, Y.; Kawabata, S.; Uedoi, T.; Honjoh, K.I.; Miyamoto, T. Construction of Leaderless-Bacteriocin-Producing Bacteriophage Targeting, E. coli and Neighboring Gram-Positive Pathogens. Microbiol. Spectr. 2021, 9, e0014121. [Google Scholar] [CrossRef]
- Haft, D.H.; Selengut, J.; Mongodin, E.F.; Nelson, K.E. A Guild of 45 CRISPR-Associated (Cas) Protein Families and Multiple CRISPR/Cas Subtypes Exist in Prokaryotic Genomes. PLoS Comput. Biol. 2005, 1, 60. [Google Scholar] [CrossRef]
- Godde, J.S.; Bickerton, A. The Repetitive DNA Elements Called CRISPRs and Their Associated Genes: Evidence of Horizontal Transfer among Prokaryotes. J. Mol. Evol. 2006, 62, 718–729. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Hatoum-Aslan, A. Phage Genetic Engineering Using CRISPR⁻Cas Systems. Viruse 2018, 10, 335. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Martel, B.; Moineau, S. CRISPR-Cas: An efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 2014, 42, 9504–9513. [Google Scholar] [CrossRef] [PubMed]
- Box, A.M.; McGuffie, M.J.; O’Hara, B.J.; Seed, K.D. Functional analysis of bacteriophage immunity through a Type I-E CRISPR-Cas system in Vibrio cholerae and its application in bacteriophage genome engineering. J. Bacteriol. 2015, 198, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Erdmann, S.; Mojica, F.J.M.; Garrett, R.A. Protospacer recognition motifs: Mixed identities and functional diversity. RNA Biol. 2013, 10, 891–899. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef]
- Mohanraju, P.; Makarova, K.S.; Zetsche, B.; Zhang, F.; Koonin, E.V.; van der Oost, J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 2016, 353, 5147. [Google Scholar] [CrossRef]
- Lemay, M.L.; Tremblay, D.M.; Moineau, S. Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth. Biol. 2017, 21, 1351–1358. [Google Scholar] [CrossRef]
- Tao, P.; Wu, X.; Tang, W.C.; Zhu, J.; Rao, V. Engineering of Bacteriophage T4 Genome Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1952–1961. [Google Scholar] [CrossRef]
- Lee, C.M.; Davis, T.H.; Bao, G. Examination of CRISPR/Cas9 design tools and the effect of target site accessibility on Cas9 activity. Exp. Physiol. 2017, 103, 456–460. [Google Scholar] [CrossRef]
- Mohr, S.E.; Hu, Y.; Ewen-Campen, B.; Housden, B.E.; Viswanatha, R.; Perrimon, N. CRISPR guide RNA design for research applications. FEBS J. 2016, 283, 3232–3238. [Google Scholar] [CrossRef]
- Dang, Y.; Jia, G.; Choi, J.; Ma, H.; Anaya, E.; Ye, C.; Shankar, P.; Wu, H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015, 16, 280. [Google Scholar] [CrossRef]
- Sharan, S.K.; Thomason, L.C.; Kuznetsov, S.G.; Court, D.L. Recombineering: A homologous recombination-based method of genetic engineering. Nat. Protoc. 2009, 4, 206–223. [Google Scholar] [CrossRef]
- Kosuri, S.; Church, G.M. Large-scale de novo DNA synthesis: Technologies and applications. Nat. Methods 2014, 11, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Hupfeld, M.; Trasanidou, D.; Ramazzini, L.; Klumpp, J.; Loessner, M.J.; Kilcher, S. A functional type II-A CRISPR-Cas system from Listeria enables efficient genome editing of large non-integrating bacteriophage. Nucleic Acids Res. 2018, 46, 6920–6933. [Google Scholar] [CrossRef]
- Bari, S.M.N.; Walker, F.C.; Cater, K.; Aslan, B.; Hatoum-Aslan, A. Strategies for Editing Virulent Staphylococcal Phages Using CRISPR-Cas10. ACS Synth. Biol. 2017, 6, 2316–2325. [Google Scholar] [CrossRef]
- Deng, X.; Wang, L.; You, X.; Dai, P.; Zeng, Y. Advances in the T7 phage display system. Mol. Med. Rep. 2018, 17, 714–720. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paramasivam, K.; Shen, Y.; Yuan, J.; Waheed, I.; Mao, C.; Zhou, X. Advances in the Development of Phage-Based Probes for Detection of Bio-Species. Biosensors 2022, 12, 30. https://doi.org/10.3390/bios12010030
Paramasivam K, Shen Y, Yuan J, Waheed I, Mao C, Zhou X. Advances in the Development of Phage-Based Probes for Detection of Bio-Species. Biosensors. 2022; 12(1):30. https://doi.org/10.3390/bios12010030
Chicago/Turabian StyleParamasivam, Kameshpandian, Yuanzhao Shen, Jiasheng Yuan, Ibtesam Waheed, Chuanbin Mao, and Xin Zhou. 2022. "Advances in the Development of Phage-Based Probes for Detection of Bio-Species" Biosensors 12, no. 1: 30. https://doi.org/10.3390/bios12010030
APA StyleParamasivam, K., Shen, Y., Yuan, J., Waheed, I., Mao, C., & Zhou, X. (2022). Advances in the Development of Phage-Based Probes for Detection of Bio-Species. Biosensors, 12(1), 30. https://doi.org/10.3390/bios12010030