1. Introduction
For basic and applied research of disease, efficient development of new drugs, innovative development of precision medicine, and reduction of animal experimentation, it is necessary to construct an in vitro cell model that precisely imitates the in vivo tumor environment. The conventional two-dimensional (2D) cell models significantly differ from the in vivo environment, producing cellular behavior dissimilar to that of natural conditions. This is because the cell-to-cell and cell-to-extracellular matrix (ECM) interactions within the actual body, as well as the expression of genes, are not properly achieved [
1,
2,
3]. Thereby, three-dimensional (3D) cell culture models have recently been introduced and great advancements have been made in the field [
4,
5,
6]. Out of the numerous types of 3D cell culture models, spheroid, which is a 3D aggregate of tumor cells in a spherical shape, is being actively explored due to its relatively simple construction and wide applicability in terms of cell types and uses [
7,
8]. Since spheroids can also be manipulated to mimic the 3D in vivo tumor structures, they have been used as a tumor model for various applications, such as investigating the mechanisms of drug response and delivery, testing anti-cancer effects, or even simulating a perfusable blood vessel network using microfluidic technologies [
3,
9,
10,
11]. In addition, many tumor spheroid-embedded hydrogel models have been built using collagen and matrigel to recapitulate the features and environment of the ECM that structurally and biochemically support the tumor cells [
12,
13,
14,
15,
16,
17]. By varying the composition, type, and organization of the protein-based hydrogel, the tumor spheroid-embedded hydrogel models serve as a useful platform for investigating invasive and metastatic characteristics, effects of drug treatment, and other biochemical cues that regulate tumor cell behavior [
18].
Despite the wide applicability and use of the spheroid-embedded hydrogel models, the process of culturing spheroids and handling them for embedding into hydrogel has several limitations. Currently, the most common method of constructing a spheroid is the hanging drop method, which simply requires micro-sized wells where the liquid is deposited and hung as a drop. However, the operational procedures of this conventional technique are inefficient and complex because the relocation of the spheroid and the replacement of the medium are carried out using a pipette. Several studies have reported that the spheroids are collected using a pipette tip and embedded into the hydrogel for further analysis [
3,
12,
19]. However, such a method of handling spheroids often results in well-to-well variation and spheroids being damaged or even lost [
20,
21,
22].
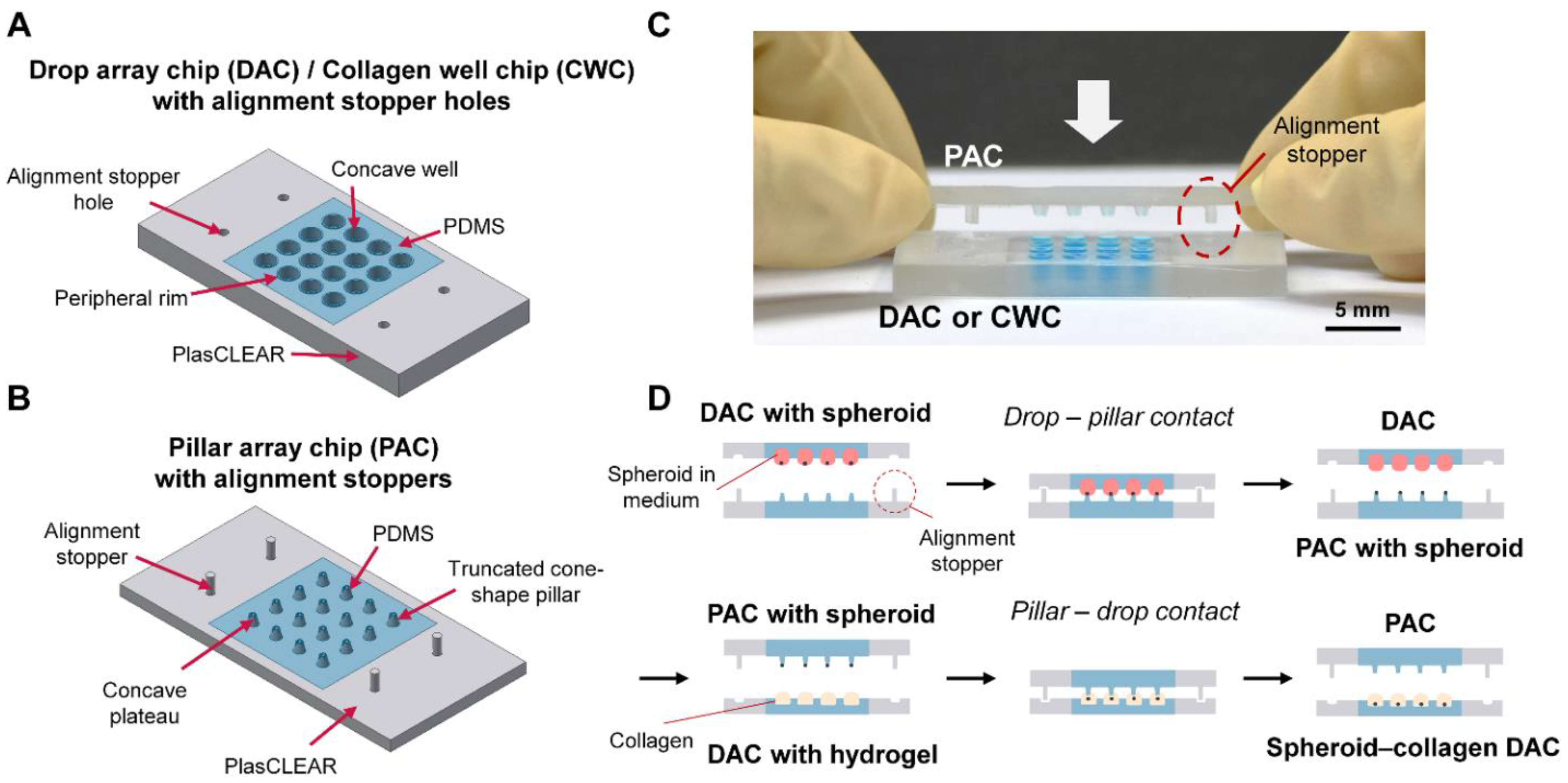
Few studies were reported to overcome the labor-intensive and time-consuming problem of handling the spheroids by constructing agarose microwells with slopes of different angles to exchange the medium without damaging the spheroids [
20]. Our group has also previously reported a novel droplet contact-based spheroid transfer (DCST) technique and array chips for simultaneous collection and transplantation of spheroids [
23,
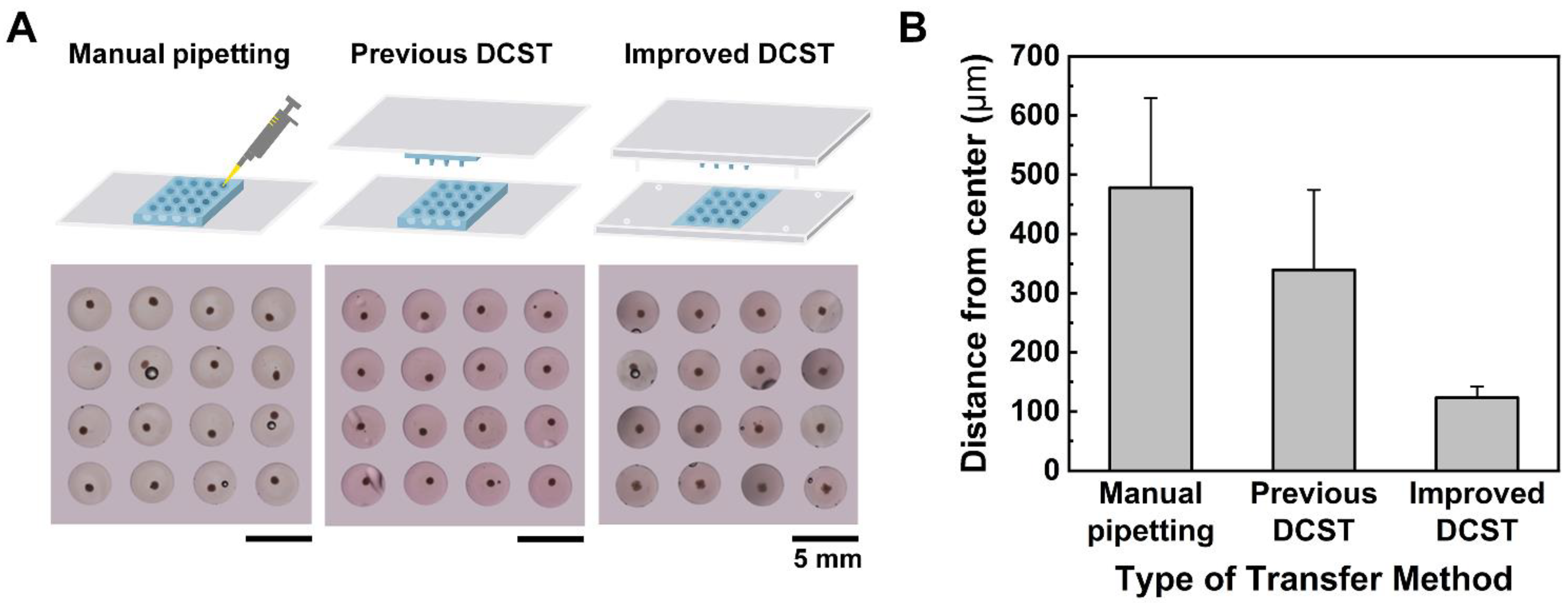
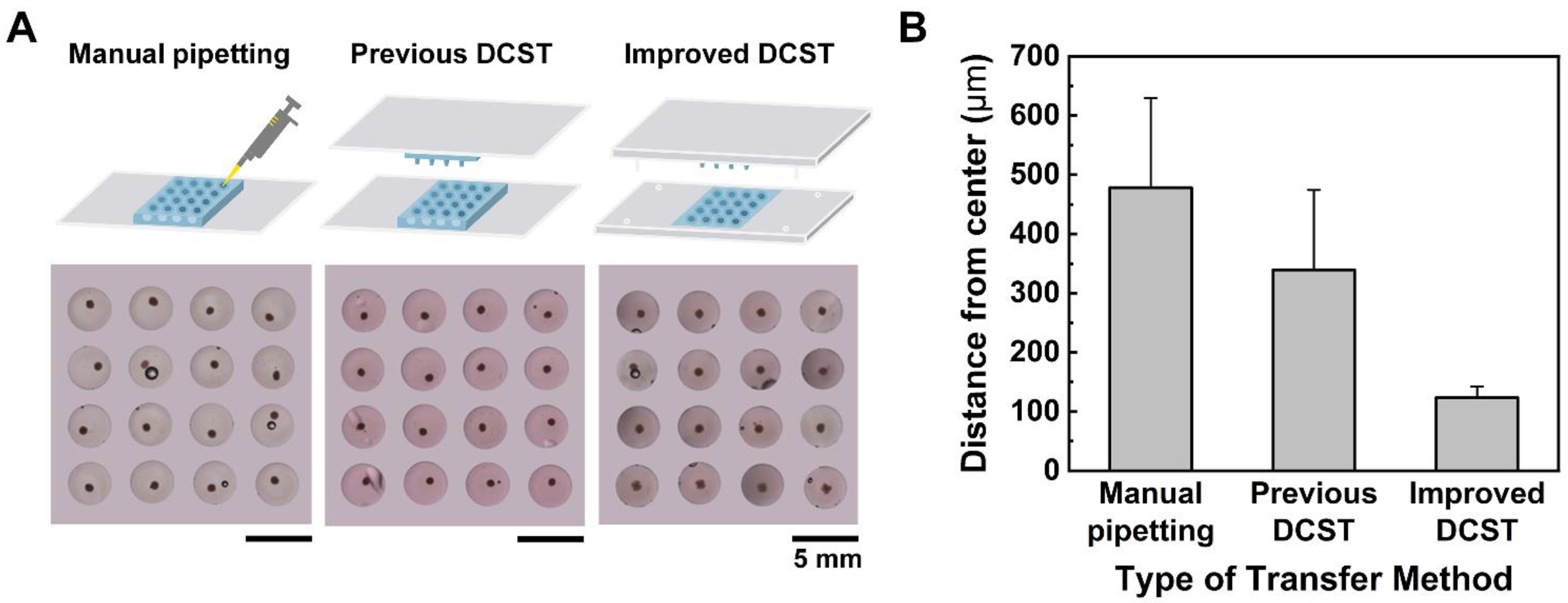
24]. Despite research, the problem was not resolved as it was still difficult to remove the old cell medium using the slanted microwells while not contacting the spheroids with a pipette tip. In the case of the DCST, the array chips were appropriately made for exchanging spheroid medium without damaging the spheroid. However, the array chips still did not have any supporting frame, resulting in unstable alignment and an inconsistent shape of the spheroid-embedded hydrogel model. In addition, for a successful spheroid embedment into the hydrogel, the transfer procedure must be performed under magnifying equipment, which is tedious and time-consuming.
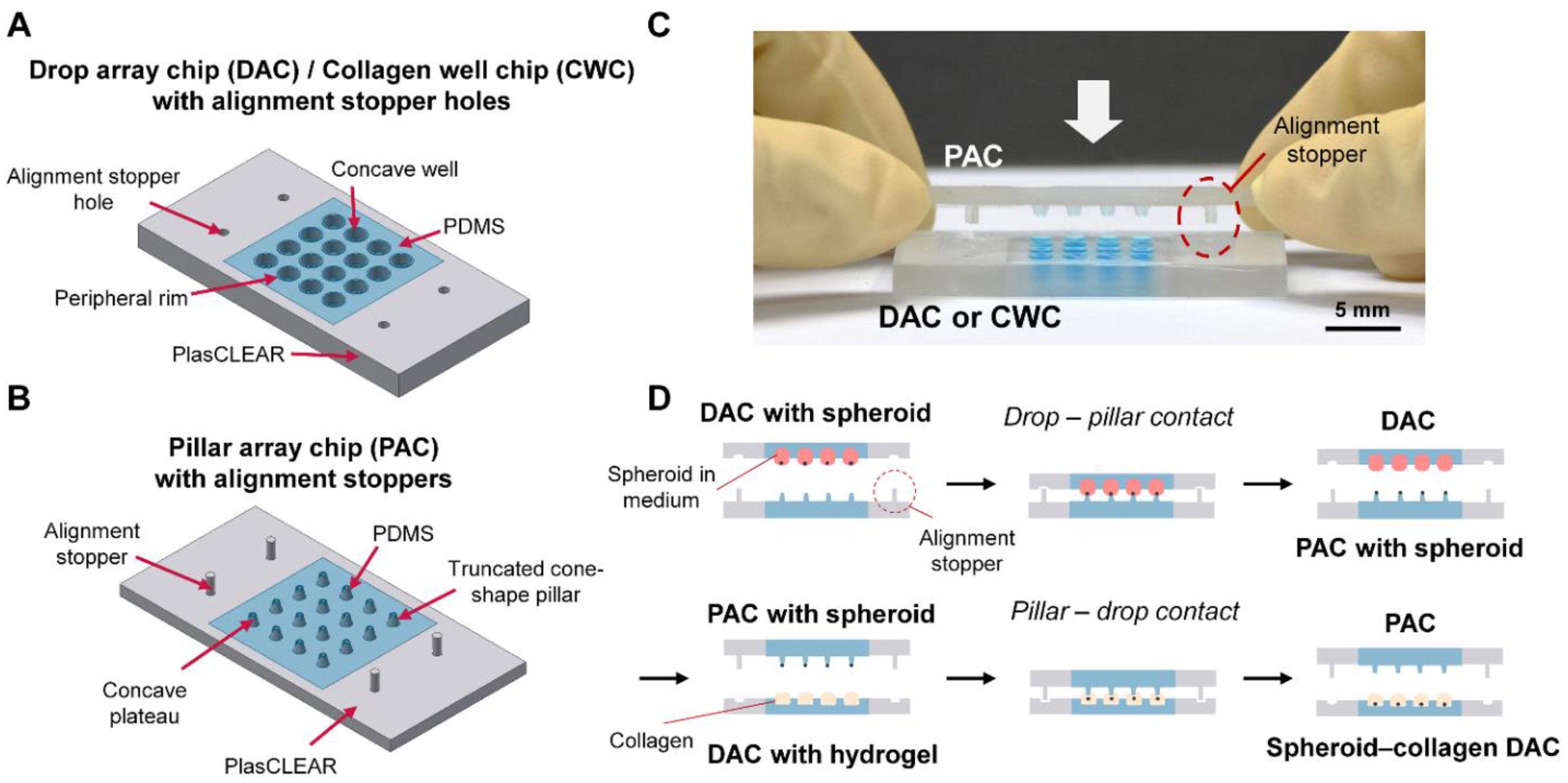
On this basis, this study presents an improved DCST method that simply and uniformly constructs an array of fibroblast-associated glioblastoma multiform spheroid models. Four sets of alignment stoppers have been developed and integrated into the original DCST method previously introduced [
23,
24] to culture and embed spheroids into hydrogel with perfect alignment. The performance of the improved DCST method with alignment stoppers is compared to that of the original DCST and conventional manual pipetting methods in the areas of spheroid shape, retention rate, and uniformity of the spheroid models. Then, the drop array chip (DAC) with alignment holes is used to co-culture both glioblastoma spheroids and fibroblast-associated glioblastoma spheroids using the chip-based hanging drop method. The spheroids are simultaneously relocated to new medium drops or into collagen drops using the pillar array chip (PAC) with alignment stoppers with stability and uniformity. Using the alignment stoppers, the alignment between the two chips is enhanced, eliminating the need to use magnifying equipment, such as a stereomicroscope, while the spheroids are being transferred. In addition, we have used the more invasive cell type and also associated fibroblast to investigate the invasive behavior of the tumor cells in our device, and since we have used a more invasive cell type with fibroblast, it is important that the alignment stoppers keep our spheroids in the center of the DAC well. Furthermore, the growth and invasion morphology of the glioblastoma spheroids are investigated under various conditions using the cell viability test or the collagen well chip (CWC). Finally, the effects of the ratio of co-cultured fibroblast cells and drug treatment are demonstrated and analyzed with the fibroblast-associated glioblastoma spheroid models.
2. Materials and Methods
2.1. Materials
For the fabrication of array chips for DCST, poly(dimethylsiloxane) (PDMS) monomers and curing agents were purchased from Dow Corning (Midland, MI, USA). Poly(methyl methacrylate) (PMMA, 3 mm thickness) plates were purchased from YM Tech (Daejeon, Korea). Phosphate-buffered saline (PBS), Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin (P/S) were purchased from Corning (Corning, NY, USA). Rat tail collagen type I was purchased from Corning. Calcein-AM and ethidium homodimer-1 (EthD-1) and Hoechst 33342 solution were purchased from Invitrogen (Carlsbad, CA, USA). CellTrackerTM Green CMFDA and CellTrackerTM Red CMTPX dyes for cell tracking were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Doxorubicin hydrochloride for drug treatment was purchased from Sigma-Aldrich (St. Louis, MO, USA). Other chemicals and reagents were purchased from Sigma-Aldrich unless otherwise stated.
2.2. Device Fabrication
The molds of a DAC with alignment stopper holes and a PAC with alignment stoppers were all designed using the AutoCAD and Inventor program (Autodesk, San Rafael, CA, USA). Next, the objects were fabricated with clear photopolymer resins (plasCLEAR) using a 3D printer (The PICO2; Asiga, Alexandria, Australia). The PMMA plates for the 3D printed molds were fabricated by etching the PMMA plates into an appropriate pre-designed shape using a laser cutter (C40–60W; Coryart, Anyang, Korea). Then, to make a complete master mold, the 3D printed objects were attached to the etched PMMA plates with a double-sided adhesive tape (Kyodo Giken Chemical, Saitama, Japan).
To fabricate the PDMS-based array chips, including the DAC with alignment stopper holes and the PAC with alignment stoppers, the master molds were positioned using the alignment stoppers and held together in a place by placing two sets of magnets on each side of the master mold. Then, the master molds were covered using aluminum foil as a support structure. A PDMS mixture, which consists of a PDMS monomer and a curing agent at a ratio of 10:1, was poured onto the patterned mold. The molds were then degassed in a vacuum chamber for 30 min to remove all the air bubbles that could cause the surface of the final PDMS product to have an uneven surface. After placing the molds in a 65 °C dry oven and curing overnight, the DAC with alignment stopper holes and the PAC with alignment stoppers were peeled off gently from the supporting molds. Before use, the array chips were autoclaved for sterilization (
Supporting Figure S1).
2.3. Two-Dimensional (2D) Cell Culture
For spheroid cell culture and analysis, the human glioblastoma multiforme cancer cell line U87-MG was purchased from the Korean Cell Line Bank (KCLB; Seoul, Korea). U87-MG cells were 2D cultured in a cell culture dish as a monolayer in DMEM supplemented with 10% FBS and 1% P/S. Then, the cells were maintained in a humidified incubator of 37 °C and 5% CO2 to be cultured. The medium was replaced every 48 h and the cells were subcultured when approximately 70% of the dish was full. The normal human lung fibroblast (NHLF) (Lonza; Basel, Switzerland) cell line was 2D cultured in a Fibroblast Growth Medium-2 (FGM-2), which consists of Fibroblast Basal Medium (FBM) and a reagent pack (HEPES, TNS, Trypsin/EDTA) (Lonza). The NHLF cell line was also 2D cultured in a humidified incubator of the same condition as U87-MG and the medium was replaced every 48 h.
2.4. Three-Dimensional (3D) Cell Culture and Embedment into Collagen
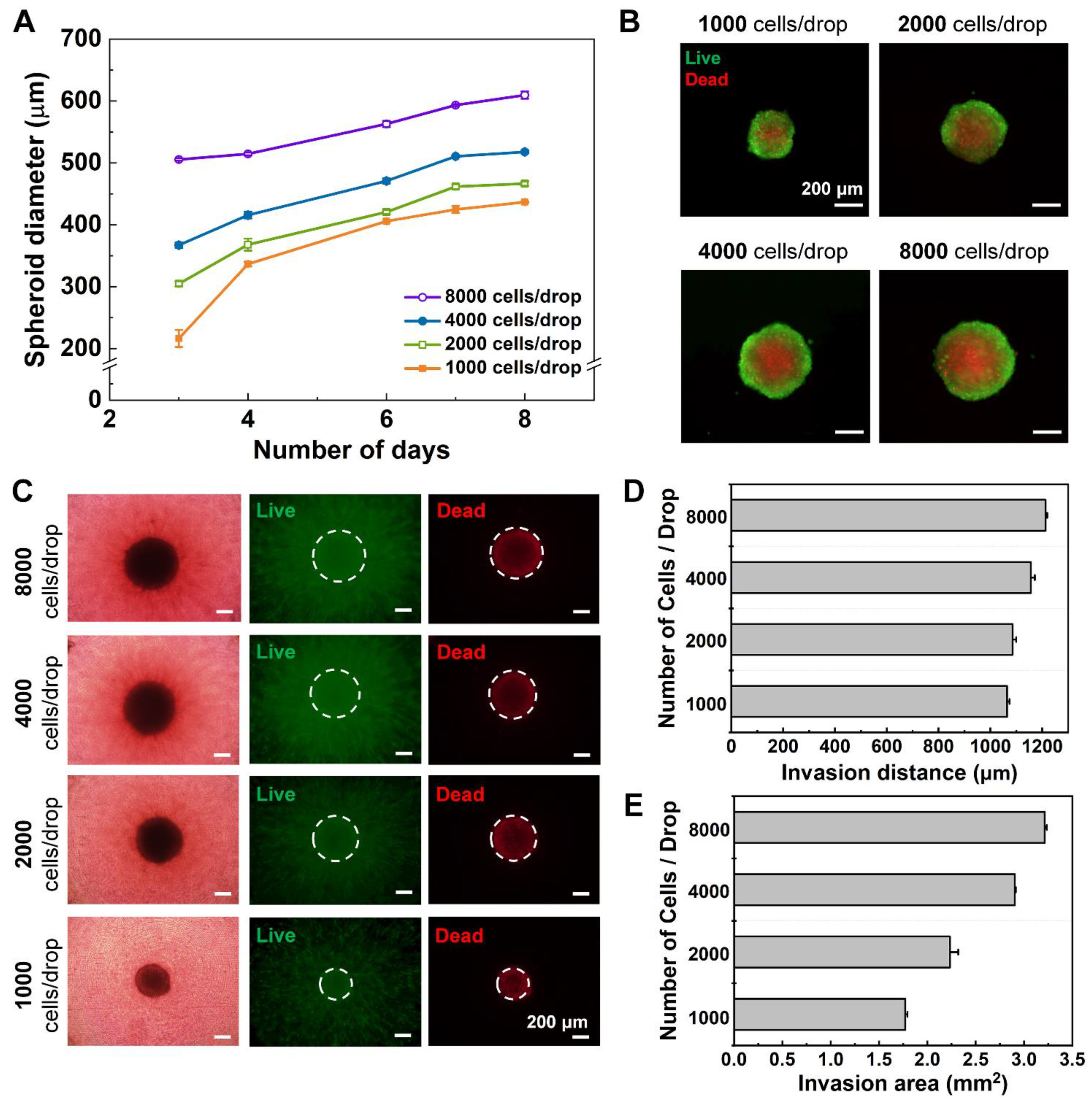
The two-dimensionally cultured U87-MG cells were suspended using a trypsinization process for spheroid formation, then were counted and diluted with an extra medium to meet the desired initial seeding concentrations (1000/2000/4000/8000 cells per well). Using a pipette, 24 μL of the cell suspensions of desired concentration were each loaded to the well of the DAC with alignment stopper holes. Then, the DAC with alignment stopper holes was positioned upside down on the two PMMA columns adhered to the culture dish as a hanging drop method. 7 mL of PBS was loaded at the bottom of the culture dish to prevent drop evaporation. The cell culture dish with the DAC with alignment stopper holes was placed in a humidified incubator of 37 °C and 5% CO2. The medium of the droplets, in which spheroids are constructed, is replaced every 48 h using the PAC with alignment stoppers.
Rat tail collagen type I was diluted to 3 mg/mL to 0.5 mg/mL using a NaOH solution, and 5 μL was loaded to each of the wells in a Collagen Well Chip (CWC) with alignment holes. The CWC was developed by modifying the height of the DAC and flattening the bottom of the well to enhance the observation of the invasive nature of the tumor spheroid under bright field conditions without using any chemical treatment (
Supporting Figure S2). The PAC with alignment stoppers was used to transfer the spheroids in the DAC with alignment stopper holes to the collagen-filled well of the CWC. After contacting the PAC with CWC for 30 s to settle down the spheroids, the spheroid-embedded collagen was placed inside a cell culture dish and put inside the incubator for 30 min to polymerize the collagen drops. Subsequently, the entire cell culture dish with the CWC was loaded with 5 mL of cell culture medium, enough to immerse the entire chip. The medium was changed every 48 h.
2.5. Fibroblast-Associated Glioblastoma Spheroid Culture and Invasion Assay
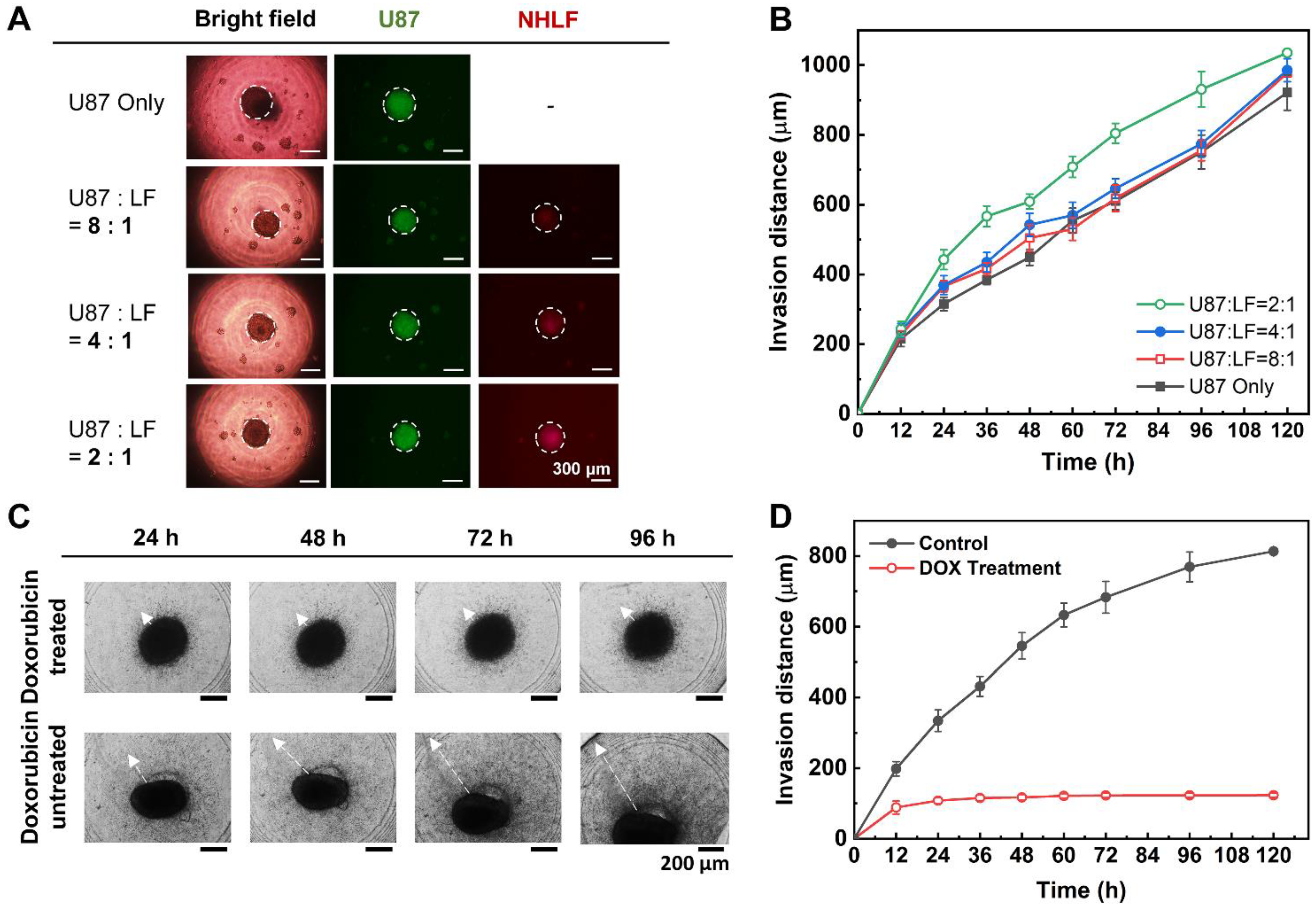
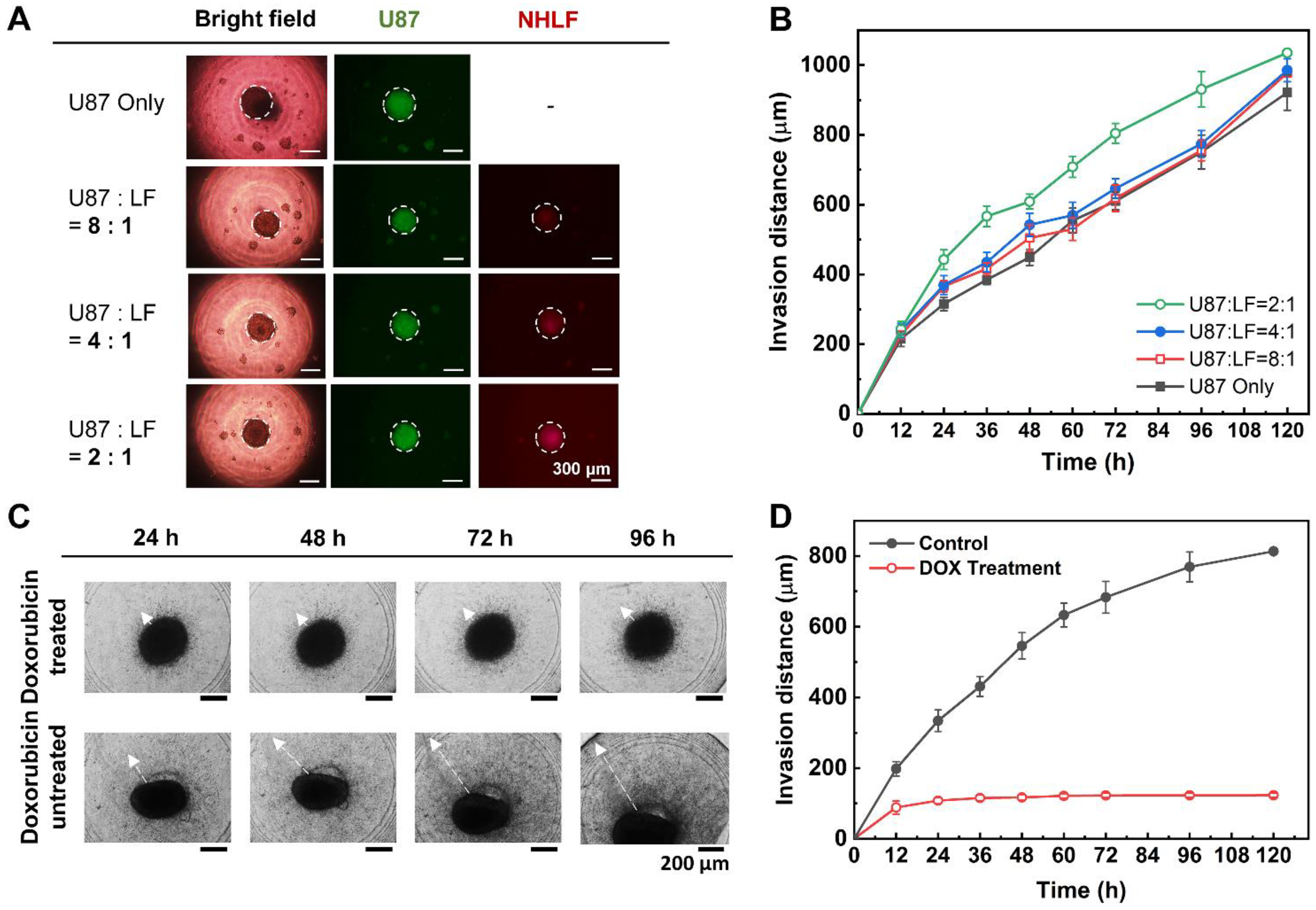
The fibroblast-associated glioblastoma spheroids were similarly constructed as the glioblastoma spheroids. Both the two-dimensionally cultured U87-MG and NHLF cell lines were suspended by the trypsinization process, counted, and diluted with an appropriate medium to meet the seeding concentration ratios (e.g., U87-MG:NHLF = 2:1, 4:1, or 8:1). The cell suspensions of U87-MG and NHLF cells were gently mixed using a pipette accordingly, and 24 μL of the cell suspension mixture was seeded to each well of the DAC. Similar to the mono-cultured spheroids, the medium of the co-culture spheroids was also replaced every 48 h using the DAC with alignment stopper holes and the PAC with alignment stoppers. The spheroid-seeded DAC with alignment stopper holes was placed upside down using a spacer inside a cell culture dish. The cell culture dish was placed in a humidified incubator of 37 °C and 5% CO2. To prevent the evaporation of the DAC droplet, the bottom of the cell culture dish was loaded with 7 mL of PBS. Then, after the spheroid cultivation, the fibroblast-associated glioblastoma spheroids were embedded into collagen using the PAC with alignment stoppers to make a spheroid–hydrogel model.
2.6. Cell Viability Assay
For the live/dead assay, the glioblastoma multiforme U87-MG spheroids in the DAC were transferred to a new DAC with each of its well filled with 24 μL of PBS containing 20 μM Calcein-AM and 10 μM EthD-1 using the PAC. After incubating the spheroids for 30 min in a humidified incubator of 37 °C and 5% CO2, the spheroids were retrieved using the PAC and returned to a new DAC with each of its well filled with 24 μL of DMEM medium for further imaging. To stain the spheroids already embedded in a collagen drop for the cell viability assay, the cell medium used to immerse a CWC was entirely removed, and 5 mL of PBS was loaded into the cell culture dish to wash the cell models. Then, 1 mL of PBS containing 20 μM Calcein-AM and 10 μM EthD-1 was gently loaded onto the top surface of the CWC so that it penetrates through the type I collagen and stain the spheroids. The bright-field and fluorescent images were taken using a charge-coupled device (CCD) camera (DP72; Olympus, Tokyo, Japan) on a fluorescence microscope (IX51; Olympus).
2.7. Nuclei Staining of Spheroids
The Hoechst 33342 solution, which emits blue fluorescence when bound to double-stranded DNA, was used to stain the nucleic acid of the spheroids to distinguish the live cells. To make a working solution, 0.5 μL of Hoechst 33342 solution was mixed with 1 mL of cell medium. The spheroids cultured in the DAC were transferred using a PAC to a different DAC with each of its well filled with the Hoechst working solution. The transferred spheroids were incubated for 10 min in a humidified incubator of 37 °C and 5% CO2, then the spheroids were retrieved, washed with a PBS-loaded DAC, and finally reloaded into a different DAC with a fresh new media. The bright-field and fluorescent images were taken using a CCD camera on a fluorescence microscope.
2.8. Staining of the Fibroblast-Associated Glioblastoma Spheroid
The fibroblast-associated glioblastoma spheroids were imaged using a CCD camera on a fluorescence microscope by staining the cells at suspension level with CellTrackerTM before the formation of spheroids. The U87-MG cells were stained using CellTrackerTM Green CMFDA dye and the NHLF cells were stained using CellTrackerTM Red CMTPX dye. The working dye solutions of both dyes were made by dissolving the dye product with dimethyl sulfoxide to a final concentration of 10 mM, then mixing 1 μL of the mixture with 1 mL of serum-free DMEM medium. After removing the culture media of the suspended cells, the prepared working dye solutions were gently added, and the suspended cells with the working dye solutions were incubated for 30 min in a humidified incubator of 37 °C and 5% CO2. The working dye solutions were removed later, and the culture medium was added again. After that, the stained cells of both cell types were counted, diluted with an extra medium to meet the desired initial seeding concentration ratio, and loaded to the well of the DAC.
2.9. Invasion Assay
Since the human glioblastoma multiforme U87-MG cell line is very pro-invasive, the spheroids extend themselves to the surrounding microenvironment as they are cultured. In this research, the rat tail type I collagen, at which the spheroids were embedded, acted as the ECM surrounding the spheroids. All mono-cultured spheroids consisting of only U87-MG cells, and co-cultured spheroids consisting of both U87-MG and NHLF cells at different seeding concentrations were monitored using a CCD camera on a fluorescence microscope. In addition, the invasive behavior of the mono-cultured spheroids in different collagen concentrations was also monitored using a CCD camera on a fluorescence microscope (
Supporting Figure S3). The bright-field images of the spheroids were captured for further image analysis of the invasion distance and area using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2.10. Spheroid Drug Treatment
The fibroblast-associated glioblastoma spheroid, which was cultured for 7 days in the DAC using the chip-based hanging drop method and embedded into a CWC with a PAC, was treated with 1 μM doxorubicin. The normal medium was replaced with a doxorubicin-containing medium, and the spheroids were monitored for up to 120 h. Another array of spheroids embedded in the collagen drops with a normal medium was also monitored up to 120 h for the control experiment.
2.11. Image Analysis
All the bright-field and fluorescence images were taken with a CCD camera on a fluorescence microscope were further analyzed using the ImageJ software. The scale of the images was set to convert the pixel unit to a micrometer. The diameter and area of the spheroid were measured by assuming the spheroid as a sphere. In the case of nucleic acid-stained spheroids, the color threshold was adjusted and the “analyze particles” tool was selected to automatically draw the outer circumference of the spheroid. The program was also used to measure the distance of how far the migratory glioblastoma cell invaded the ECM. The invasion distance is the length between the surface of the spheroid and the single cell that radiated off the most from the spheroid surface.
4. Conclusions
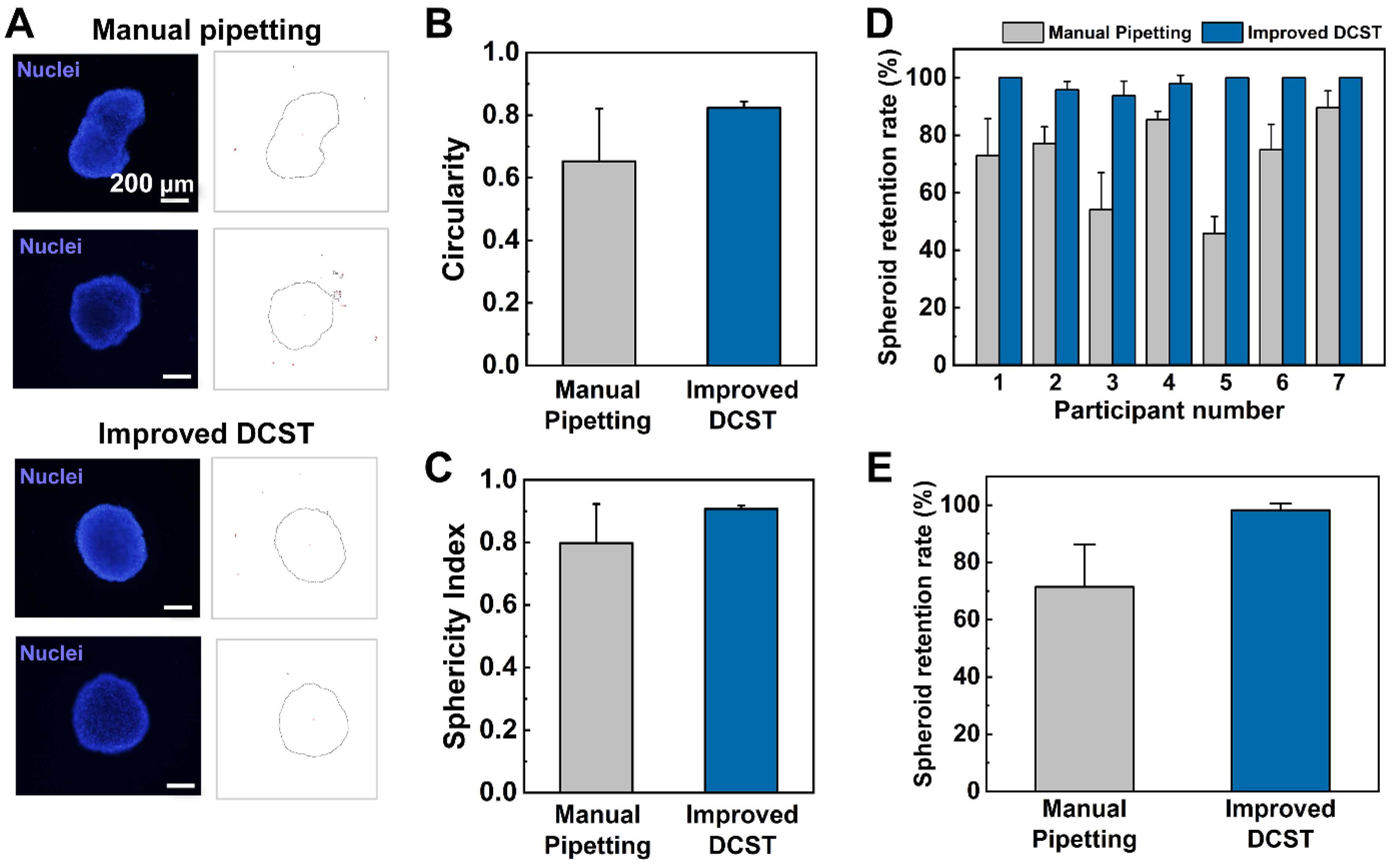
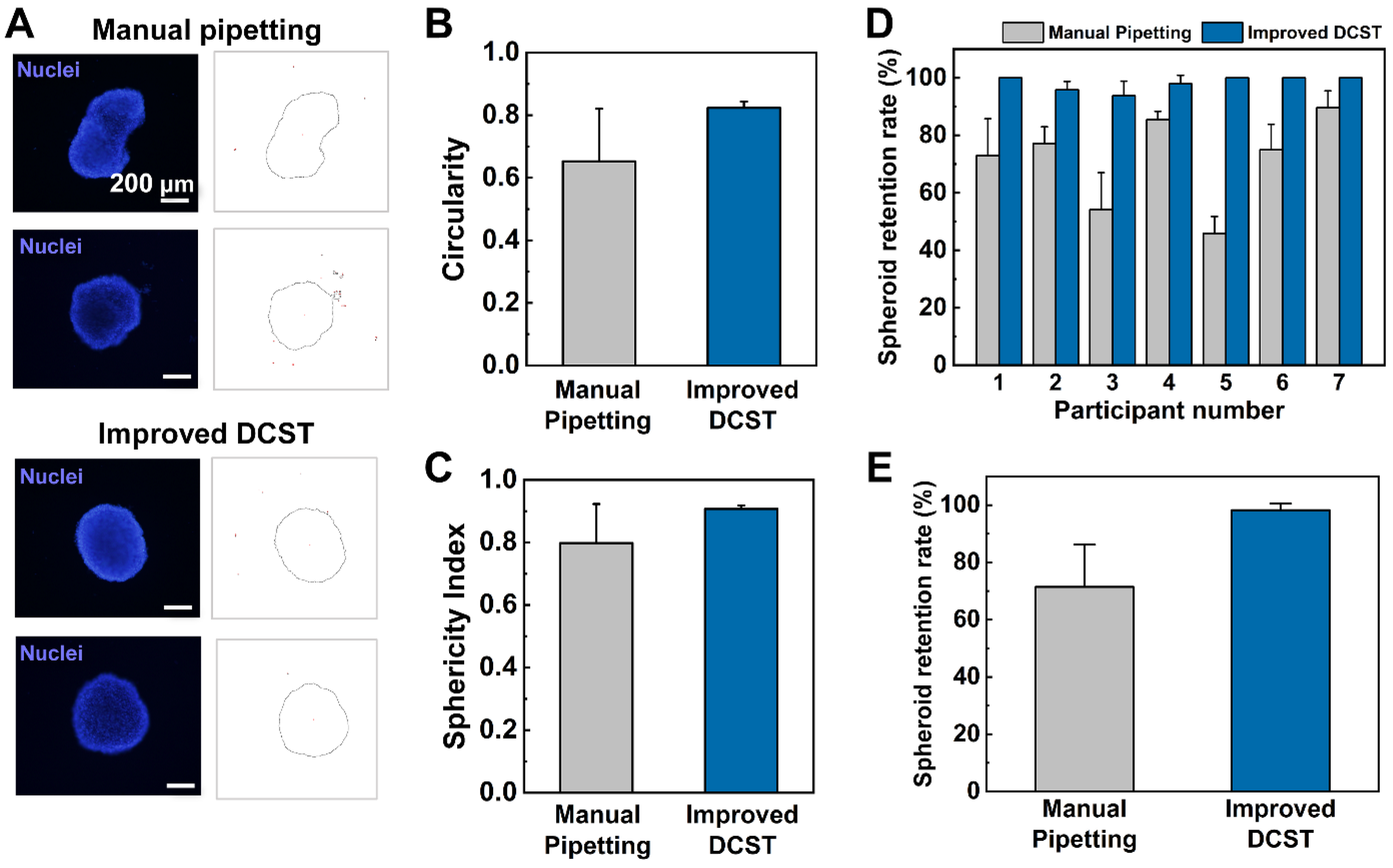
In this study, the uniform construction of an array of fibroblast-associated glioblastoma multiforme 3D cell culture models was demonstrated. The spheroids were grown in a medium droplet using the DAC with alignment stopper holes and transferred using the PAC with alignment stoppers by a DAC–PAC contacting process. This contacting process was used repetitively throughout the experiment for medium change, embedment into collagen as well as cell staining, and drug treatment. With the help of the alignment stoppers, the process of DAC–PAC contacting became more convenient by providing support and the constructed spheroid models were uniform. We were able to quantitatively demonstrate the enhanced performance of the improved DCST method in terms of the sphericity of the spheroid shape, higher retention rate, lower well-to-well variation. In addition, the experimental process of using the improved DCST method took less time than using the manual pipetting method.
Using the improved DCST method, U87-MG spheroids were cultured and then analyzed both quantitatively and qualitatively with fluorescent microscope images to confirm proper spheroid growth and the existence of a necrotic core. Furthermore, fibroblast-associated tumor spheroid models were successfully cultured by mixing the glioblastoma multiforme U87-MG cell with the human lung fibroblast cell NHLF in different ratios. The fibroblast-associated tumor spheroid models were then embedded in collagen droplets and treated with a single concentration of doxorubicin to demonstrate the effects of drug treatment on spheroid invasion and to show the applicability of the spheroid models constructed using the improved DCST method as a drug assay platform. This study is expected to be applied in areas such as new drug development as a drug screening platform. Since the improved DCST method has wide applicability, it may also be used to conveniently construct and handle patient-derived tumor spheroids during the process of investigating a personalized cancer treatment.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}