Native llama Nanobody Library Panning Performed by Phage and Yeast Display Provides Binders Suitable for C-Reactive Protein Detection

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Phage Display Nanobody Library Panning
2.2. Phage Screening by ELISA
2.3. Yeast Display Panning and Output Evaluation
2.4. VHH Small- and Large-Scale Purification
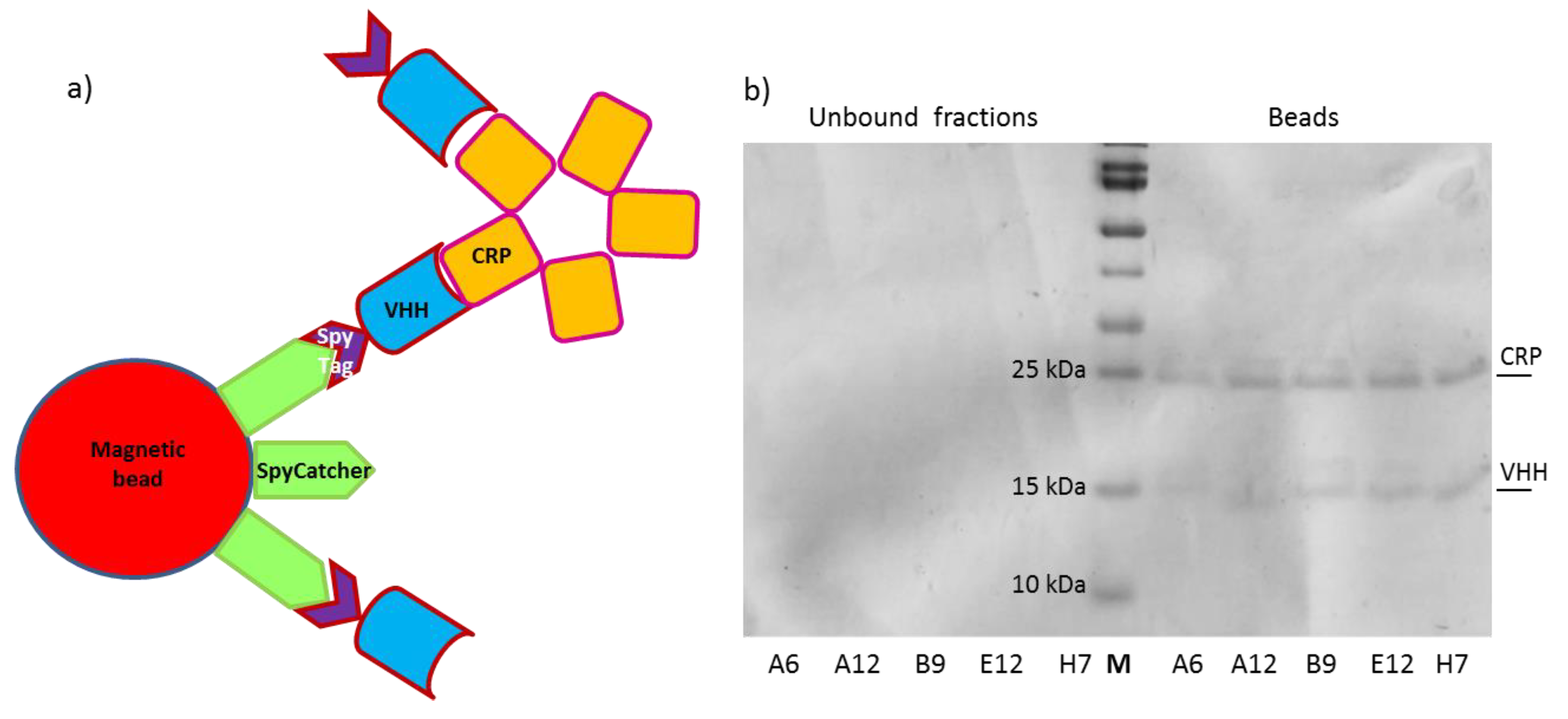
2.5. Reconstitution of the SpyTag-SpyCatcher Complex
2.6. ELISA
2.7. Immunoprecipitation Assay (IP)
2.8. Bio-Layer Interferometry (BLI) Measurement
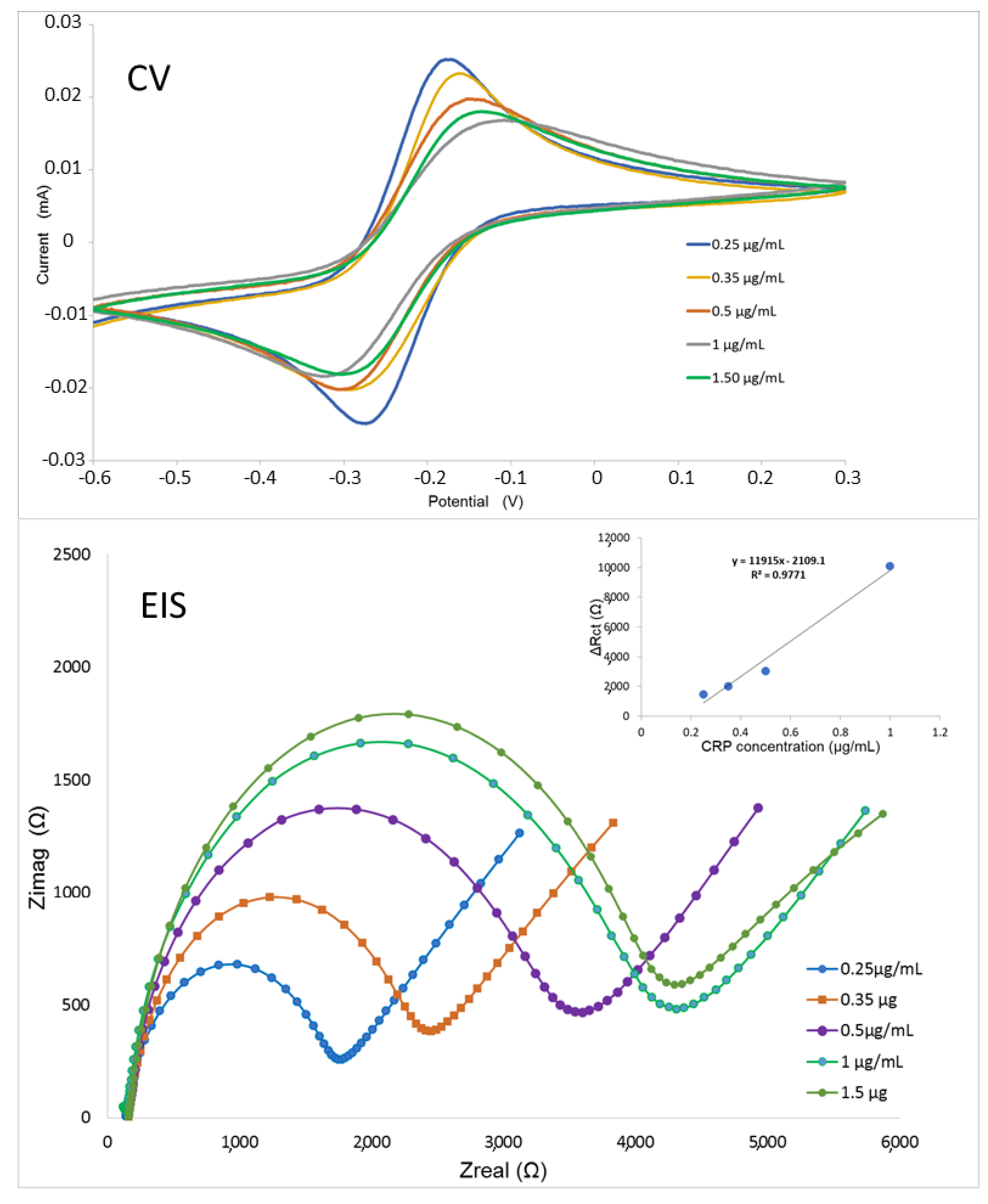
2.9. CRP Detection by Means of An Electrochemical Impedance Biosensor
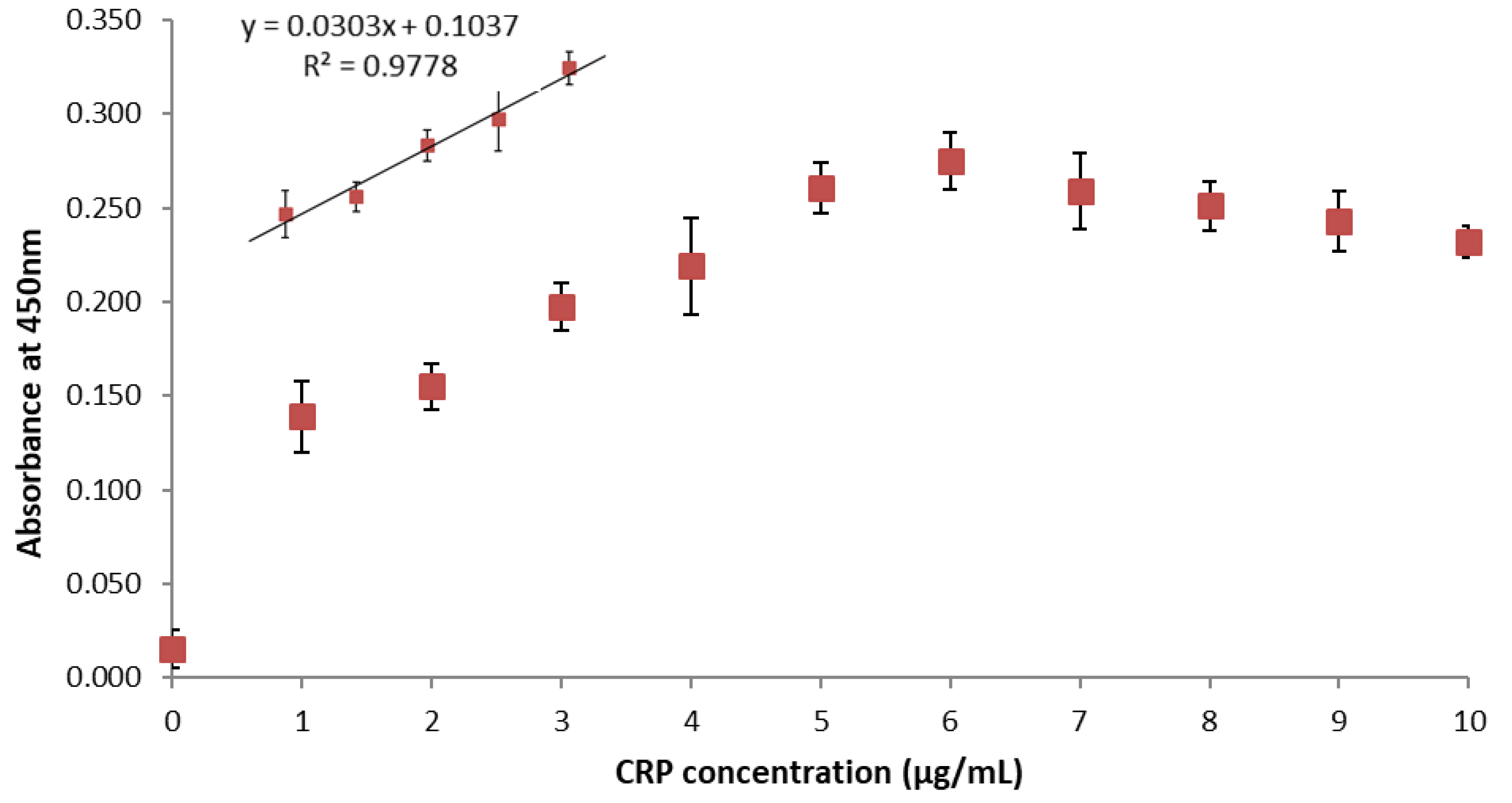
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Black, S.; Kushner, I.; Samols, D. C-reactive Protein. J. Biol. Chem. 2004, 279, 48487–48490. [Google Scholar] [CrossRef] [PubMed]
- Marnell, L.; Mold, C.; Du Clos, T.W. C-reactive protein: Ligands, receptors and role in inflammation. Clin. Immunol. 2005, 117, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Hermans, M.P.; Ahn, S.A.; Rousseau, M.F. Increased CRP: An extended biomarker of microvascular risk in men with type 2 diabetes. J. Diabetes Complicat. 2019, 33, 107413. [Google Scholar] [CrossRef]
- Schimmack, S.; Yang, Y.; Felix, K.; Herbst, M.; Li, Y.; Schenk, M.; Bergmann, F.; Hackert, T.; Strobel, O. C-reactive protein (CRP) promotes malignant properties in pancreatic neuroendocrine neoplasms. Endocr. Connect. 2019, 8, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Herold, T.; Jurinovic, V.; Arnreich, C.; Lipworth, B.J.; Hellmuth, J.C.; von Bergwelt-Baildon, M.; Klein, M.; Weinberger, T. Elevated levels of IL-6 and CRP predict the need for mechanical ventilation in COVID-19. J. Allergy Clin. Immunol. 2020, 146, 128–136.e4. [Google Scholar] [CrossRef]
- Eckschlager, C.; Schwenoha, K.; Roth, C.; Bogner, B.; Oostingh, G.J. Comparative analysis of high CRP-levels in human blood using point-of-care and laboratory-based methods. Pract. Lab. Med. 2019, 17, e00137. [Google Scholar] [CrossRef]
- Bryan, T.; Luo, X.; Bueno, P.R.; Davis, J.J. An optimised electrochemical biosensor for the label-free detection of C-reactive protein in blood. Biosens. Bioelectron. 2013, 39, 94–98. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, J.; Yim, G.; Ahn, H.J.; Lee, M.; Kim, T.H.; Park, C.; Min, J.; Jang, H.; Lee, T. Fabrication of a surface-enhanced Raman spectroscopy-based analytical method consisting of multifunctional DNA three-way junction-conjugated porous gold nanoparticles and Au-Te nanoworm for C-reactive protein detection. Anal. Bioanal. Chem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Szot-Karpińska, K.; Kudła, P.; Szarota, A.; Narajczyk, M.; Marken, F.; Niedziółka-Jönsson, J. CRP-binding bacteriophage as a new element of layer-by-layer assembly carbon nanofiber modified electrodes. Bioelectrochemistry 2020, 136, 107629. [Google Scholar] [CrossRef]
- Piccoli, J.P.; Soares, A.C.; Oliveira, O.N., Jr.; Cilli, E.M. Nanostructured functional peptide films and their application in C-reactive protein immunosensors. Bioelectrochemistry 2021, 138, 107692. [Google Scholar] [CrossRef]
- Veggiani, G.; Ossolengo, G.; Aliprandi, M.; Cavallaro, U.; de Marco, A. Single-domain antibodies that compete with the natural ligand fibroblast growth factor block the internalization of the fibroblast growth factor receptor 1. Biochem. Biophys. Res. Commun. 2011, 408, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Veggiani, G.; Giabbai, B.; Semrau, M.S.; Medagli, B.; Riccio, V.; Bajc, G.; Storici, P.; de Marco, A. Comparative analysis of fusion tags used to functionalize recombinant antibodies. Protein Expr. Purif. 2020, 166, 105505. [Google Scholar] [CrossRef]
- Oloketuyi, S.; Mazzega, E.; Zavašnik, J.; Pungjunun, K.; Kalcher, K.; de Marco, A.; Mehmeti, E. Electrochemical immunosensor functionalized with nanobodies for the detection of the toxic microalgae Alexandrium minutum using glassy carbon electrode modified with gold nanoparticles. Biosens. Bioelectron. 2020, 154, 112052. [Google Scholar] [CrossRef]
- Monegal, A.; Ami, D.; Martinelli, C.; Huang, H.; Aliprandi, M.; Capasso, P.; Francavilla, C.; Ossolengo, G.; de Marco, A. Immunological applications of single-domain llama recombinant antibodies isolated from a naïve library. Protein Eng. Des. Sel. 2009, 22, 273–280. [Google Scholar] [CrossRef]
- De Marco, A. Isolation of recombinant antibodies that recognize native and accessible membrane biomarkers. In Nanotechnology to Aid Chemical and Biological Defense, NATO Science for Peace and Security Series A: Chemistry and Biology; Camesano, T.A., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 49–66. [Google Scholar]
- Bowley, D.R.; Labrijn, A.F.; Zwick, M.B.; Burton, D.R. Antigen selection from an HIV-1 immune antibody library displayed on yeast yields many novel antibodies compared to selection from the same library displayed on phage. Protein Eng. Des. Sel. 2007, 20, 81–90. [Google Scholar] [CrossRef]
- Pandya, P.; Sayers, R.O.; Ting, J.P.; Morshedian, S.; Torres, C.; Cudal, J.S.; Zhang, K.; Fitchett, J.R.; Zhang, Q.; Zhang, F.F.; et al. Integration of phage and yeast display platforms: A reliable and cost effective approach for binning of peptides as displayed on-phage. PLoS ONE 2020, 15, e0233961. [Google Scholar] [CrossRef] [PubMed]
- Meiring, S.M.; Setlai, B.D.P.; Theron, C.; Bragg, R. The use of phage display and yeast based expression system for the development of a Von Willebrand factor propeptide assay: Development of a Von Willebrand factor propeptide assay. Biomed. Res. Int. 2018, 2018, 6232091. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Wen, K.; Mou, Z.; Zou, L.; Che, X.; Ni, B.; Wu, Y. Antibody binding site mapping of SARS-CoV spike protein receptor-binding domain by a combination of yeast surface display and phage peptide library screening. Viral Immunol. 2009, 22, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Grzeschik, J.; Yanakieva, D.; Roth, L.; Krah, S.; Hinz, S.C.; Elter, A.; Zollmann, T.; Schwall, G.; Zielonka, S.; Kolmar, H. Yeast surface display in combination with fluorescence-activated cell sorting enables the rapid isolation of antibody fragments derived from immunized chickens. Biotechnol. J. 2019, 14, e1800466. [Google Scholar] [CrossRef]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef]
- Benatuil, L.; Perez, J.M.; Belk, J.; Hsieh, C.M. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng. Des. Sel. 2010, 23, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.C.; Elter, A.; Grzeschik, J.; Habermann, J.; Becker, B.; Kolmar, H. Simplifying the detection of surface presentation levels in yeast surface display by intracellular tGFP Expression. Methods Mol. Biol. 2020, 2070, 211–222. [Google Scholar] [PubMed]
- Veggiani, G.; de Marco, A. Improved quantitative and qualitative production of single-domain intrabodies mediated by the co-expression of Erv1p sulfhydryl oxidase. Protein Expr. Purif. 2011, 79, 111–114. [Google Scholar] [CrossRef]
- Djender, S.; Schneider, A.; Beugnet, A.; Crepin, R.; Even Desrumeaux, K.; Moutel, S.; Perez, F.; de Marco, A. Bacterial cytoplasm as an effective cell compartment for producing functional VHH-based affinity reagents and Camelidae IgG-like recombinant antibodies. Microb. Cell Fact. 2014, 13, 140. [Google Scholar] [CrossRef]
- Keeble, A.H.; Howarth, M. Insider information on successful covalent protein coupling with help from SpyBank. Methods Enzymol. 2019, 617, 443–461. [Google Scholar]
- Kunik, V.; Ashkenazi, S.; Ofran, Y. Paratome: An online tool for systematic identification of antigen binding regions in antibodies based on sequence or structure. Nucleic Acids Res. 2012, 40, W521–W524. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.P.; Liu, J.L.; Shriver-Lake, L.C.; Zabetakis, D.; Sugiharto, V.A.; Chen, H.W.; Lee, C.R.; Defang, G.N.; Wu, S.L.; Venkateswaran, N.; et al. Oriented immobilization of single-domain antibodies using SpyTag/SpyCatcher yields improved limits of detection. Anal. Chem. 2019, 91, 9424–9429. [Google Scholar] [CrossRef]
- Guo, K.; Wustoni, S.; Koklu, A.; Díaz-Galicia, E.; Moser, M.; Hama, A.; Alqahtani, A.A.; Ahmad, A.N.; Alhamlan, F.S.; Shuaib, M.; et al. Rapid single-molecule detection of COVID-19 and MERS antigens via nanobody-functionalized organic electrochemical transistors. Nat. Biomed. Eng. 2021, 5, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Vashist, S.K.; Venkatesh, A.G.; Schneider, E.M.; Beaudoin, C.; Luppa, P.B.; Luong, J.H. Bioanalytical advances in assays for C-reactive protein. Biotechnol. Adv. 2016, 34, 272–290. [Google Scholar] [CrossRef]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. published correction appears in. J. Clin. Investig. 2003, 111, 1805–1812. [Google Scholar] [CrossRef]
- Johnson, A.; Song, Q.; Ferrigno, P.K.; Bueno, P.R.; Davis, J.J. Sensitive affimer and antibody based impedimetric label-free assays for C-reactive protein. Anal. Chem. 2012, 84, 6553–6560. [Google Scholar] [CrossRef]
- Ahn, J.S.; Choi, S.; Jang, S.H.; Chang, H.J.; Kim, J.H.; Nahm, K.B.; Oh, S.W.; Choi, E.Y. Development of a point-of-care assay system for high-sensitivity C-reactive protein in whole blood. Clin. Chim. Acta 2003, 332, 51–59. [Google Scholar] [CrossRef]
- Oh, Y.K.; Joung, H.A.; Han, H.S.; Suk, H.J.; Kim, M.G. A three-line lateral flow assay strip for the measurement of C-reactive protein covering a broad physiological concentration range in human sera. Biosens. Bioelectron. 2014, 61, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Vashist, S.K.; Czilwik, G.; van Oordt, T.; von Stetten, F.; Zengerle, R.; Schneider, E.M.; Luong, J.H. One-step kinetics-based immunoassay for the highly sensitive detection of C-reactive protein in less than 30 min. Anal. Biochem. 2014, 456, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.F.; Gao, B.Z.; Tsai, H.Y.; Fuh, C.B. Detection of c-reactive protein based on a magnetic immunoassay by using functional magnetic and fluorescent nanoparticles in microplates. Analyst 2014, 139, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Thangamuthu, M.; Santschi, C.J.F.; Martin, O. Label-Free Electrochemical Immunoassay for C-Reactive Protein. Biosensors 2018, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.A.; Medagli, B.; Semrau, M.S.; Storici, P.; Bajc, G.; de Marco, A.; Laio, A.; Fortuna, S. A consensus protocol for the In Silico optimisation of antibody fragments. Chem. Commun. 2019, 55, 14043–14046. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wang, J.; Kang, G.; Hu, M.; Yuan, B.; Zhang, Y.; Huang, H. Homology modeling-based In Silico affinity maturation improves the affinity of a nanobody. Int. J. Mol. Sci. 2019, 20, 4187. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Kang, G.; Cheng, X.; Wang, J.; Li, R.; Bai, Z.; Yang, D.; Huang, H. In Vitro affinity maturation to improve the efficacy of a hypoxia-inducible factor 1α single-domain intrabody. Biochem. Biophys. Res. Commun. 2020, 529, 936–942. [Google Scholar] [CrossRef] [PubMed]
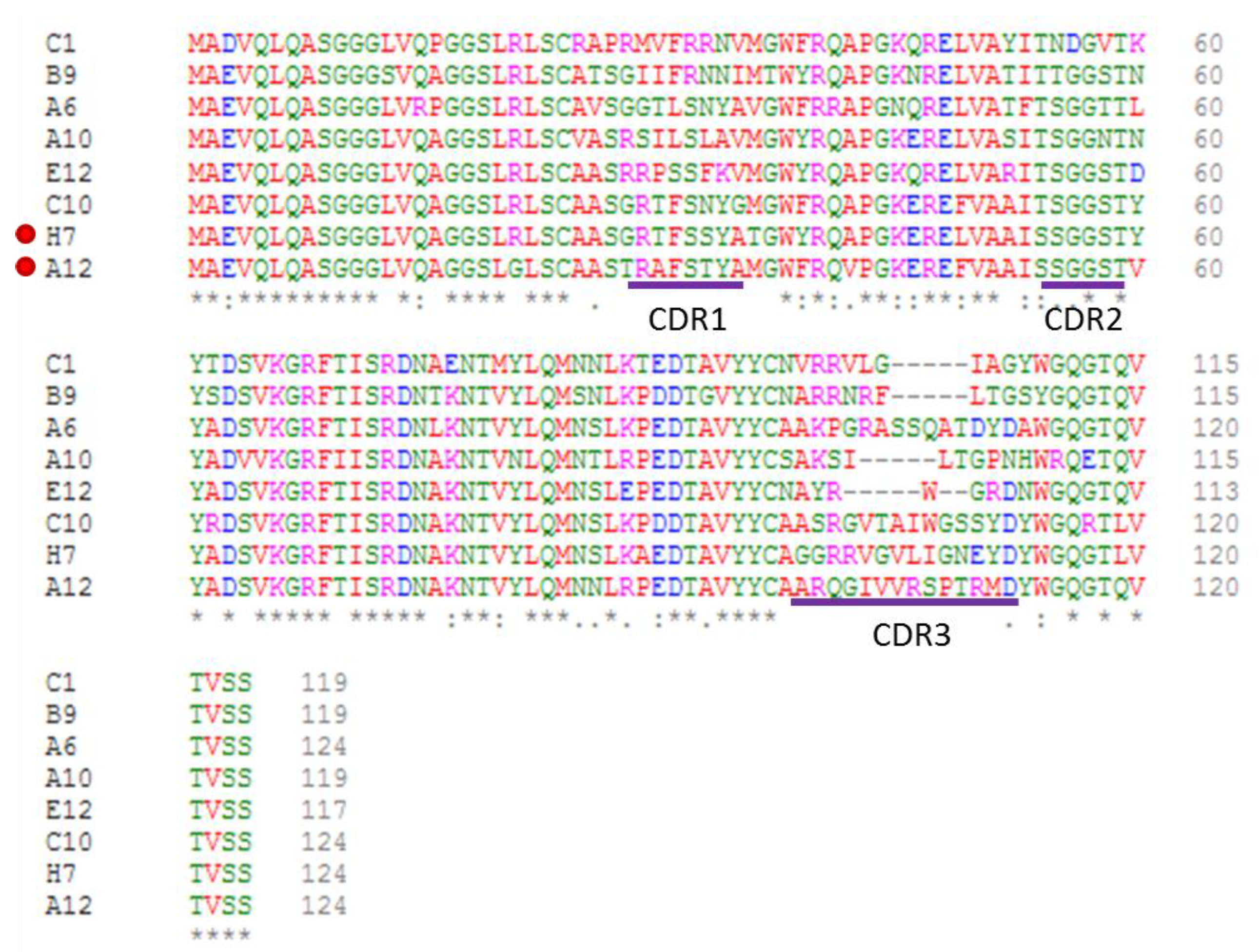
- Orlando, M.; Fortuna, S.; Oloketuyi, S.; Bajc, G.; Goldenzweig, A.; de Marco, A. CDR1 composition can affect nanobody recombinant expression yields. Biomolecules 2021, 11, 1362. [Google Scholar] [CrossRef] [PubMed]
- Ansar, W.; Ghosh, S. C-reactive protein and the biology of disease. Immunol. Res. 2013, 56, 131–142. [Google Scholar] [CrossRef]
- Yeh, E.T.; Willerson, J.T. Coming of age of C-reactive protein: Using inflammation markers in cardiology. Circulation 2003, 107, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Derda, R.; Tang, S.K.; Li, S.C.; Ng, S.; Matochko, W.; Jafari, M.R. Diversity of phage-displayed libraries of peptides during panning and amplification. Molecules 2011, 16, 1776–1803. [Google Scholar] [CrossRef]
- Matochko, W.L.; Li, S.C.; Tang, S.K.; Derda, R. Prospective identification of parasitic sequences in phage display screens. Nucleic Acids Res. 2014, 42, 1784–1798. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Barmina, O.; Burgoon, M.; Gilden, D. Identification of measles virus epitopes using an ultra-fast method of panning phage-displayed random peptide libraries. J. Virol. Meth. 2009, 156, 169–173. [Google Scholar] [CrossRef][Green Version]
- Egloff, P.; Zimmermann, I.; Arnold, F.M.; Hutter, C.A.J.; Morger, D.; Opitz, L.; Poveda, L.; Keserue, H.A.; Panse, C.; Roschitzki, B.; et al. Engineered peptide barcodes for in-depth analyses of binding protein libraries. Nat. Methods 2019, 16, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gentili, M.; Hacohen, N.; Regev, A. A cell-free nanobody engineering platform rapidly generates SARS-CoV-2 neutralizing nanobodies. Nat. Commun. 2021, 12, 5506. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clones | MW kDa | pI | Screening ELISA (CRP/BSA Signal) | Yield mg/L | Affinity nM |
|---|---|---|---|---|---|
| A6 | 13.1 | 9.30 | 11.8 | 9.0 | 51 |
| B9 | 12.9 | 9.86 | 14.6 | 8.8 | - |
| E12 | 12.8 | 9.51 | 13.3 | 11.1 | 13 |
| C10 | 13.4 | 9.43 | 14.6 | 0 | - |
| A12 | 13.2 | 9.34 | 15.5 | 5.3 | 122 |
| H7 | 13.1 | 8.95 | 7.1 | 9.8 | 154 |
| A10 | 12.8 | 9.39 | 12.3 | - | - |
| C1 | 13.2 | 9.85 | 12.5 | - | - |
| Overnight Coating at 4 °C | 2 h Coating at 37 °C | |||
|---|---|---|---|---|
| CRP | BSA | CRP | BSA | |
| A6 | 0.476 ± 0.101 | 0.066 ± 0.007 | 0.231 ± 0.010 | 0.067 ± 0.005 |
| A12 | 1.232 ± 0.420 | 0.070 ± 0.008 | 0.974 ± 0.006 | 0.063 ± 0.003 |
| B9 | 1.561 ± 0.008 | 0.214 ± 0.014 | 2.284 ± 0.227 | 0.111 ± 0.005 |
| E12 | 2.842 ± 0.136 | 0.283 ± 0.020 | 2.602 ± 0.450 | 0.202 ± 0.016 |
| H7 | 1.468 ± 0.057 | 0.080 ± 0.014 | 0.904 ± 0.100 | 0.066 ± 0.002 |
| antiHis+ antimouse HRP | 0.070 ± 0.005 | 0.069 ± 0.022 | 0.066 ± 0.001 | 0.066 ± 0.005 |
| antimouse HRP | 0.063 ± 0.004 | 0.060 ± 0.013 | 0.062 ± 0.003 | 0.062 ± 0.003 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oloketuyi, S.; Bernedo, R.; Christmann, A.; Borkowska, J.; Cazzaniga, G.; Schuchmann, H.W.; Niedziółka-Jönsson, J.; Szot-Karpińska, K.; Kolmar, H.; de Marco, A. Native llama Nanobody Library Panning Performed by Phage and Yeast Display Provides Binders Suitable for C-Reactive Protein Detection. Biosensors 2021, 11, 496. https://doi.org/10.3390/bios11120496
Oloketuyi S, Bernedo R, Christmann A, Borkowska J, Cazzaniga G, Schuchmann HW, Niedziółka-Jönsson J, Szot-Karpińska K, Kolmar H, de Marco A. Native llama Nanobody Library Panning Performed by Phage and Yeast Display Provides Binders Suitable for C-Reactive Protein Detection. Biosensors. 2021; 11(12):496. https://doi.org/10.3390/bios11120496
Chicago/Turabian StyleOloketuyi, Sandra, Robert Bernedo, Andreas Christmann, Justyna Borkowska, Giulia Cazzaniga, Horst Wilhelm Schuchmann, Joanna Niedziółka-Jönsson, Katarzyna Szot-Karpińska, Harald Kolmar, and Ario de Marco. 2021. "Native llama Nanobody Library Panning Performed by Phage and Yeast Display Provides Binders Suitable for C-Reactive Protein Detection" Biosensors 11, no. 12: 496. https://doi.org/10.3390/bios11120496
APA StyleOloketuyi, S., Bernedo, R., Christmann, A., Borkowska, J., Cazzaniga, G., Schuchmann, H. W., Niedziółka-Jönsson, J., Szot-Karpińska, K., Kolmar, H., & de Marco, A. (2021). Native llama Nanobody Library Panning Performed by Phage and Yeast Display Provides Binders Suitable for C-Reactive Protein Detection. Biosensors, 11(12), 496. https://doi.org/10.3390/bios11120496