DNA-Based Assembly of Quantum Dots into Dimers and Helices


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
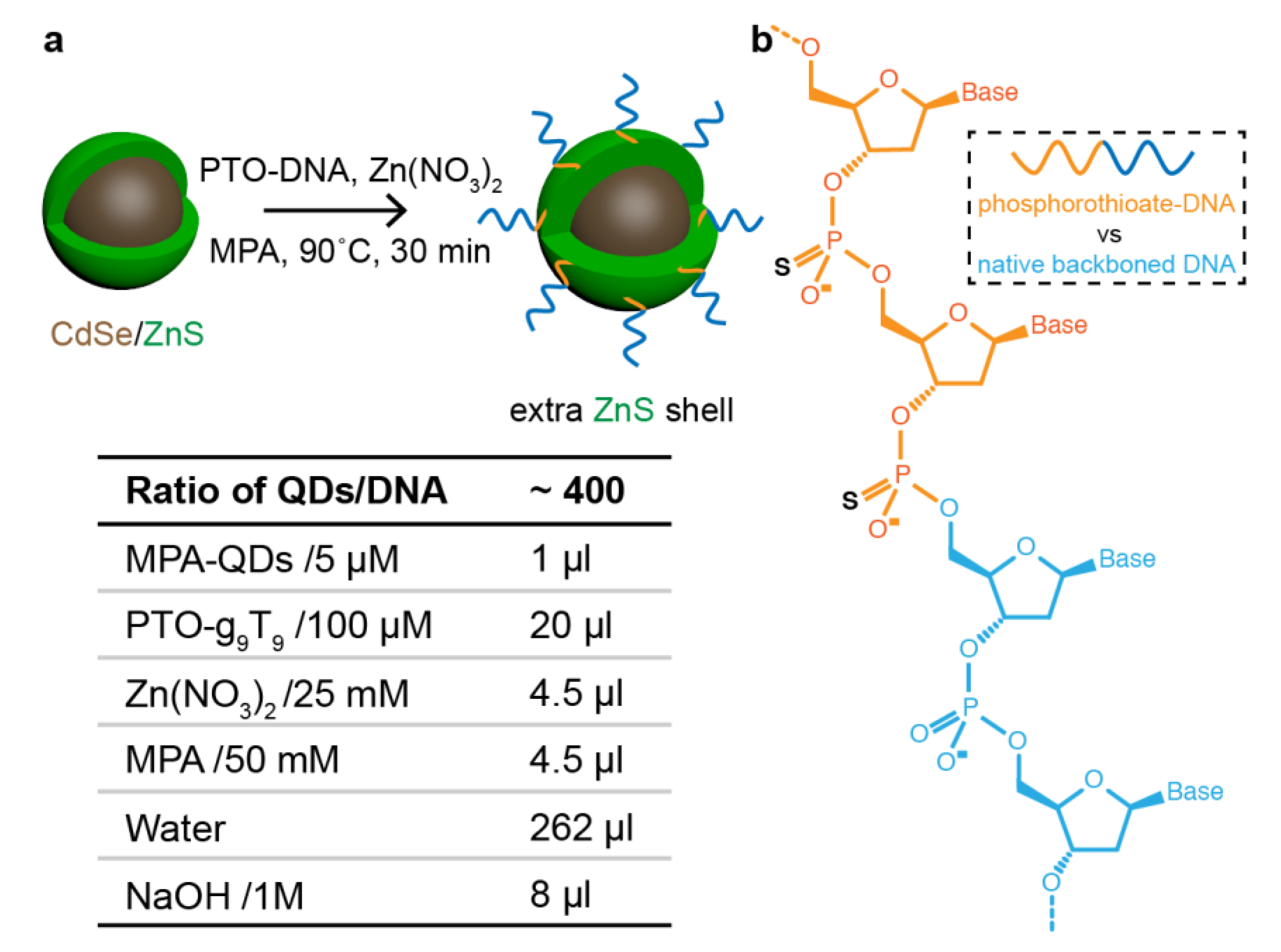
2.1. Quantum Dots
2.2. DNA
2.3. DNA–QD Modification
2.4. DNA Origami
2.5. QD Assembly on DNA Nanostructures
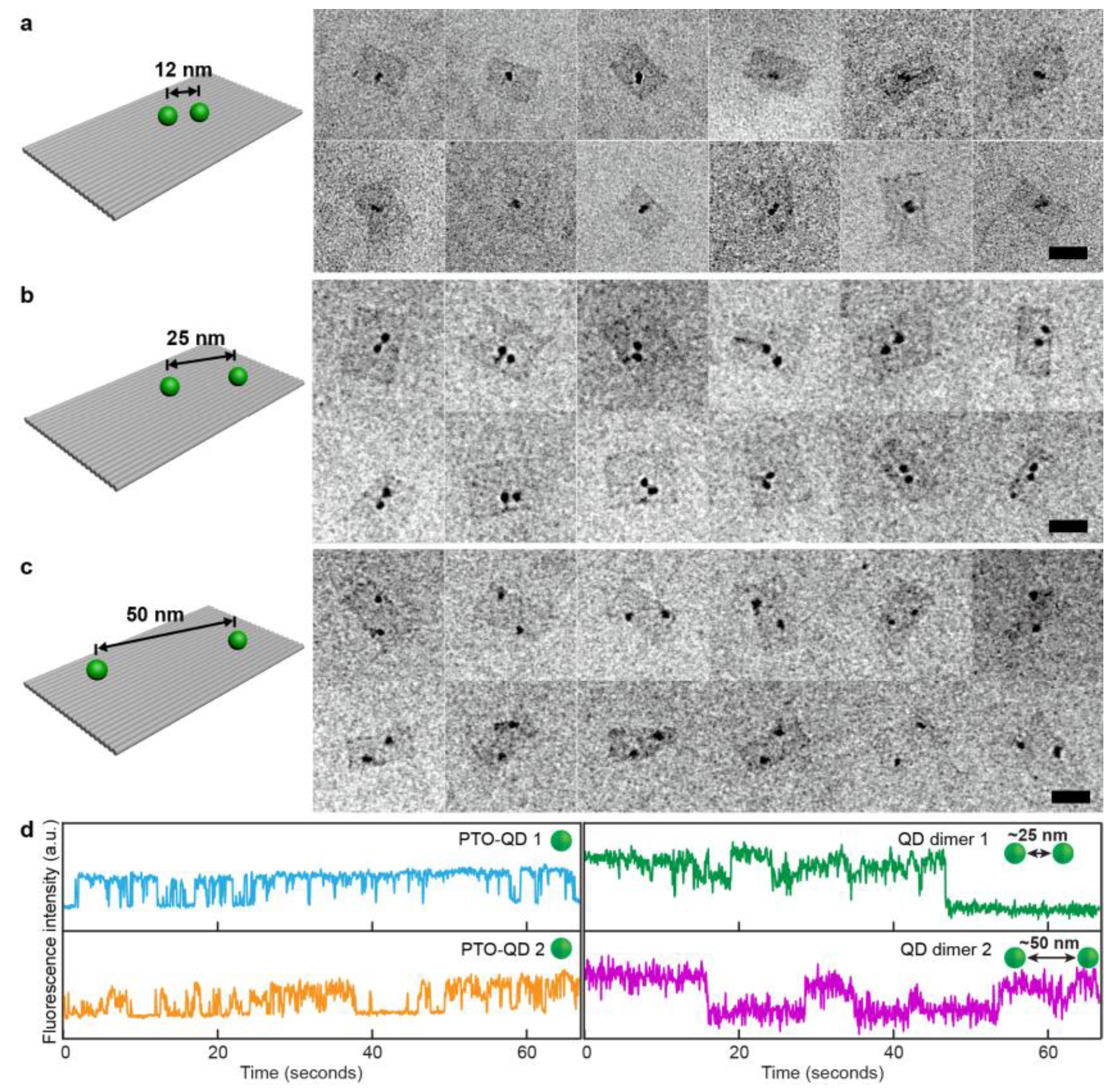
2.6. TEM Imaging
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Resch-Genger, U.; Grabolle, M.; Cavaliere-Jaricot, S.; Nitschke, R.; Nann, T. Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 2008, 5, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Alivisatos, A.P. Semiconductor clusters, nanocrystals, and quantum dots. Science 1996, 271, 933–937. [Google Scholar] [CrossRef]
- Smith, A.M.; Nie, S. Semiconductor nanocrystals: Structure, properties, and band gap engineering. Acc. Chem. Res. 2010, 43, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Pietryga, J.M.; Park, Y.-S.; Lim, J.; Fidler, A.F.; Bae, W.K.; Brovelli, S.; Klimov, V.I. Spectroscopic and Device Aspects of Nanocrystal Quantum Dots. Chem. Rev. 2016, 116, 10513–10622. [Google Scholar] [CrossRef] [PubMed]
- Kagan, C.R.; Lifshitz, E.; Sargent, E.H.; Talapin, D.V. Building devices from colloidal quantum dots. Science 2016, 353, aac5523. [Google Scholar] [CrossRef] [PubMed]
- Derfus, A.M.; Chan, W.C.W.; Bhatia, S.N. Probing the Cytotoxicity of Semiconductor Quantum Dots. Nano Lett. 2004, 4, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, C.; Liedl, T.; Kudera, S.; Pellegrino, T.; Muñoz Javier, A.; Gaub, H.E.; Stölzle, S.; Fertig, N.; Parak, W.J. Cytotoxicity of Colloidal CdSe and CdSe/ZnS Nanoparticles. Nano Lett. 2005, 5, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Ruan, G.; Rhyner, M.N.; Nie, S. Engineering Luminescent Quantum Dots for In Vivo Molecular and Cellular Imaging. Ann. Biomed. Eng. 2006, 34, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Medintz, I.L.; Uyeda, H.T.; Goldman, E.R.; Mattoussi, H. Quantum dot bioconjugates for imaging, labelling and sensing. Nat. Mater. 2005, 4, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Michalet, X.; Pinaud, F.F.; Bentolila, L.A.; Tsay, J.M.; Doose, S.; Li, J.J.; Sundaresan, G.; Wu, A.M.; Gambhir, S.S.; Weiss, S. Quantum Dots for Live Cells, in Vivo Imaging, and Diagnostics. Science 2005, 307, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Kairdolf, B.A.; Smith, A.M.; Stokes, T.H.; Wang, M.D.; Young, A.N.; Nie, S. Semiconductor Quantum Dots for Bioimaging and Biodiagnostic Applications. Annu. Rev. Anal. Chem. 2013, 6, 143–162. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhang, Z.; Jin, Y.; Niu, Y.; Cao, H.; Liang, X.; Chen, L.; Wang, J.; Peng, X. Solution-processed, high-performance light-emitting diodes based on quantum dots. Nature 2014, 515, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Deng, Y.; Peng, X.; Jin, Y. Quantum-Dot Light-Emitting Diodes for Large-Area Displays: Towards the Dawn of Commercialization. Adv. Mater. 2017, 29, 1607022. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.B.; Norris, D.J.; Bawendi, M.G. Synthesis and Characterization of Nearly Monodisperse CdE (E = S, Se, Te) Semiconductor Nanocrystallites. J. Am. Chem. Soc. 1993, 115, 8706–8715. [Google Scholar] [CrossRef]
- Beberwyck, B.J.; Surendranath, Y.; Alivisatos, A.P. Cation exchange: A versatile tool for nanomaterials synthesis. J. Phys. Chem. C 2013, 117, 19759–19770. [Google Scholar] [CrossRef]
- Peng, X.; Manna, L.; Yang, W.; Wickham, J.; Scher, E.; Kadavanich, A.; Alivisatos, A.P. Shape control of CdSe nanocrystals. Nature 2000, 404, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Fisher, B.; Eisler, H.J.; Bawendi, M. Type-II quantum dots: CdTe/CdSe (core/shell) and CdSe/ZnTe (core/shell) heterostructures. J. Am. Chem. Soc. 2003, 125, 11466–11467. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Alivisatos, A.P. Colloidal nanocrystal synthesis and the organic-inorganic interface. Nature 2005, 437, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Pu, C.; Qin, H.; Gao, Y.; Zhou, J.; Wang, P.; Peng, X. Synthetic Control of Exciton Behavior in Colloidal Quantum Dots. J. Am. Chem. Soc. 2017, 139, 3302–3311. [Google Scholar] [CrossRef] [PubMed]
- Govorov, O. Spin and energy transfer in nanocrystals without tunneling. Phys. Rev. B 2003, 68, 075315. [Google Scholar] [CrossRef]
- Ouyang, M.; Awschalom, D.D. Coherent spin transfer between molecularly bridged quantum dots. Science 2003, 301, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Govorov, A.O.; Bryant, G.W.; Zhang, W.; Skeini, T.; Lee, J.; Kotov, N.A.; Slocik, J.M.; Naik, R.R. Exciton-plasmon interaction and hybrid excitons in semiconductor-metal nanoparticle assemblies. Nano Lett. 2006, 6, 984–994. [Google Scholar] [CrossRef]
- Lee, J.J.; Hernandez, P.; Lee, J.J.; Govorov, A.O.; Kotov, N.A. Exciton–plasmon interactions in molecular spring assemblies of nanowires and wavelength-based protein detection. Nat. Mater. 2007, 6, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Stangl, J.; Holý, V.; Bauer, G. Structural properties of self-organized semiconductor nanostructures. Rev. Mod. Phys. 2004, 76, 725–783. [Google Scholar] [CrossRef]
- Ye, X.; Chen, J.; Engel, M.; Millan, J.A.; Li, W.; Qi, L.; Xing, G.; Collins, J.E.; Kagan, C.R.; Li, J.; et al. Competition of shape and interaction patchiness for self-assembling nanoplates. Nat. Chem. 2013, 5, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Kalsin, A.M.; Fialkowski, M.; Paszewski, M.; Smoukov, S.K.; Bishop, K.J.M.; Grzybowski, B.A. Electrostatic self-assembly of binary nanoparticle crystals with a diamond-like lattice. Science 2006, 312, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Böker, A.; He, J.; Sill, K.; Xiang, H.; Abetz, C.; Li, X.; Wang, J.; Emrick, T.; Long, S.; et al. Self-directed self-assembly of nanoparticle/copolymer mixtures. Nature 2005, 434, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zhu, C.; Ercius, P.; Raja, S.N.; He, B.; Jones, M.R.; Hauwiller, M.R.; Liu, Y.; Xu, T.; Alivisatos, A.P. Structural diversity in binary superlattices self-assembled from polymer-grafted nanocrystals. Nat. Commun. 2015, 6, 10052. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.M.; Flynn, N.T.; Wang, C.; Tirrell, D.A.; Langer, R. Coiled-coil peptide-based assembly of gold nanoparticles. Adv. Mater. 2004, 16, 915–918. [Google Scholar] [CrossRef]
- Kostiainen, M.A.; Hiekkataipale, P.; Laiho, A.; Lemieux, V.; Seitsonen, J.; Ruokolainen, J.; Ceci, P. Electrostatic assembly of binary nanoparticle superlattices using protein cages. Nat. Nanotechnol. 2013, 8, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Nykypanchuk, D.; Maye, M.M.; Van Der Lelie, D.; Gang, O. DNA-guided crystallization of colloidal nanoparticles. Nature 2008, 451, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, R.J.; Lee, B.; Jones, M.R.; Harris, N.; Schatz, G.C.; Mirkin, C.A. Nanoparticle Superlattice Engineering with DNA. Science 2011, 334, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Seeman, N.C.; Mirkin, C.A. Programmable materials and the nature of the DNA bond. Science 2015, 347, 1260901. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C.; Sleiman, H.F. DNA nanotechnology. Nat. Rev. Mater. 2017, 3, 17068. [Google Scholar] [CrossRef]
- Mitchell, G.P.; Mirkin, C.A.; Letsinger, R.L. Programmed assembly of DNA functionalized quantum dots. J. Am. Chem. Soc. 1999, 121, 8122–8123. [Google Scholar] [CrossRef]
- Farlow, J.; Seo, D.; Broaders, K.E.; Taylor, M.J.; Gartner, Z.J.; Jun, Y.W. Formation of targeted monovalent quantum dots by steric exclusion. Nat. Methods 2013, 10, 1203–1205. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Tang, Q.; Li, L.; Li, J.; Zuo, X.; Qu, X.; Pei, H.; Wang, L.; Fan, C. Valence-Engineering of Quantum Dots Using Programmable DNA Scaffolds. Angew. Chem. Int. Ed. 2017, 56, 16077–16081. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gang, O. DNA-Functionalized Quantum Dots: Fabrication, Structural, and Physicochemical Properties. Langmuir 2013, 29, 7038–7046. [Google Scholar] [CrossRef] [PubMed]
- Bui, H.; Onodera, C.; Kidwell, C.; Tan, Y.; Graugnard, E.; Kuang, W.; Lee, J.; Knowlton, W.B.; Yurke, B.; Hughes, W.L. Programmable Periodicity of Quantum Dot Arrays with DNA Origami Nanotubes. Nano Lett. 2010, 10, 3367–3372. [Google Scholar] [CrossRef] [PubMed]
- Takabayashi, S.; Klein, W.P.; Onodera, C.; Rapp, B.; Flores-Estrada, J.; Lindau, E.; Snowball, L.; Sam, J.T.; Padilla, J.E.; Lee, J.; et al. High precision and high yield fabrication of dense nanoparticle arrays onto DNA origami at statistically independent binding sites. Nanoscale 2014, 6, 13928–13938. [Google Scholar] [CrossRef] [PubMed]
- Parak, W.J.; Gerion, D.; Zanchet, D.; Woerz, A.S.; Pellegrino, T.; Micheel, C.; Williams, S.C.; Seitz, M.; Bruehl, R.E.; Bryant, Z.; et al. Conjugation of DNA to silanized colloidal semiconductor nanocrystalline quantum dots. Chem. Mater. 2002, 14, 2113–2119. [Google Scholar] [CrossRef]
- Lin, C.-A.J.; Sperling, R.A.; Li, J.K.; Yang, T.-Y.; Li, P.-Y.; Zanella, M.; Chang, W.H.; Parak, W.J. Design of an Amphiphilic Polymer for Nanoparticle Coating and Functionalization. Small 2008, 4, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Parak, W.J. Surface modification, functionalization and bioconjugation of colloidal Inorganic nanoparticles. Philos. Trans. R. Soc. A 2010, 368, 1333–1383. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Ke, Y.; Yan, H. Quantum Dot Bioconjugation during Core–Shell Synthesis. Angew. Chem. 2008, 120, 322–325. [Google Scholar] [CrossRef]
- Tikhomirov, G.; Hoogland, S.; Lee, P.E.; Fischer, A.; Sargent, E.H.; Kelley, S.O. DNA-based programming of quantum dot valency, self-assembly and luminescence. Nat. Nanotechnol. 2011, 6, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Samanta, A.; Nangreave, J.; Yan, H.; Liu, Y. Robust DNA-functionalized core/shell quantum dots with fluorescent emission spanning from UV–vis to near-IR and compatible with DNA-directed self-assembly. J. Am. Chem. Soc. 2012, 134, 17424–17427. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Zhou, Y.; Zou, S.; Yan, H.; Liu, Y. Fluorescence quenching of quantum dots by gold nanoparticles: A potential long range spectroscopic ruler. Nano Lett. 2014, 14, 5052–5057. [Google Scholar] [CrossRef] [PubMed]
- Vaxenburg, R.; Rodina, A.; Lifshitz, E.; Efros, A.L. Biexciton Auger Recombination in CdSe/CdS Core/Shell Semiconductor Nanocrystals. Nano Lett. 2016, 16, 2503–2511. [Google Scholar] [CrossRef] [PubMed]
- Nickels, P.C.; Ke, Y.; Jungmann, R.; Smith, D.M.; Leichsenring, M.; Shih, W.M.; Liedl, T.; Högberg, B. DNA Origami Structures Directly Assembled from Intact Bacteriophages. Small 2014, 10, 1765–1769. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Deng, Z.; Liu, Y.; Yan, H. A perspective on functionalizing colloidal quantum dots with DNA. Nano Res. 2013, 6, 853–870. [Google Scholar] [CrossRef]
- Douglas, S.M.; Marblestone, A.H.; Teerapittayanon, S.; Vazquez, A.; Church, G.M.; Shih, W.M. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 2009, 37, 5001–5006. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Schulz, O.; Lin, S.; Ding, B.; Liu, X.; Wei, X.; Ros, R.; Yan, H.; Liu, Y. Aqueous Synthesis of Zinc Blende CdTe/CdS Magic-Core/Thick-Shell Tetrahedral-Shaped Nanocrystals with Emission Tunable to Near-Infrared. J. Am. Chem. Soc. 2010, 132, 5592–5593. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-N.; Kilchherr, F.; Dietz, H.; Bathe, M. Quantitative prediction of 3D solution shape and flexibility of nucleic acid nanostructures. Nucleic Acids Res. 2012, 40, 2862–2868. [Google Scholar] [CrossRef] [PubMed]
- Kuzyk, A.; Schreiber, R.; Fan, Z.; Pardatscher, G.; Roller, E.-M.M.; Högele, A.; Simmel, F.C.; Govorov, A.O.; Liedl, T. DNA-based self-assembly of chiral plasmonic nanostructures with tailored optical response. Nature 2012, 483, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Efros, A.L.; Nesbitt, D.J. Origin and control of blinking in quantum dots. Nat. Nanotechnol. 2016, 11, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Empedocles, S.A.; Neuhauser, R.; Bawendi, M.G. Three-dimensional orientation measurements of symmetric single chromophores using polarization microscopy. Nature 1999, 399, 126–130. [Google Scholar] [CrossRef]
- Lethiec, C.; Laverdant, J.; Vallon, H.; Javaux, C.; Dubertret, B.; Frigerio, J.M.; Schwob, C.; Coolen, L.; Maître, A. Measurement of three-dimensional dipole orientation of a single fluorescent nanoemitter by emission polarization analysis. Phys. Rev. X 2014, 4, 021037. [Google Scholar] [CrossRef]
- Hu, J.; Li, L.; Yang, W.; Manna, L.; Wang, L.; Alivisatos, A.P. Linearly Polarized Emission from Colloidal Semiconductor Quantum Rods. Science 2001, 292, 2060–2063. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Alivisatos, A.P. Origin and Scaling of the Permanent Dipole Moment in CdSe Nanorods. Phys. Rev. Lett. 2003, 90, 097402. [Google Scholar] [CrossRef] [PubMed]
- Shabaev, A.; Efros, A.L. 1D exciton spectroscopy of semiconductor nanorods. Nano Lett. 2004, 4, 1821–1825. [Google Scholar] [CrossRef]
- Eshet, H.; Grünwald, M.; Rabani, E. The Electronic Structure of CdSe/CdS Core/Shell Seeded Nanorods: Type-I or Quasi-Type-II? Nano Lett. 2013, 13, 5880–5885. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Hill, L.J.; Chen, J.; McBride, J.R.; Pavlopolous, N.G.; Richey, N.E.; Pyun, J.; Lian, T. Universal Length Dependence of Rod-to-Seed Exciton Localization Efficiency in Type I and Quasi-Type II CdSe@CdS Nanorods. ACS Nano 2015, 9, 4591–4599. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Liedl, T. DNA-Based Assembly of Quantum Dots into Dimers and Helices. Nanomaterials 2019, 9, 339. https://doi.org/10.3390/nano9030339
Zhang T, Liedl T. DNA-Based Assembly of Quantum Dots into Dimers and Helices. Nanomaterials. 2019; 9(3):339. https://doi.org/10.3390/nano9030339
Chicago/Turabian StyleZhang, Tao, and Tim Liedl. 2019. "DNA-Based Assembly of Quantum Dots into Dimers and Helices" Nanomaterials 9, no. 3: 339. https://doi.org/10.3390/nano9030339
APA StyleZhang, T., & Liedl, T. (2019). DNA-Based Assembly of Quantum Dots into Dimers and Helices. Nanomaterials, 9(3), 339. https://doi.org/10.3390/nano9030339