Effects of Transition Metal Substituents on Interfacial and Electronic Structure of CH3NH3PbI3/TiO2 Interface: A First-Principles Comparative Study

 ,
, 

Abstract
:1. Introduction
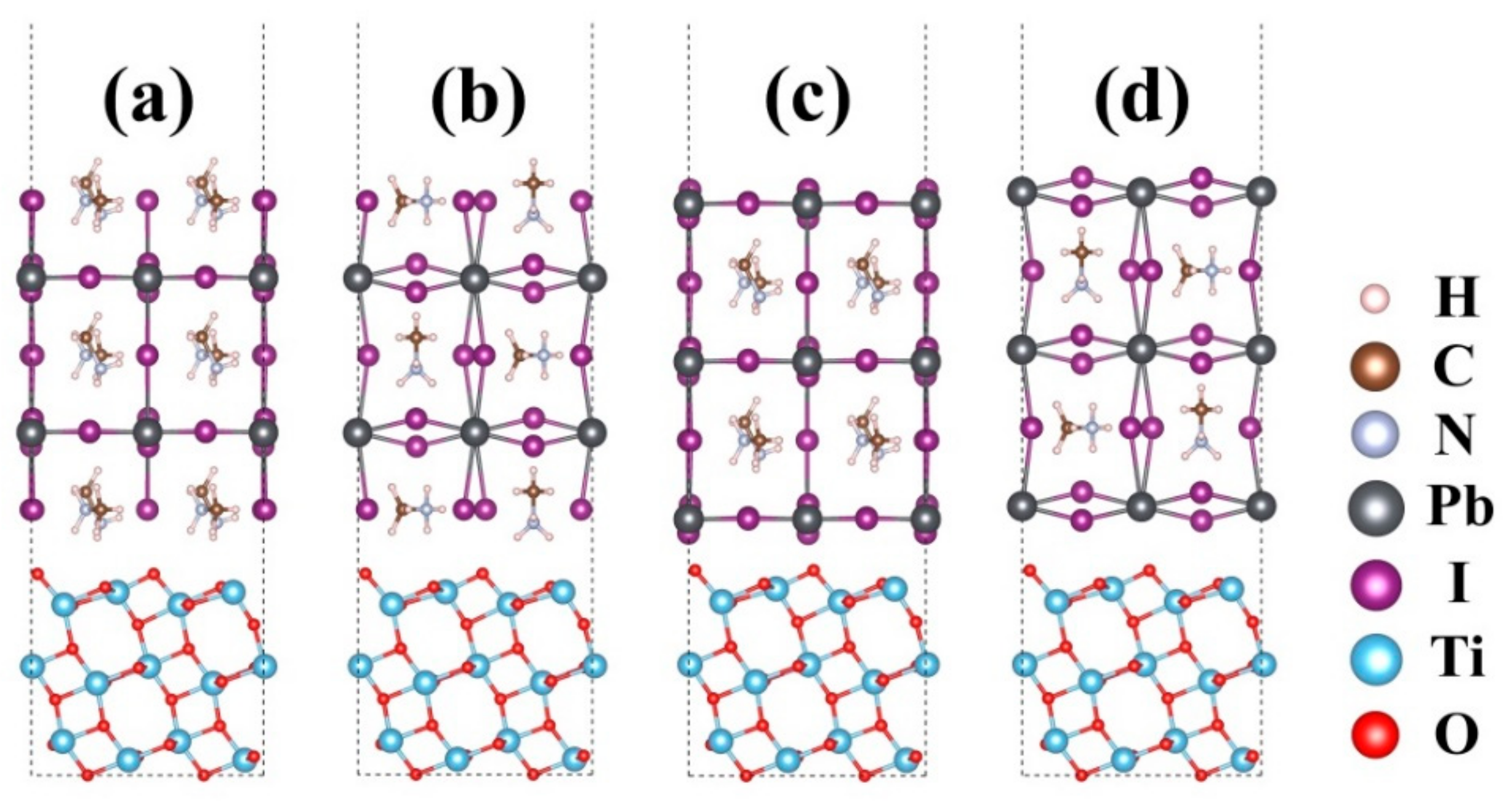
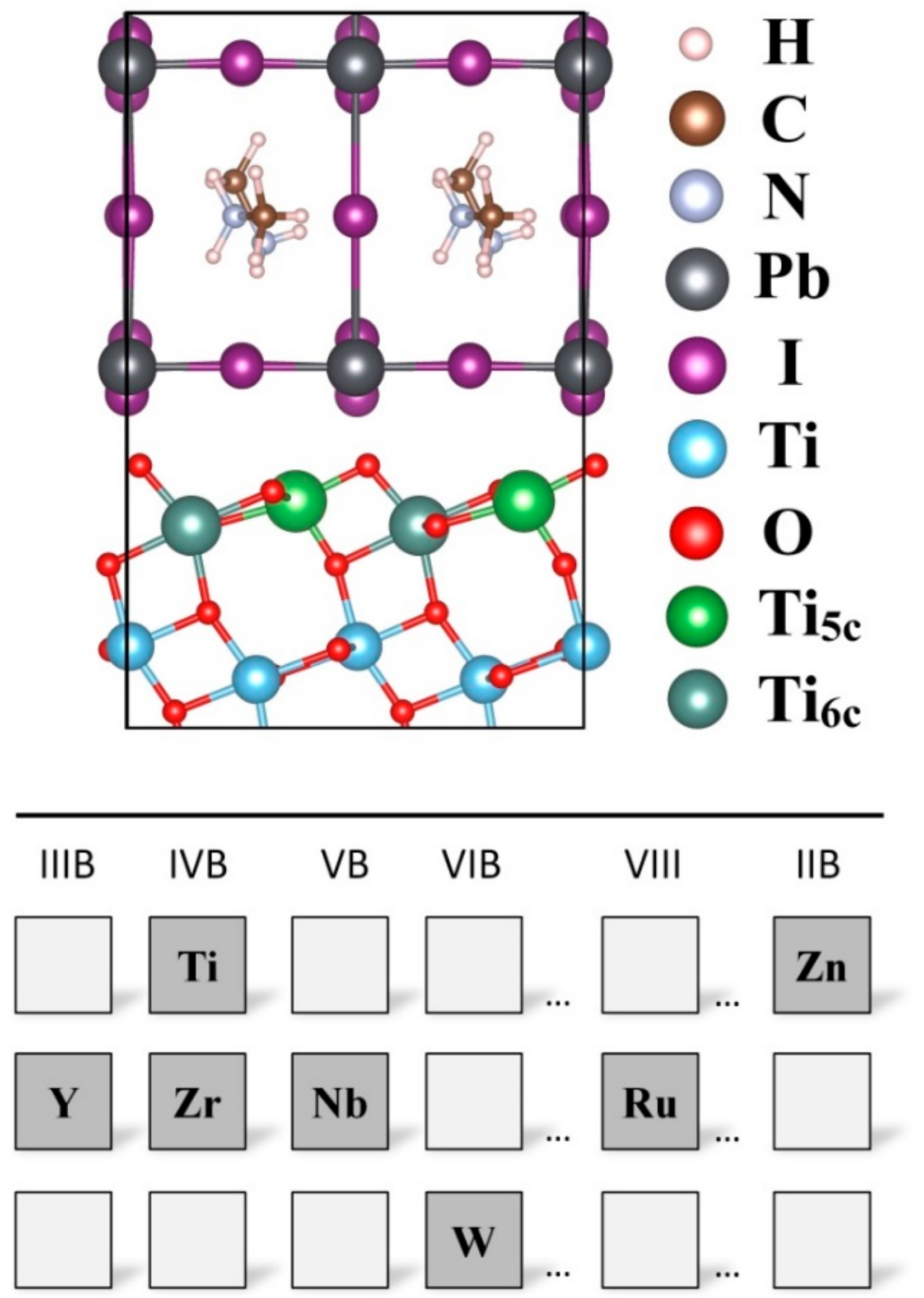
2. Methods
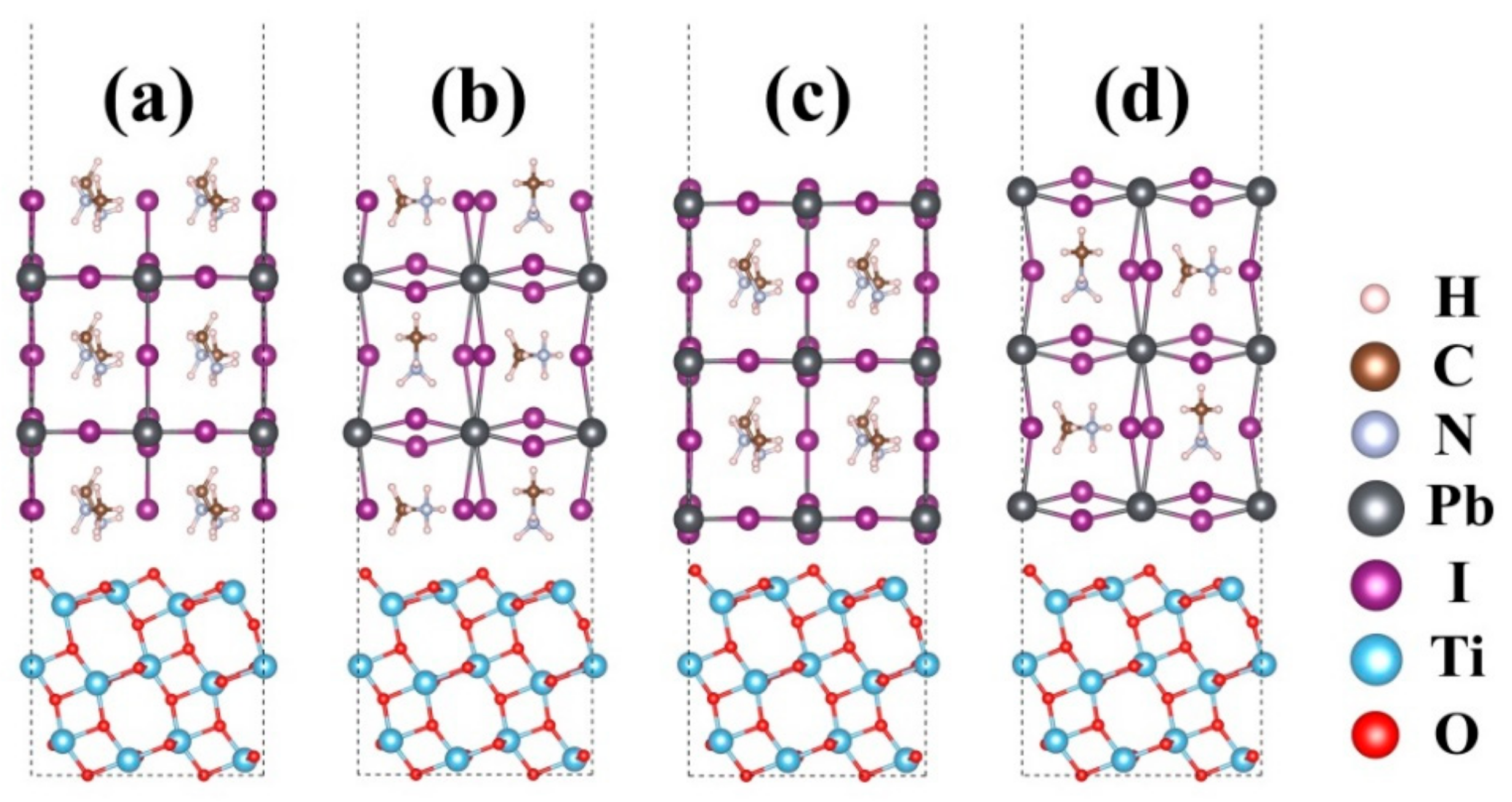
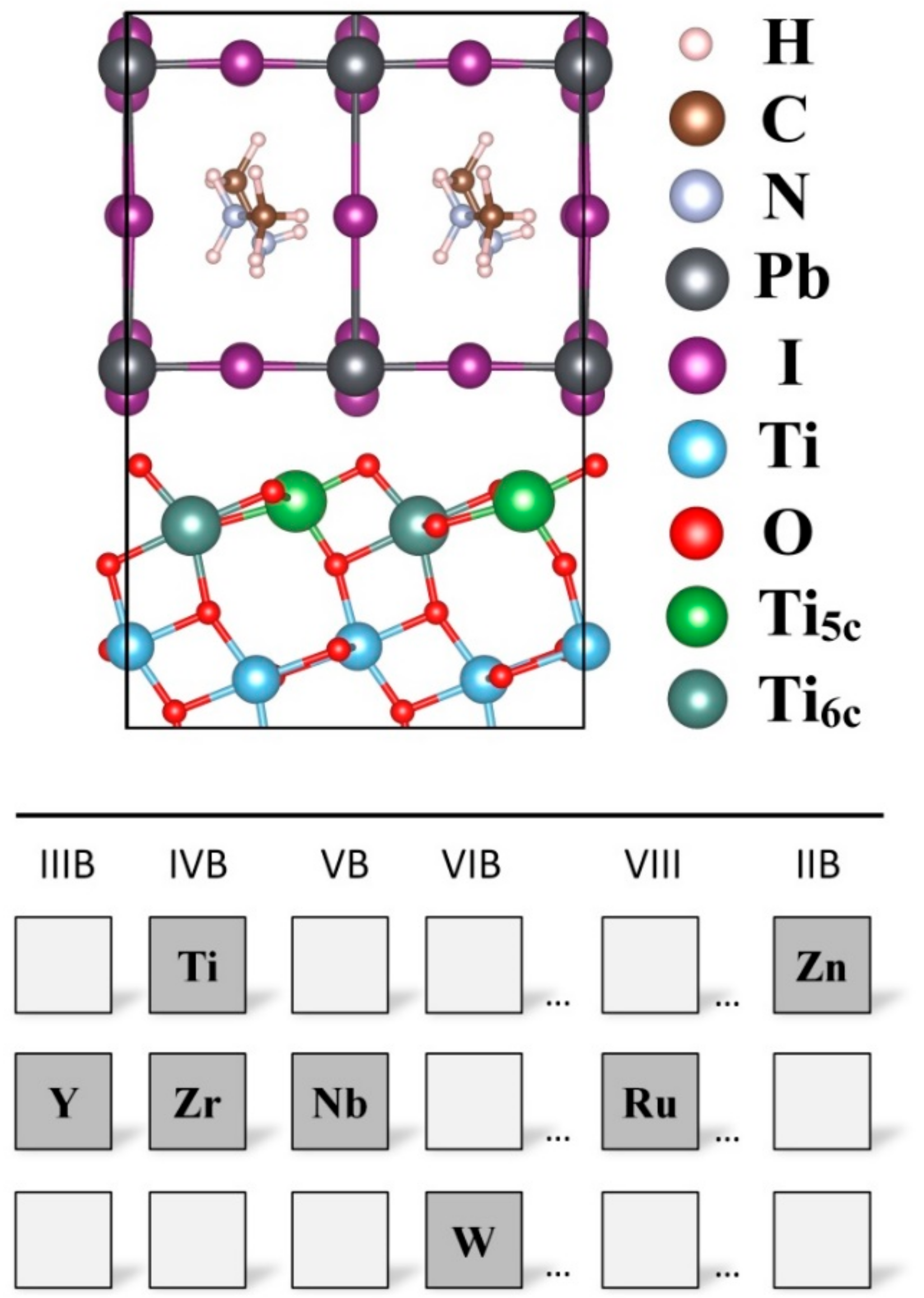
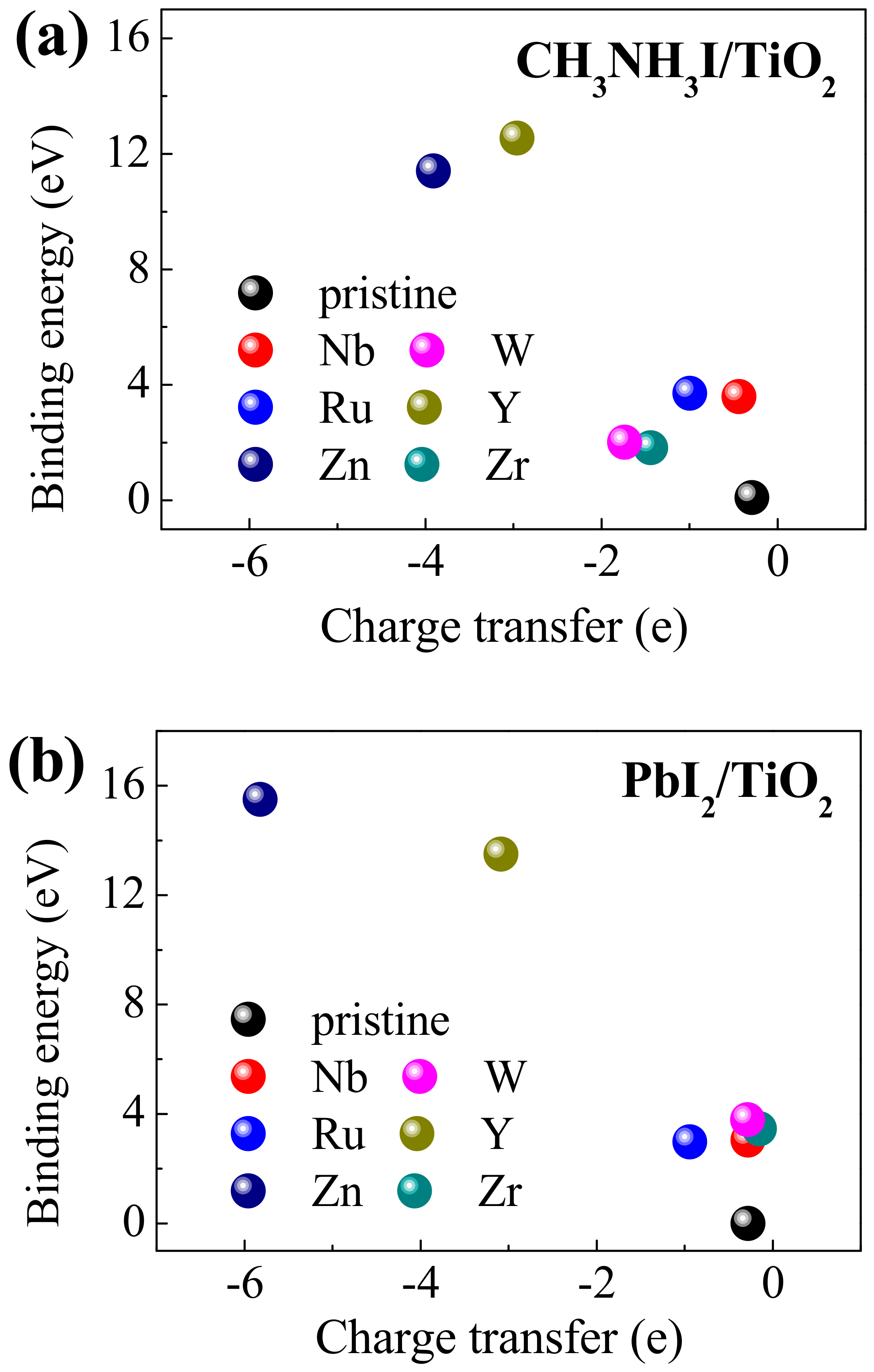
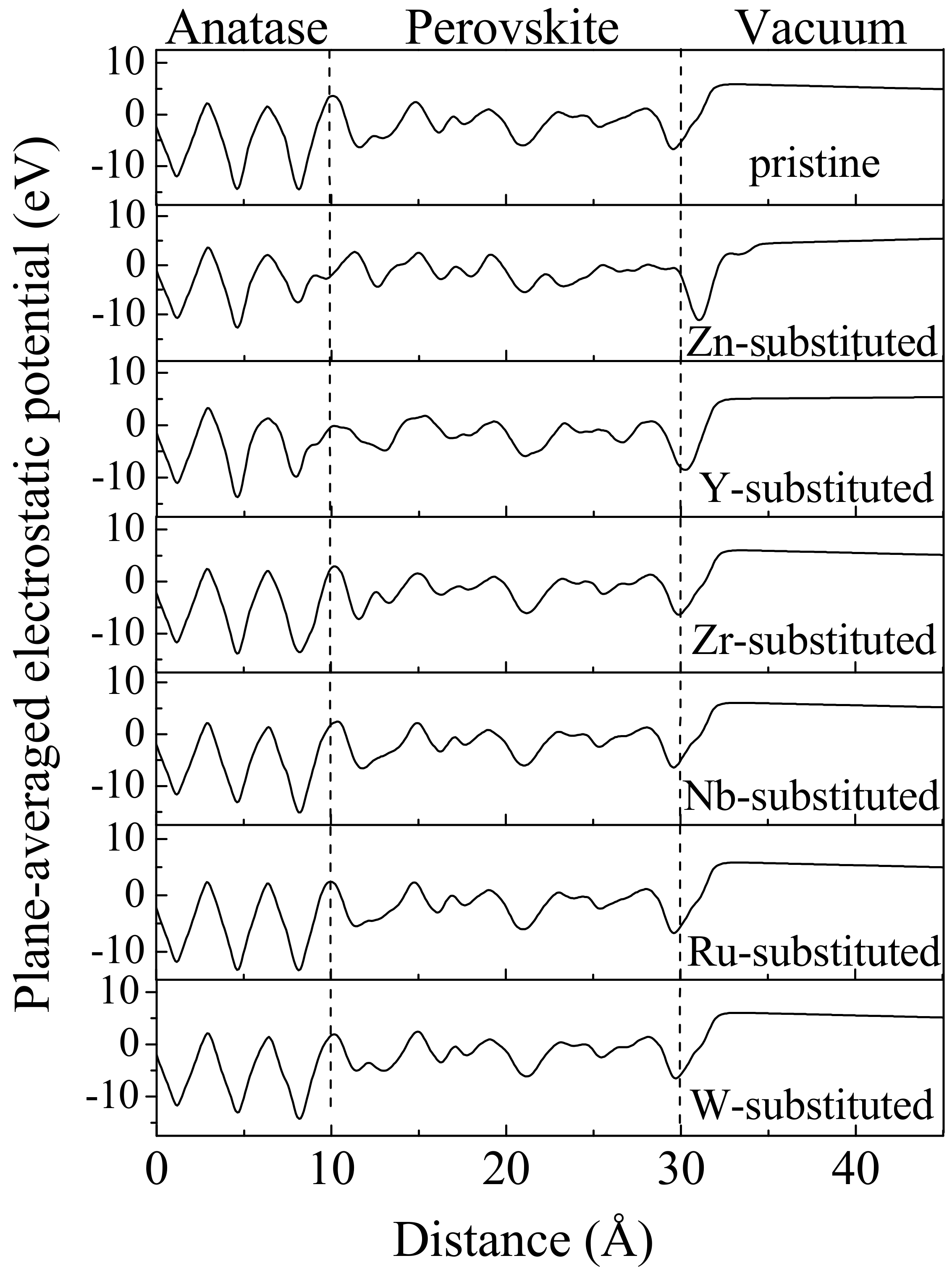
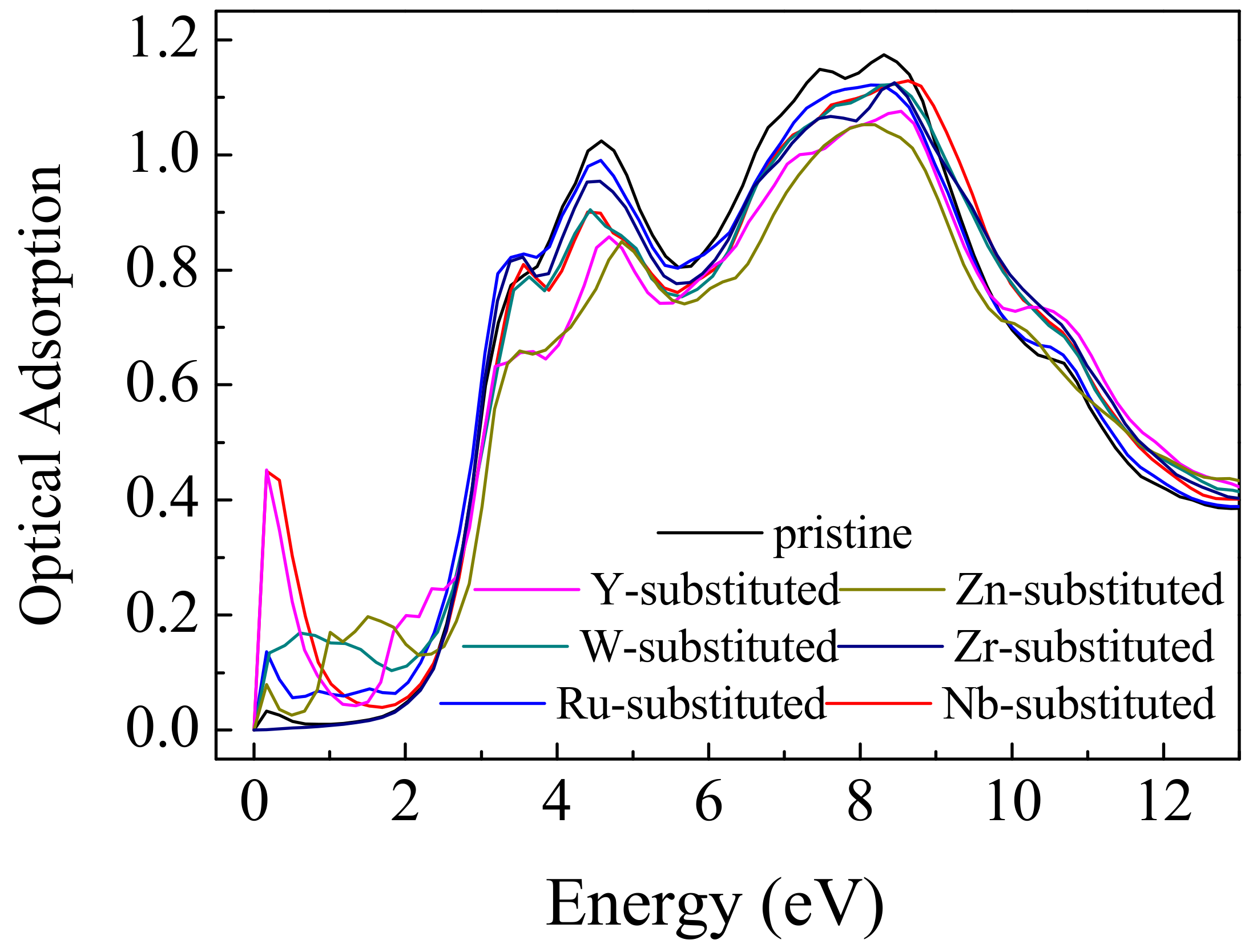
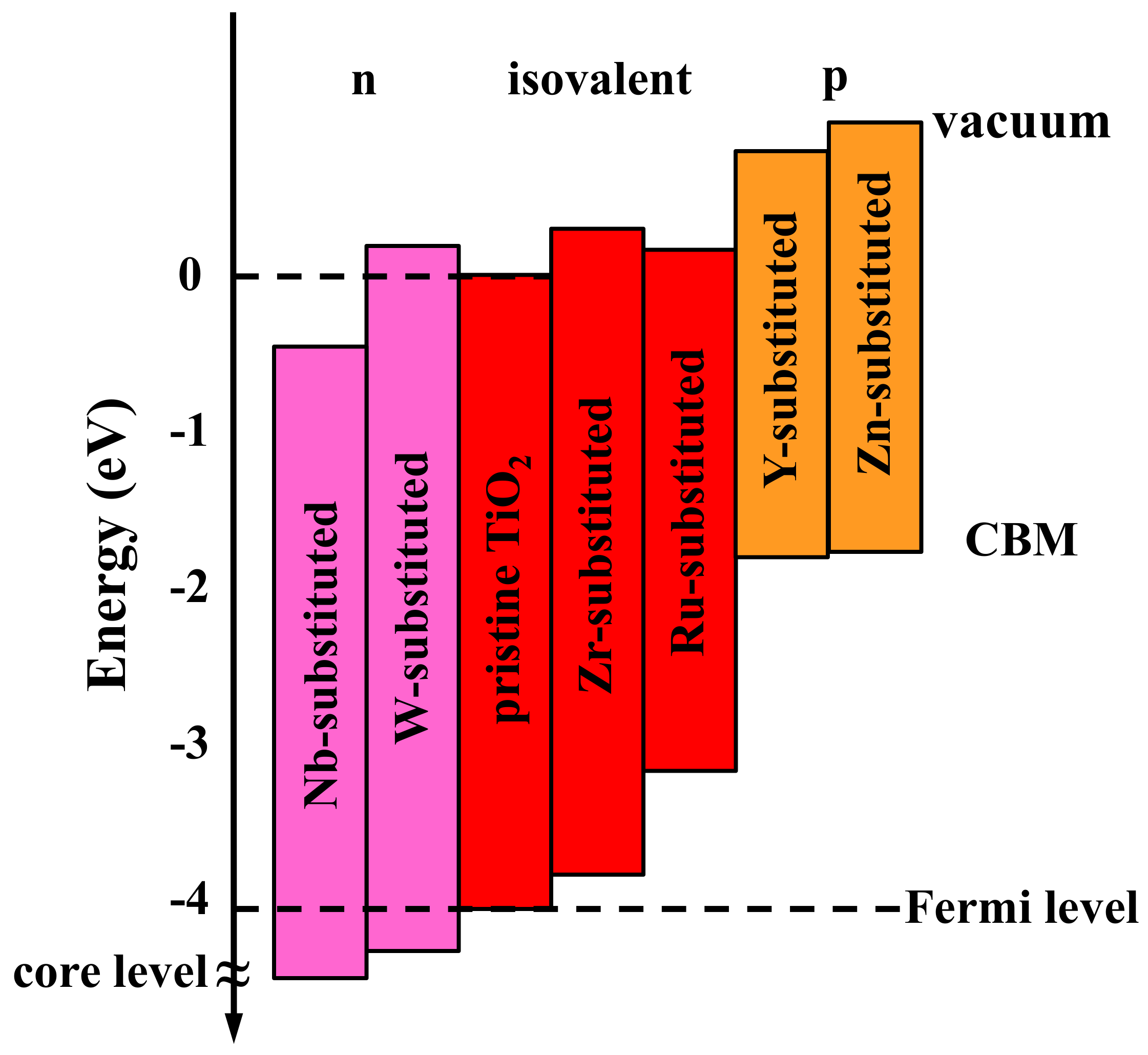
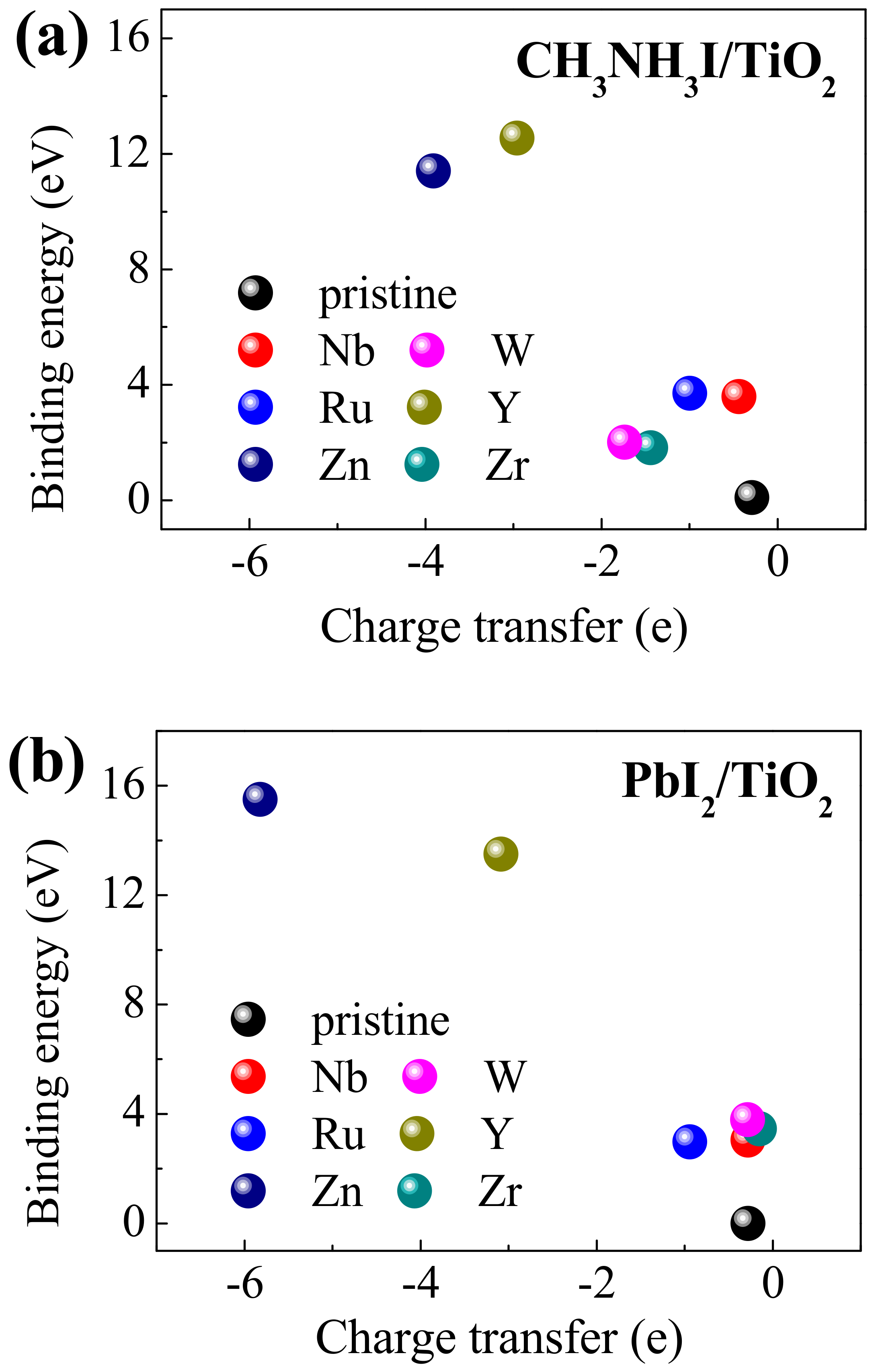
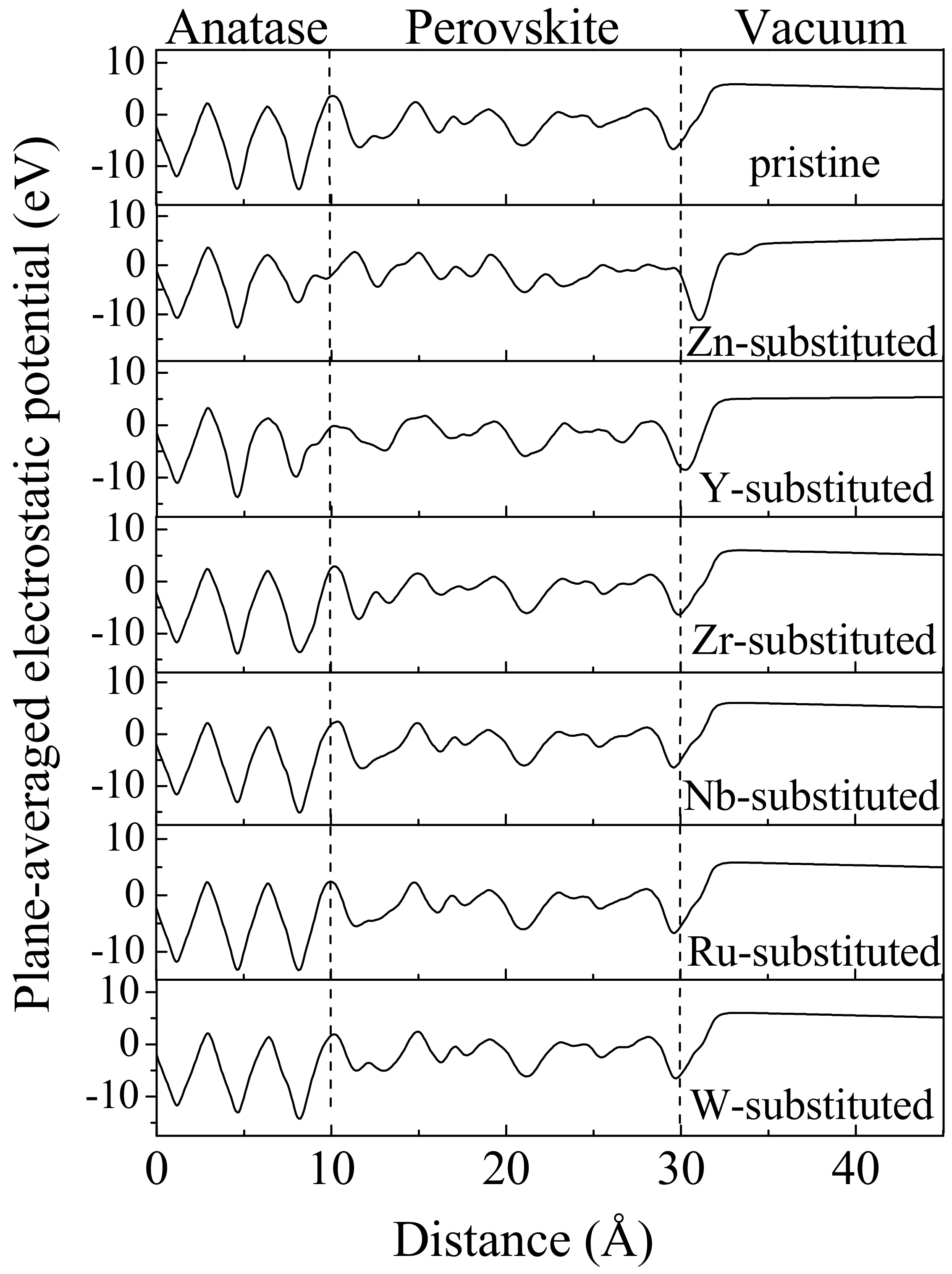
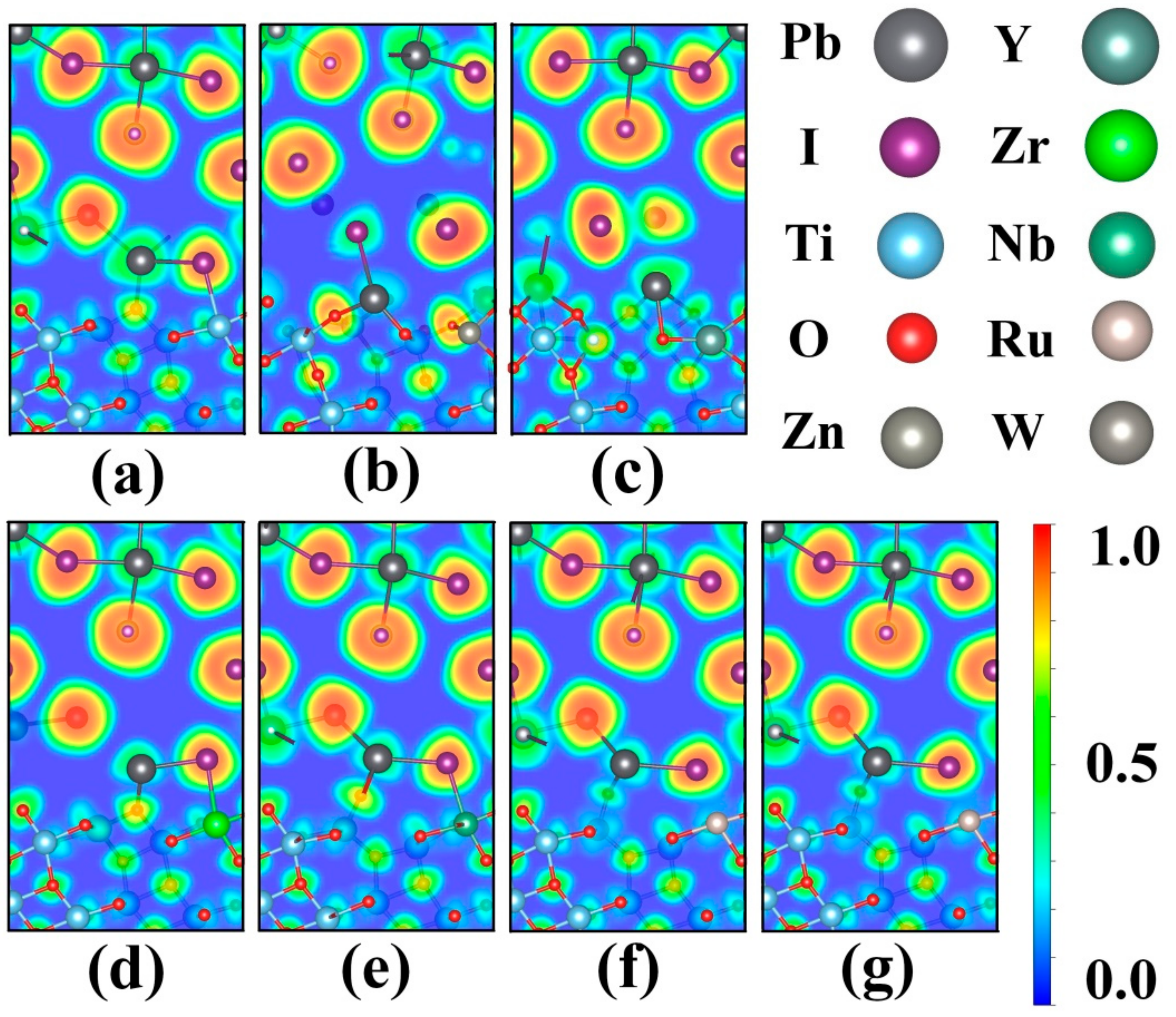
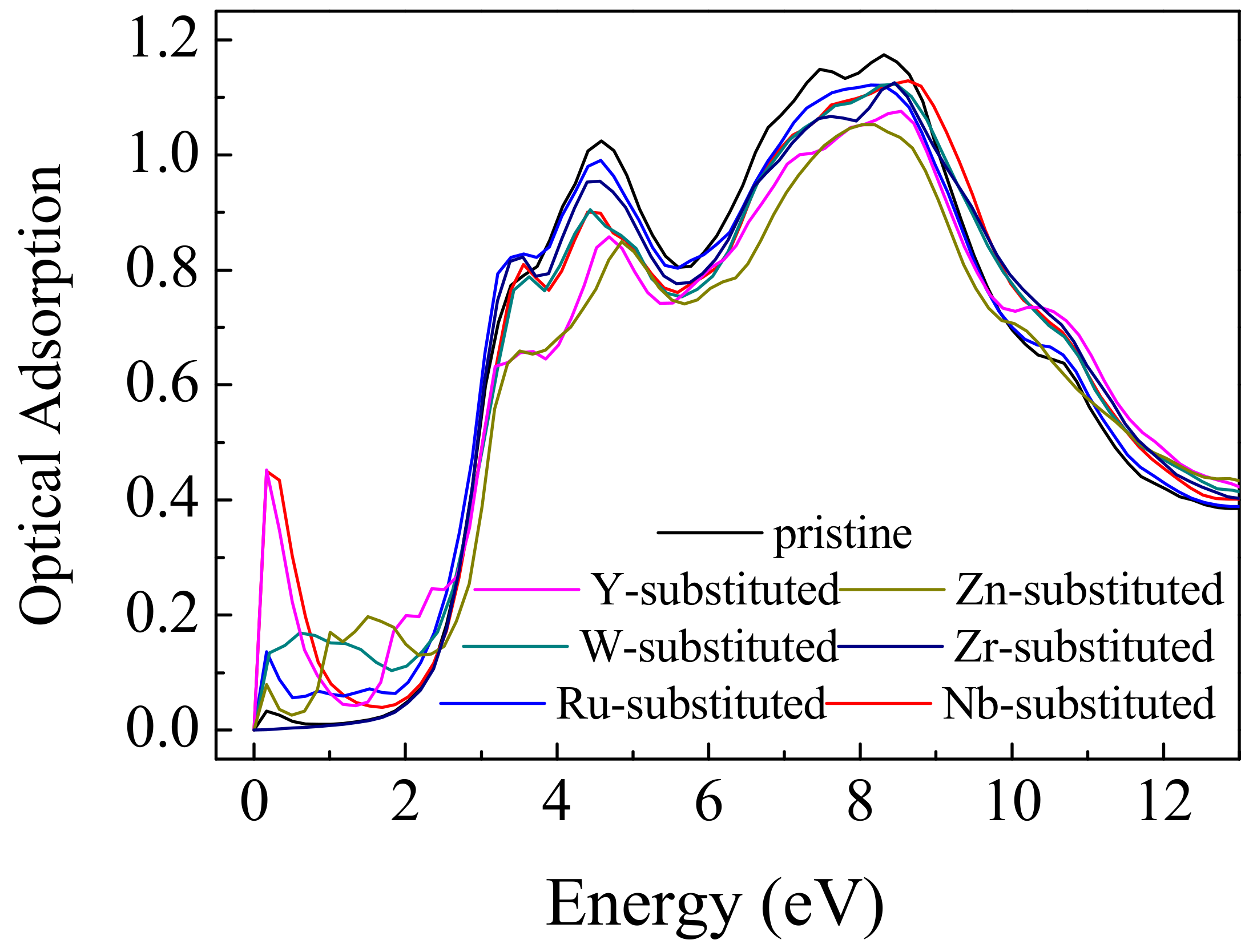
3. Results and Discussions
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, J.; Zhou, S.; Jin, S.; Li, H.; Zhai, T. Crystal Organometal Halide Perovskites with Promising Optoelectronic Applications. J. Mater. Chem. C 2016, 4, 11–27. [Google Scholar] [CrossRef]
- Yang, M.; Li, Z.; Reese, M.O.; Reid, O.G.; Kim, D.H.; Siol, S.; Klein, T.R.; Yan, Y.; Berry, J.J.; van Hest, M.F.A.M.; et al. Perovskite Ink with Wide Processing Window for Scalable High-Efficiency Solar Cells. Nat. Energy 2017, 2, 17038. [Google Scholar]
- Chen, W.; Wu, Y.; Yue, Y.; Liu, J.; Zhang, W.; Yang, X.; Chen, H.; Bi, E.; Ashraful, I.; Grätzel, M.; et al. Efficient and Stable Large-Area Perovskite Solar Cells with Inorganic Charge Extraction Layers. Science 2015, 350, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.H.; Han, H.J.; Kim, D.; Ahn, T.K.; Im, S.H. Hysteresis-Less Inverted CH3NH3PbI3 Planar Perovskite Hybrid Solar Cells with 18.1% Power Conversion Efficiency. Energy Environ. Sci. 2015, 8, 1602–1608. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, S.; Liu, H.; Feng, F.; Li, W. Improved Efficiency of Perovskite Solar Cells by the Interfacial Modification of the Active Layer. Nanomaterials 2019, 9, 204. [Google Scholar] [CrossRef] [PubMed]
- Mohamad Noh, M.F.; Teh, C.H.; Daik, R.; Lim, E.L.; Yap, C.C.; Ibrahim, M.A.; Ahmad Ludin, N.; Mohd Yusoff, A.R.b.; Jang, J.; Mat Teridi, M.A. The Architecture of the Electron Transport Layer for a Perovskite Solar Cell. J. Mater. Chem. C 2018, 6, 682–712. [Google Scholar] [CrossRef]
- Yang, G.; Tao, H.; Qin, P.; Ke, W.; Fang, G. Recent Progress in Electron Transport Layers for Efficient Perovskite Solar Cells. J. Mater. Chem. A 2016, 4, 3970–3990. [Google Scholar] [CrossRef]
- Ke, W.; Fang, G.; Liu, Q.; Xiong, L.; Qin, P.; Tao, H.; Wang, J.; Lei, H.; Li, B.; Wan, J.; et al. Low-Temperature Solution-Processed Tin Oxide as an Alternative Electron Transporting Layer for Efficient Perovskite Solar Cells. J. Am. Chem. Soc. 2015, 137, 6730–6733. [Google Scholar] [CrossRef]
- Yang, H.-Y.; Rho, W.-Y.; Lee, S.K.; Kim, S.H.; Hahn, Y.-B. TiO2 Nanoparticles/Nanotubes for Efficient Light Harvesting in Perovskite Solar Cells. Nanomaterials 2019, 9, 326. [Google Scholar] [CrossRef]
- Lindblad, R.; Bi, D.; Park, B.-W.; Oscarsson, J.; Gorgoi, M.; Siegbahn, H.; Odelius, M.; Johansson, E.M.J.; Rensmo, H. Electronic Structure of TiO2/CH3NH3PbI3 Perovskite Solar Cell Interfaces. J. Phys. Chem. Lett. 2014, 5, 648–653. [Google Scholar] [CrossRef]
- Yella, A.; Heiniger, L.-P.; Gao, P.; Nazeeruddin, M.K.; Grätzel, M. Nanocrystalline Rutile Electron Extraction Layer Enables Low-Temperature Solution Processed Perovskite Photovoltaics with 13.7% Efficiency. Nano Lett. 2014, 14, 2591–2596. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-X.; Ge, Q.-Q.; Xue, D.-J.; Ding, J.; Ma, J.-Y.; Chen, Y.-X.; Zhang, B.; Feng, Y.; Wan, L.-J.; Hu, J.-S. Tuning the Fermi-Level of TiO2 Mesoporous Layer by Lanthanum Doping towards Efficient Perovskite Solar Cells. Nanoscale 2016, 8, 16881–16885. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Ma, J.; Jiang, H.; Li, J.; Yang, D.; Gao, F.; Zeng, J.; Liu, Z.; Liu, S.F. Correction to Enhancing Efficiency and Stability of Perovskite Solar Cells through Nb-Doping of TiO2 at Low Temperature. Acs Appl. Mater. Interfaces 2017, 9, 14545. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-X.; Rao, H.-S.; Li, W.-G.; Xu, Y.-F.; Chen, H.-Y.; Kuang, D.-B.; Su, C.-Y. Achieving High-Performance Planar Perovskite Solar Cell with Nb-Doped TiO2 Compact Layer by Enhanced Electron Injection and Efficient Charge Extraction. J. Mater. Chem. A 2016, 4, 5647–5653. [Google Scholar] [CrossRef]
- Chen, S.-H.; Chan, S.-H.; Lin, Y.-T.; Wu, M.-C. Enhanced Power Conversion Efficiency of Perovskite Solar Cells Based on Mesoscopic Ag-Doped TiO2 Electron Transport Layer. Appl. Surf. Sci. 2019, 469, 18–26. [Google Scholar] [CrossRef]
- Liu, D.; Li, S.; Zhang, P.; Wang, Y.; Zhang, R.; Sarvari, H.; Wang, F.; Wu, J.; Wang, Z.; Chen, Z.D. Efficient Planar Heterojunction Perovskite Solar Cells with Li-Doped Compact TiO2 Layer. Nano Energy 2017, 31, 462–468. [Google Scholar] [CrossRef]
- Wu, M.-C.; Chan, S.-H.; Jao, M.-H.; Su, W.-F. Enhanced Short-Circuit Current Density of Perovskite Solar Cells Using Zn-Doped TiO2 as Electron Transport Layer. Sol. Energy Mater. Sol. Cells 2016, 157, 447–453. [Google Scholar] [CrossRef]
- Xu, Z.; Yin, X.; Guo, Y.; Pu, Y.; He, M. Ru-Doping in TiO2 Electron Transport Layers of Planar Heterojunction Perovskite Solar Cells for Enhanced Performance. J. Mater. Chem. C 2018, 6, 4746–4752. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, J.; Yue, G.; Lu, X.; Hu, Z.; Zhu, Y. W-Doped TiO2 Photoanode for High Performance Perovskite Solar Cell. Electrochim. Acta 2016, 195, 143–149. [Google Scholar]
- Zhou, H.; Chen, Q.; Li, G.; Luo, S.; Song, T.-B.; Duan, H.-S.; Hong, Z.; You, J.; Liu, Y.; Yang, Y. Interface Engineering of Highly Efficient Perovskite Solar Cells. Science 2014, 345, 542–546. [Google Scholar] [CrossRef]
- Cui, Q.; Zhao, X.; Lin, H.; Yang, L.; Chen, H.; Zhang, Y.; Li, X. Improved Efficient Perovskite Solar Cells Based on Ta-Doped TiO2 Nanorod Arrays. Nanoscale 2017, 9, 18897–18907. [Google Scholar] [CrossRef] [PubMed]
- García-Mota, M.; Vojvodic, A.; Abild-Pedersen, F.; Nørskov, J.K. Electronic Origin of the Surface Reactivity of Transition-Metal-Doped TiO2(110). J. Phys. Chem. C 2013, 117, 460–465. [Google Scholar] [CrossRef]
- Pathak, S.K.; Abate, A.; Ruckdeschel, P.; Roose, B.; Gödel, K.C.; Vaynzof, Y.; Santhala, A.; Watanabe, S.-I.; Hollman, D.J.; Noel, N.; et al. Performance and Stability Enhancement of Dye-Sensitized and Perovskite Solar Cells by Al Doping of TiO2. Adv. Funct. Mater. 2014, 24, 6046–6055. [Google Scholar] [CrossRef]
- Xu, X.; Li, K.; Yang, Z.; Shi, J.; Li, D.; Gu, L.; Wu, Z.; Meng, Q. Methylammonium Cation Deficient Surface for Enhanced Binding Stability at TiO2/CH3NH3PbI3 Interface. Nano Res. 2017, 10, 483–490. [Google Scholar] [CrossRef]
- Geng, W.; Tong, C.-J.; Liu, J.; Zhu, W.; Lau, W.-M.; Liu, L.-M. Structures and Electronic Properties of Different CH3NH3PbI3/TiO2 Interface: A First-Principles Study. Sci. Rep. 2016, 6, 20131. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Zheng, G.; Zhao, B.; Song, F.; Zhang, X.; Shen, K.; Yang, Y.; Xiong, Y.; Gao, X.; Cao, L.; et al. Interfacial Electronic Structures Revealed at the Rubrene/CH3NH3PbI3 Interface. Phys. Chem. Chem. Phys. 2017, 19, 6546–6553. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.-J. Ferroelectric Polarization Driven Optical Absorption and Charge Carrier Transport in CH3NH3PbI3/TiO2-Based Photovoltaic Cells. J. Power Sources 2015, 291, 58–65. [Google Scholar] [CrossRef]
- Hu, J.; Ji, G.; Ma, X.; He, H.; Huang, C. Probing Interfacial Electronic Properties of Graphene/CH3NH3PbI3 Heterojunctions: A Theoretical Study. Appl. Surf. Sci. 2018, 440, 35–41. [Google Scholar] [CrossRef]
- Nemnes, G.A.; Goehry, C.; Mitran, T.L.; Nicolaev, A.; Ion, L.; Antohe, S.; Plugaru, N.; Manolescu, A. Band Alignment and Charge Transfer in Rutile-TiO2/CH3NH3PbI3-xClx Interfaces. Phys. Chem. Chem. Phys. 2015, 17, 30417–30423. [Google Scholar] [CrossRef]
- Haruyama, J.; Sodeyama, K.; Hamada, I.; Han, L.; Tateyama, Y. First-Principles Study of Electron Injection and Defects at the TiO2/CH3NH3PbI3 Interface of Perovskite Solar Cells. J. Phys. Chem. Lett. 2017, 8, 5840–5847. [Google Scholar] [CrossRef]
- Mosconi, E.; Grancini, G.; Roldán-Carmona, C.; Gratia, P.; Zimmermann, I.; Nazeeruddin, M.K.; De Angelis, F. Enhanced TiO2/MAPbI3 Electronic Coupling by Interface Modification with PbI2. Chem. Mater. 2016, 28, 3612–3615. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, W. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Mosconi, E.; Umari, P.; De Angelis, F. Electronic and Optical Properties of MAPbX3 Perovskites (X = I, Br, Cl): A Unified DFT and GW Theoretical Analysis. Phys. Chem. Chem. Phys. 2016, 18, 27158–27164. [Google Scholar] [CrossRef] [PubMed]
- Sumita, M.; Sodeyama, K.; Jono, R.; Han, L.; Tateyama, Y. Electronic Structure of Acetonitrile Adsorbed on the Anatase TiO2 (101) Surface. Chem. Phys. Lett. 2013, 556, 225–229. [Google Scholar]
- Sun, R.; Wang, Z.; Shibata, N.; Ikuhara, Y. A Dislocation Core in Titanium Dioxide and Its Electronic Structure. Rsc Adv. 2015, 5, 18506–18510. [Google Scholar] [CrossRef]
- Fang, Y.; Cheng, D.; Niu, M.; Yi, Y.; Wu, W. Tailoring the Electronic and Optical Properties of Rutile TiO2 by (Nb+Sb, C) Codoping from DFT+U Calculations. Chem. Phys. Lett. 2013, 567, 34–38. [Google Scholar]
- Hou, Y.S.; Xiang, H.J.; Gong, X.G. Unveiling Magnetic Interactions of Ruthenium Trichloride via Constraining Direction of Orbital Moments: Potential Routes to Realize a Quantum Spin Liquid. Phys. Rev. B 2017, 96, 054410. [Google Scholar] [CrossRef]
- Gong, J.; Yang, C.; Zhang, J.; Pu, W. Origin of photocatalytic activity of W/N-codoped TiO2:H2 production and DFT calculation with GGA+U. Appl. Catal. B Environ. 2014, 152–153, 73–81. [Google Scholar] [CrossRef]
- Zasada, F.; Gryboś, J.; Indyka, P.; Piskorz, W.; Kaczmarczyk, J.; Sojka, Z. Surface Structure and Morphology of M[CoM′]O4 (M = Mg, Zn, Fe, Co and M′ = Ni, Al, Mn, Co) Spinel Nanocrystals—DFT+U and TEM Screening Investigations. J. Phys. Chem. C 2014, 118, 19085–19097. [Google Scholar] [CrossRef]
- Chen, H.-Y.T.; Tosoni, S.; Pacchioni, G. Adsorption of Ruthenium Atoms and Clusters on Anatase TiO2 and Tetragonal ZrO2(101) Surfaces: A Comparative DFT Study. J. Phys. Chem. C 2015, 119, 10856–10868. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-Precision Sampling for Brillouin-Zone Integration in Metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [PubMed]
- Even, J.; Pedesseau, L.; Jancu, J.-M.; Katan, C. Importance of Spin–Orbit Coupling in Hybrid Organic/Inorganic Perovskites for Photovoltaic Applications. J. Phys. Chem. Lett. 2013, 4, 2999–3005. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A Three-Dimensional Visualization System for Electronic and Structural Analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Ke, W.; Fang, G.; Wang, J.; Qin, P.; Tao, H.; Lei, H.; Liu, Q.; Dai, X.; Zhao, X. Perovskite Solar Cell with an Efficient TiO2 Compact Film. Acs Appl. Mater. Interfaces 2014, 6, 15959–15965. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, E.; Ronca, E.; De Angelis, F. First-Principles Investigation of the TiO2/Organohalide Perovskites Interface: The Role of Interfacial Chlorine. J. Phys. Chem. Lett. 2014, 5, 2619–2625. [Google Scholar] [CrossRef]
- Li, Z.; Wang, X.; Chi, B. First Principles Investigation of the Conversion of N2O and CO to N2 and CO2 on a Modified N + Fe/TiO2 (101) Surface. Rsc Adv. 2014, 4, 17896–17901. [Google Scholar] [CrossRef]
- Chen, Q.L.; Li, B.; Zheng, G.; He, K.H.; Zheng, A.S. First-Principles Calculations on Electronic Structures of Fe-Vacancy-Codoped TiO2 Anatase (101) Surface. Phys. B Condens. Matter 2011, 406, 3841–3846. [Google Scholar] [CrossRef]
- Ma, J.-G.; Zhang, C.-R.; Gong, J.-J.; Wu, Y.-Z.; Kou, S.-Z.; Yang, H.; Chen, Y.-H.; Liu, Z.-J.; Chen, H.-S. The Electronic Structures and Optical Properties of Alkaline-Earth Metals Doped Anatase TiO2: A Comparative Study of Screened Hybrid Functional and Generalized Gradient Approximation. Materials 2015, 8, 5508–5525. [Google Scholar] [CrossRef] [PubMed]
- Cromer, D.T.; Herrington, K. The Structures of Anatase and Rutile. J. Am. Chem. Soc. 1955, 77, 4708–4709. [Google Scholar] [CrossRef]
- Feng, J.; Xiao, B. Crystal Structures, Optical Properties, and Effective Mass Tensors of CH3NH3PbX3 (X = I and Br) Phases Predicted from HSE06. J. Phys. Chem. Lett. 2014, 5, 1278–1282. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.; Mashiyama, H.; Hasebe, K. Structural Study on Cubic-Tetragonal Transition of CH3NH3PbI3. J. Phys. Soc. Jpn. 2002, 71, 1694–1697. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, Y.; Liu, Y. Stability and Charge Separation of Different CH3NH3SnI3/TiO2 Interface: A First-Principles Study. Appl. Surf. Sci. 2018, 441, 394–400. [Google Scholar] [CrossRef]
- Si, F.; Hu, W.; Tang, F.; Cheng, Y.; Xue, H. Electronic and Optical Properties of the Wurtzite- ZnO/CH3NH3PbI3 Interface: First-Principles Calculations. J. Mater. Sci. 2017, 52, 13841–13851. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A Fast and Robust Algorithm for Bader Decomposition of Charge Density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar]
- Feng, H.-J.; Paudel, T.R.; Tsymbal, E.Y.; Zeng, X.C. Tunable Optical Properties and Charge Separation in CH3NH3SnxPb1−xI3/TiO2-Based Planar Perovskites Cells. J. Am. Chem. Soc. 2015, 137, 8227–8236. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, X.; Yang, F.; Yu, Y. Combination Study of DFT Calculation and Experiment for Photocatalytic Properties of S-Doped Anatase TiO2. Appl. Surf. Sci. 2014, 319, 50–59. [Google Scholar] [CrossRef]
- Navas, J.; Sánchez-Coronilla, A.; Aguilar, T.; Hernández, N.C.; de los Santos, D.M.; Sánchez-Márquez, J.; Zorrilla, D.; Fernández-Lorenzo, C.; Alcántara, R.; Martín-Calleja, J. Experimental and theoretical study of the electronic properties of Cu-doped anatase TiO2. Phys. Chem. Chem. Phys. 2014, 16, 3835–3845. [Google Scholar] [CrossRef]
- Navas, J.; Sánchez-Coronilla, A.; Gallardo, J.J.; Cruz Hernández, N.; Piñero, J.C.; Alcántara, R.; Fernández-Lorenzo, C.; De los Santos, D.M.; Aguilar, T.; Martín-Calleja, J. New Insights into Organic–Inorganic Hybrid Perovskite CH3NH3PbI3 Nanoparticles. An Experimental and Theoretical Study of Doping in Pb2+ Sites with Sn2+, Sr2+, Cd2+ and Ca2+. Nanoscale 2015, 7, 6216–6229. [Google Scholar] [CrossRef] [PubMed]
- Li, R.-W.; Wang, Z.-H.; Chen, X.; Sun, J.-R.; Shen, B.-G. Magnetic Properties and Colossal Magnetoresistance of the Perovskites La2/3Ca1/3Mn1−xTi xO. J. Appl. Phys. 2000, 87, 5597–5599. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Yu, X.; Li, J. Enhanced Photoluminescence Properties of Zn2SiO4:Mn2+ Co-Activated with Y3+/Li+ under VUV Excitation. J. Lumin. 2010, 130, 2171–2174. [Google Scholar] [CrossRef]
- Yang, J.Y.; Liu, L.H.; Tan, J.Y. First-principles Molecular Dynamics Study on Temperature-Dependent Dielectric Function of Bulk 3C and 6H SiC in the Energy Range 3–8eV. Phys. B Condens. Matter 2014, 436, 182–187. [Google Scholar] [CrossRef]
- Lin, C.; Li, S.; Zhang, W.; Shao, C.; Yang, Z. Effect of Bromine Substitution on the Ion Migration and Optical Absorption in MAPbI3 Perovskite Solar Cells: The First-Principles Study. Acs Appl. Energy Mater. 2018, 1, 1374–1380. [Google Scholar] [CrossRef]
- Khan, M.; Xu, J.; Chen, N.; Cao, W. First Principle Calculations of the Electronic and Optical Properties of Pure and (Mo, N) Co-Doped Anatase TiO2. J. Alloy. Compd. 2012, 513, 539–545. [Google Scholar] [CrossRef]
- Dashora, A.; Patel, N.; Kothari, D.C.; Ahuja, B.L.; Miotello, A. Formation of an Intermediate Band in the Energy Gap of TiO2 by Cu–N-Codoping: First Principles Study and Experimental Evidence. Sol. Energy Mater. Sol. Cells 2014, 125, 120–126. [Google Scholar] [CrossRef]
- Jia, L.; Wu, C.; Han, S.; Yao, N.; Li, Y.; Li, Z.; Chi, B.; Pu, J.; Jian, L. Theoretical Study on the Electronic and Optical Properties of (N, Fe)-Codoped Anatase TiO2 Photocatalyst. J. Alloy. Compd. 2011, 509, 6067–6071. [Google Scholar] [CrossRef]
- Long, R.; Prezhdo, O.V. Dopants Control Electron–Hole Recombination at Perovskite-TiO2 Interfaces: Ab Initio Time-Domain Study. ACS Nano 2015, 9, 11143–11155. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
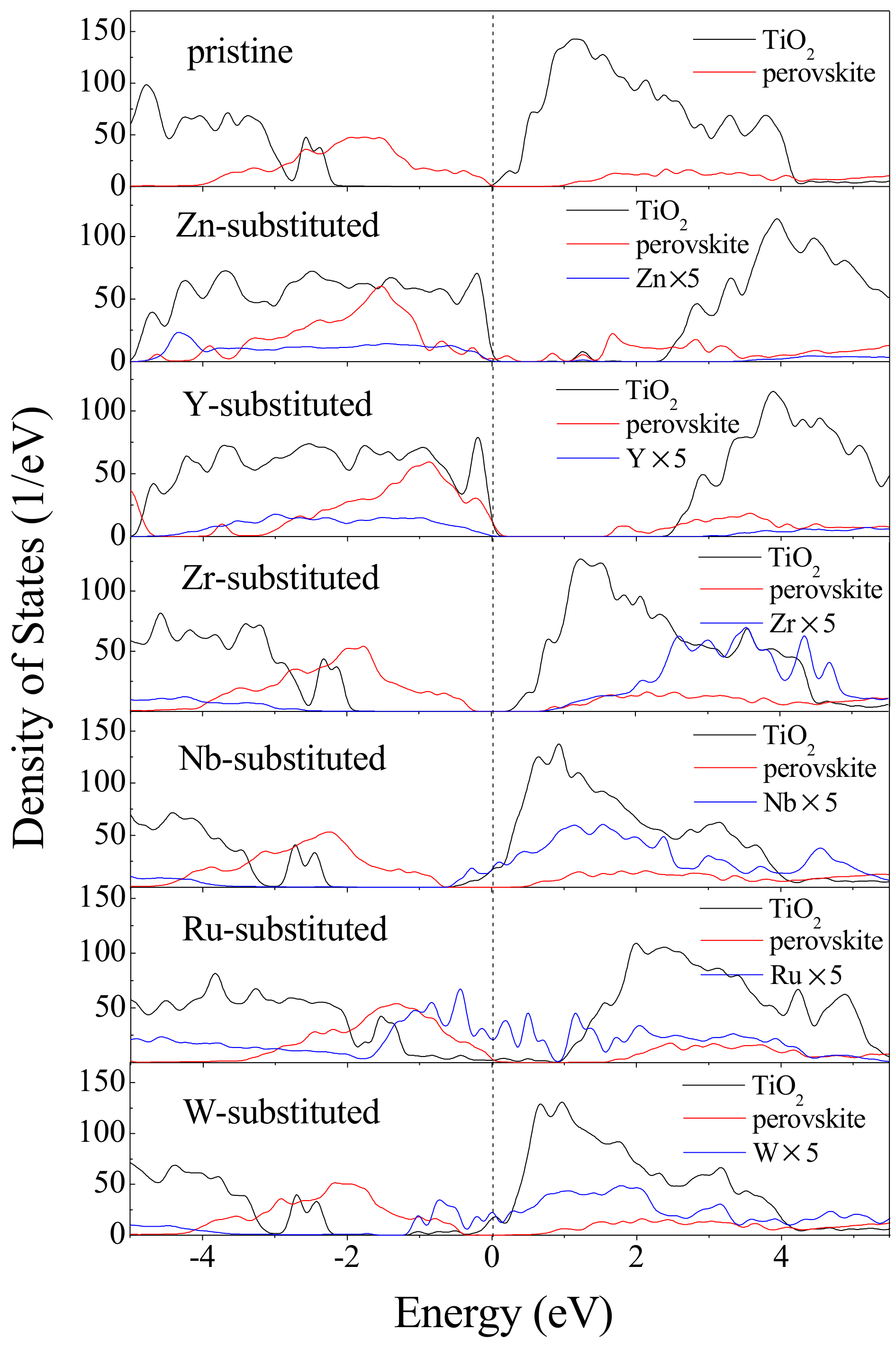{kind=link}
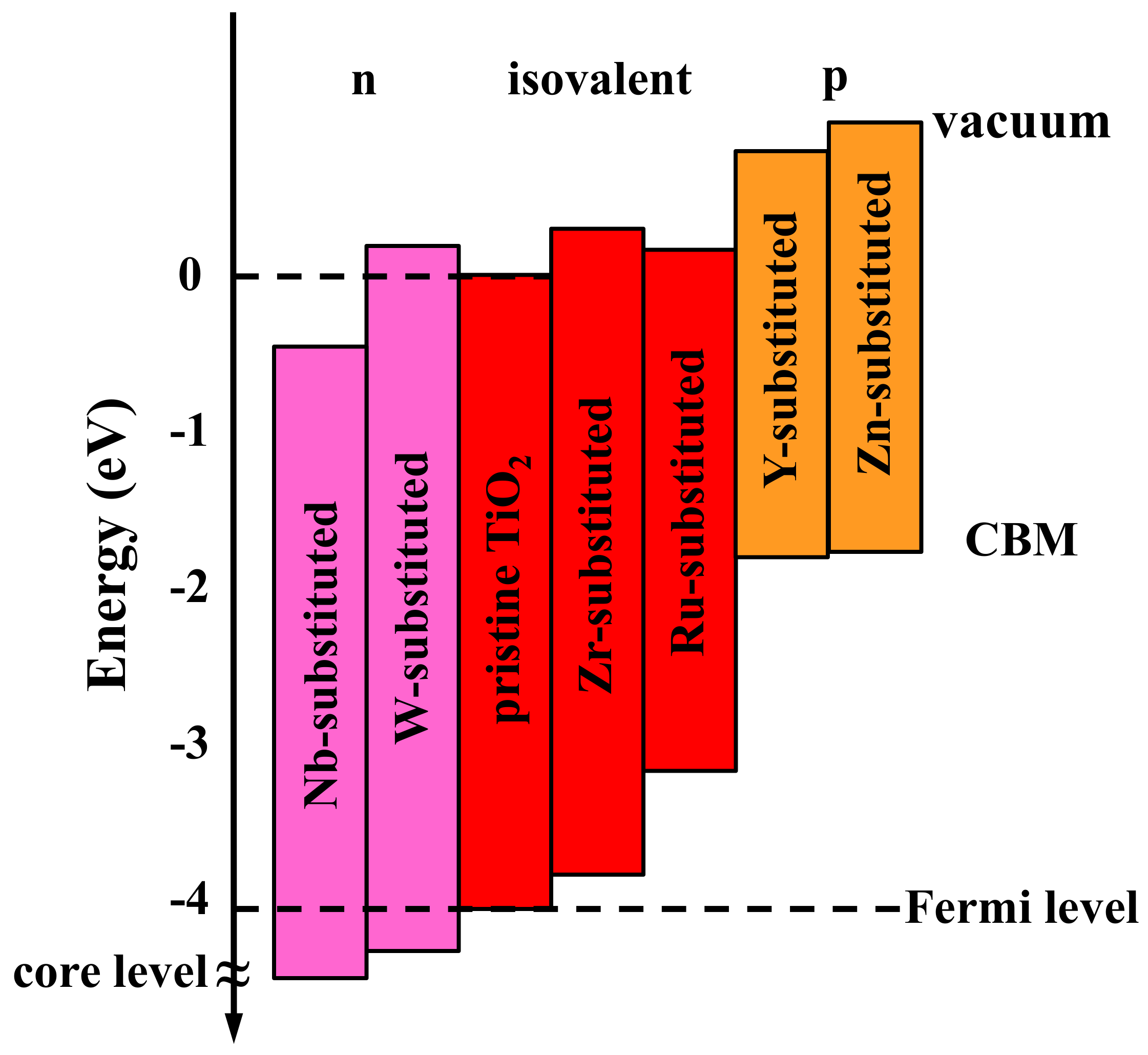{kind=link}
| Anatase TiO2 | Tetragonal -MAPbI3 | |||
|---|---|---|---|---|
| a | c | a | c | |
| Experimental | 3.785 | 9.514 | 8.80 | 12.685 |
| Calculated | 3.79 | 9.53 | 8.80 | 13.05 |
| Deviation | 0.13% | 0.17% | - | 2.8% |
| CH3NH3I/TiO2 | PbI2/TiO2 | CH3NH3I/TiO2 With Rotation | PbI2/TiO2 With Rotation | |
|---|---|---|---|---|
| Binding energy | 2.16 | 2.07 | 0.00 | 1.44 |
| Lattice mismatch | −12.0% | −12.0% | −13.8% | −13.8% |
| Charge transfer | −0.29 | −0.28 | −0.16 | −0.16 |
| Position | Surface (Ti5c) | Sub-Surface (Ti6c) | Inner-Surface (Third Ti Layer) | Inner-Surface (Fourth Ti Layer) |
|---|---|---|---|---|
| Total energy | −1401.71 | −1401.94 | −1401.52 | −1401.72 |
| Ti5c Site | Ti6c Site | CH3NH3I/TiO2 | PbI2/TiO2 | |||
|---|---|---|---|---|---|---|
| CH3NH3I/TiO2 | PbI2/TiO2 | CH3NH3I/TiO2 | PbI2/TiO2 | |||
| Binding energy | 3.59 | 3.05 | 2.77 | 2.54 | 0.09 | 0.00 |
| Charge transfer | −0.44 | −0.28 | −0.26 | −0.15 | −0.29 | −0.28 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Xue, Y.; Li, X.; Li, C.; Song, H.; Niu, Y.; Liu, H.; Mai, X.; Zhang, J.; Guo, Z. Effects of Transition Metal Substituents on Interfacial and Electronic Structure of CH3NH3PbI3/TiO2 Interface: A First-Principles Comparative Study. Nanomaterials 2019, 9, 966. https://doi.org/10.3390/nano9070966
Guo Y, Xue Y, Li X, Li C, Song H, Niu Y, Liu H, Mai X, Zhang J, Guo Z. Effects of Transition Metal Substituents on Interfacial and Electronic Structure of CH3NH3PbI3/TiO2 Interface: A First-Principles Comparative Study. Nanomaterials. 2019; 9(7):966. https://doi.org/10.3390/nano9070966
Chicago/Turabian StyleGuo, Yao, Yuanbin Xue, Xianchang Li, Chengbo Li, Haixiang Song, Yongsheng Niu, Hu Liu, Xianmin Mai, Jiaoxia Zhang, and Zhanhu Guo. 2019. "Effects of Transition Metal Substituents on Interfacial and Electronic Structure of CH3NH3PbI3/TiO2 Interface: A First-Principles Comparative Study" Nanomaterials 9, no. 7: 966. https://doi.org/10.3390/nano9070966
APA StyleGuo, Y., Xue, Y., Li, X., Li, C., Song, H., Niu, Y., Liu, H., Mai, X., Zhang, J., & Guo, Z. (2019). Effects of Transition Metal Substituents on Interfacial and Electronic Structure of CH3NH3PbI3/TiO2 Interface: A First-Principles Comparative Study. Nanomaterials, 9(7), 966. https://doi.org/10.3390/nano9070966