The Electronic and Magnetic Properties of Multi-Atom Doped Black Phosphorene




Abstract
1. Introduction
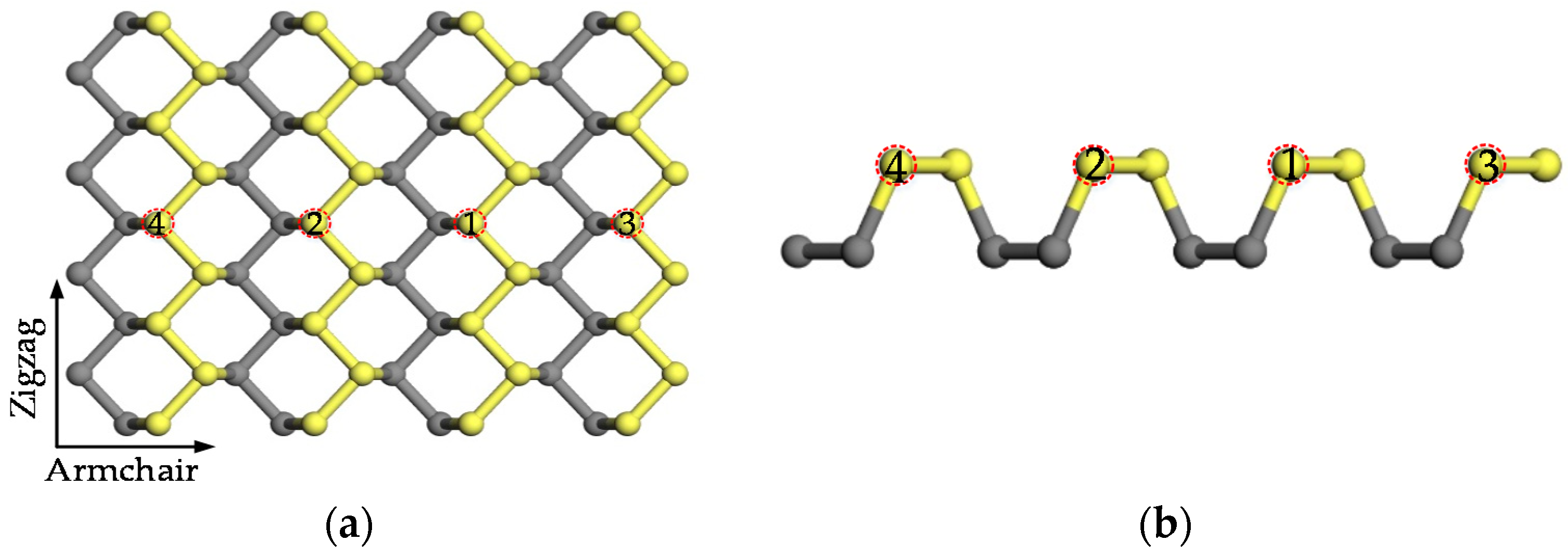
2. Physical Model and Computational Method
3. Results and Discussion
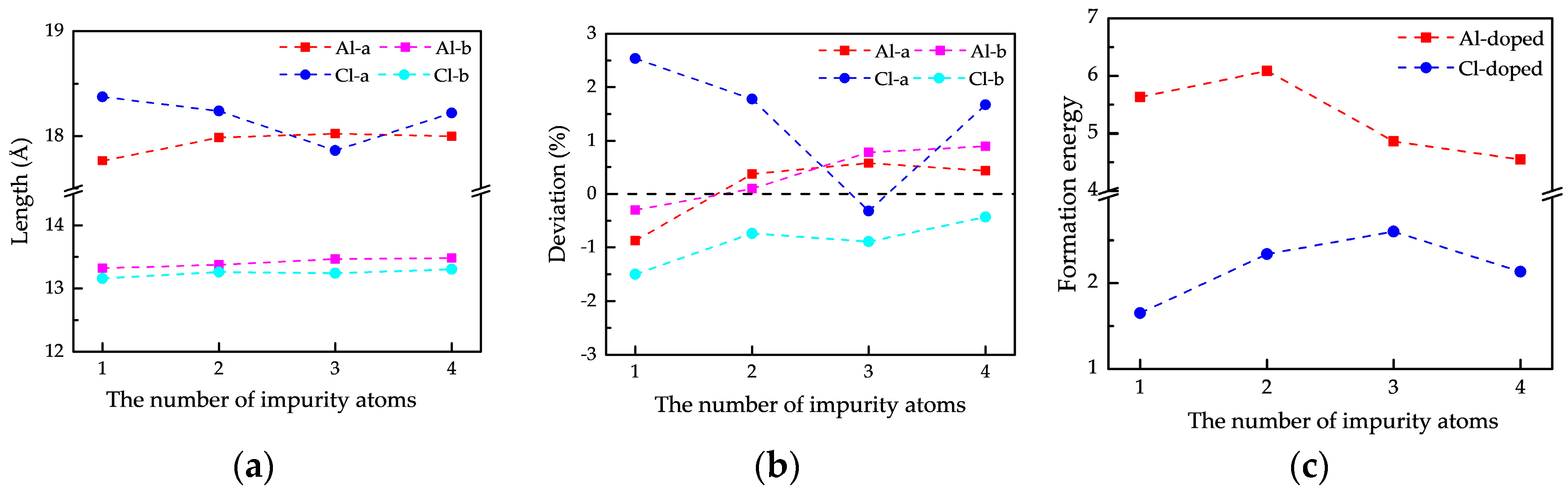
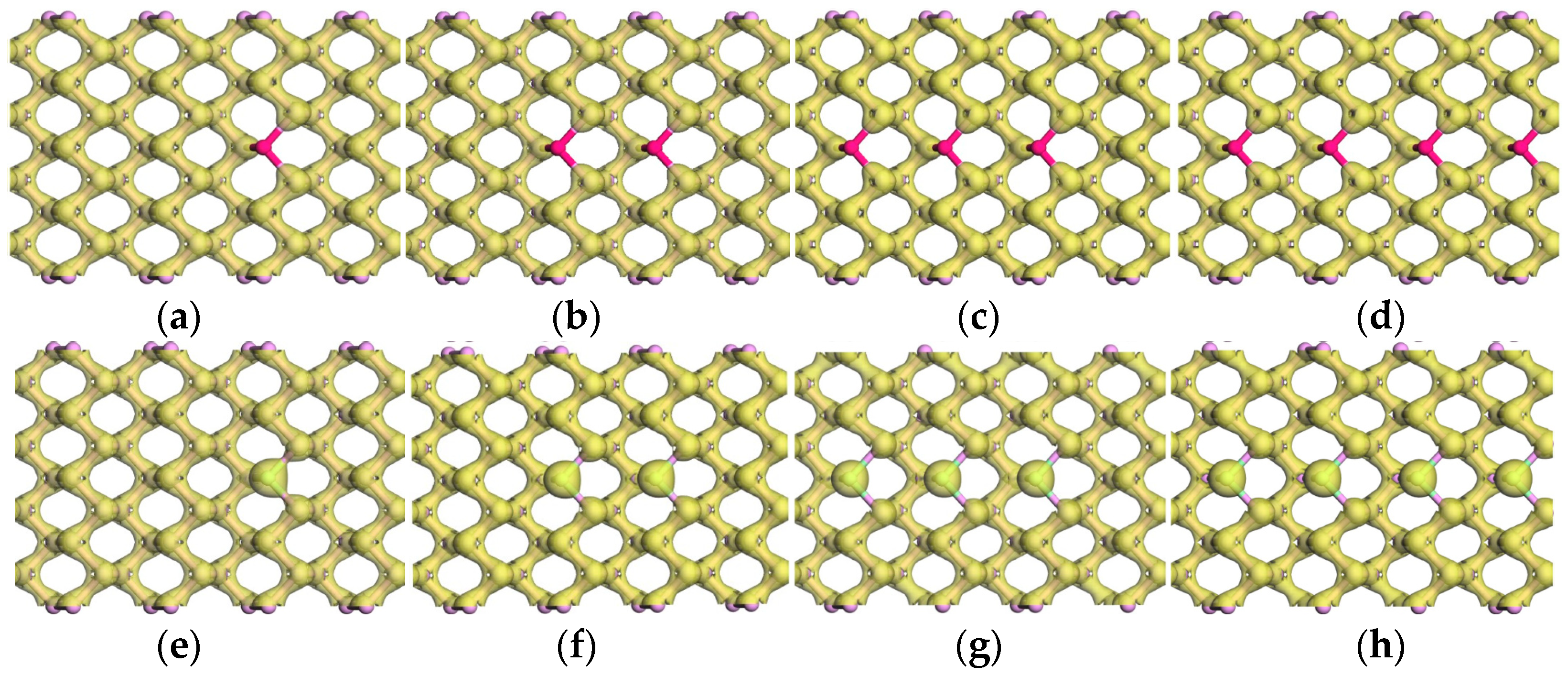
3.1. Geometrical Structure and Stability
3.2. Magnetic Properties
3.2.1. Magnetism and Magnetic Moment
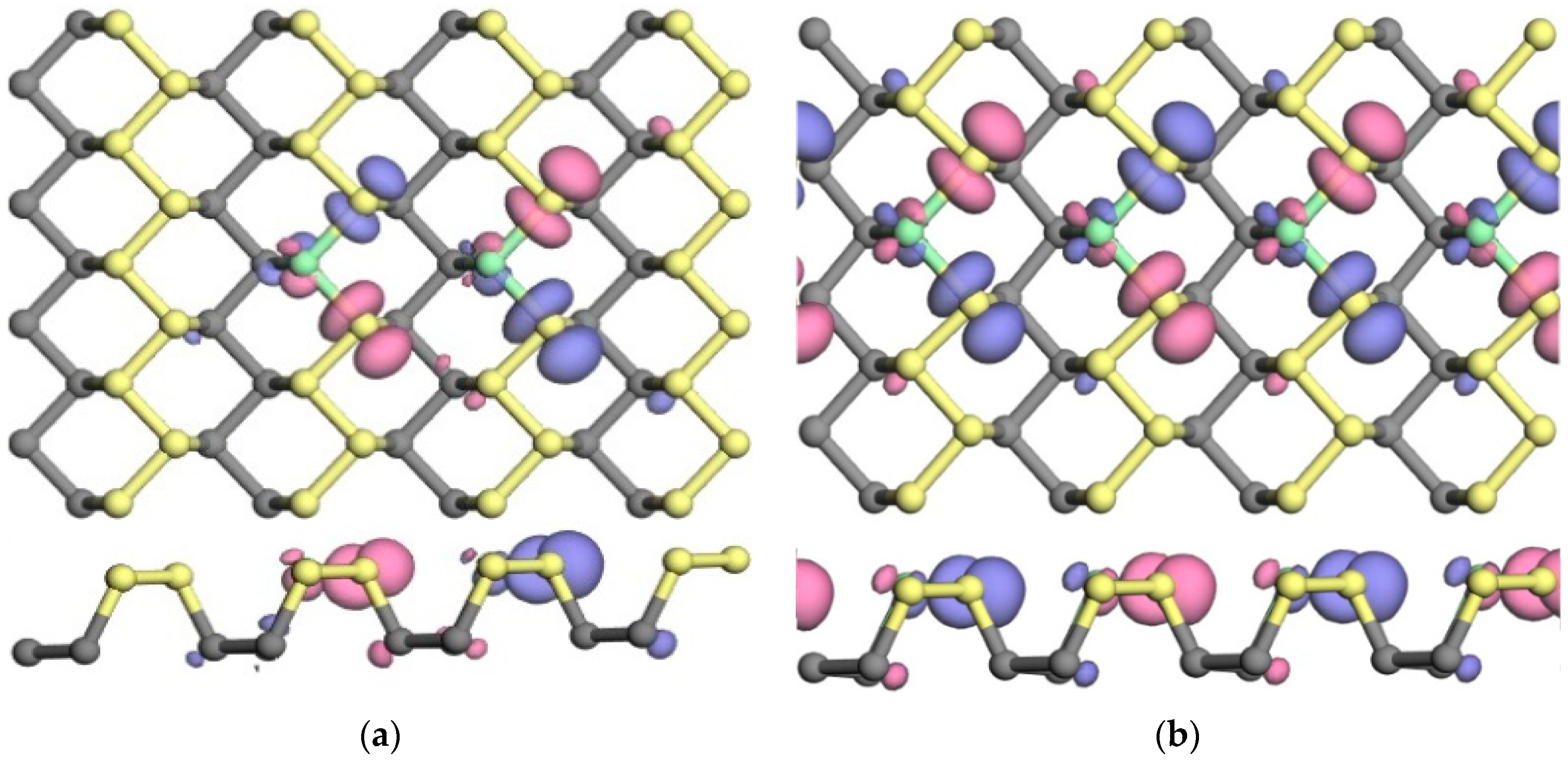
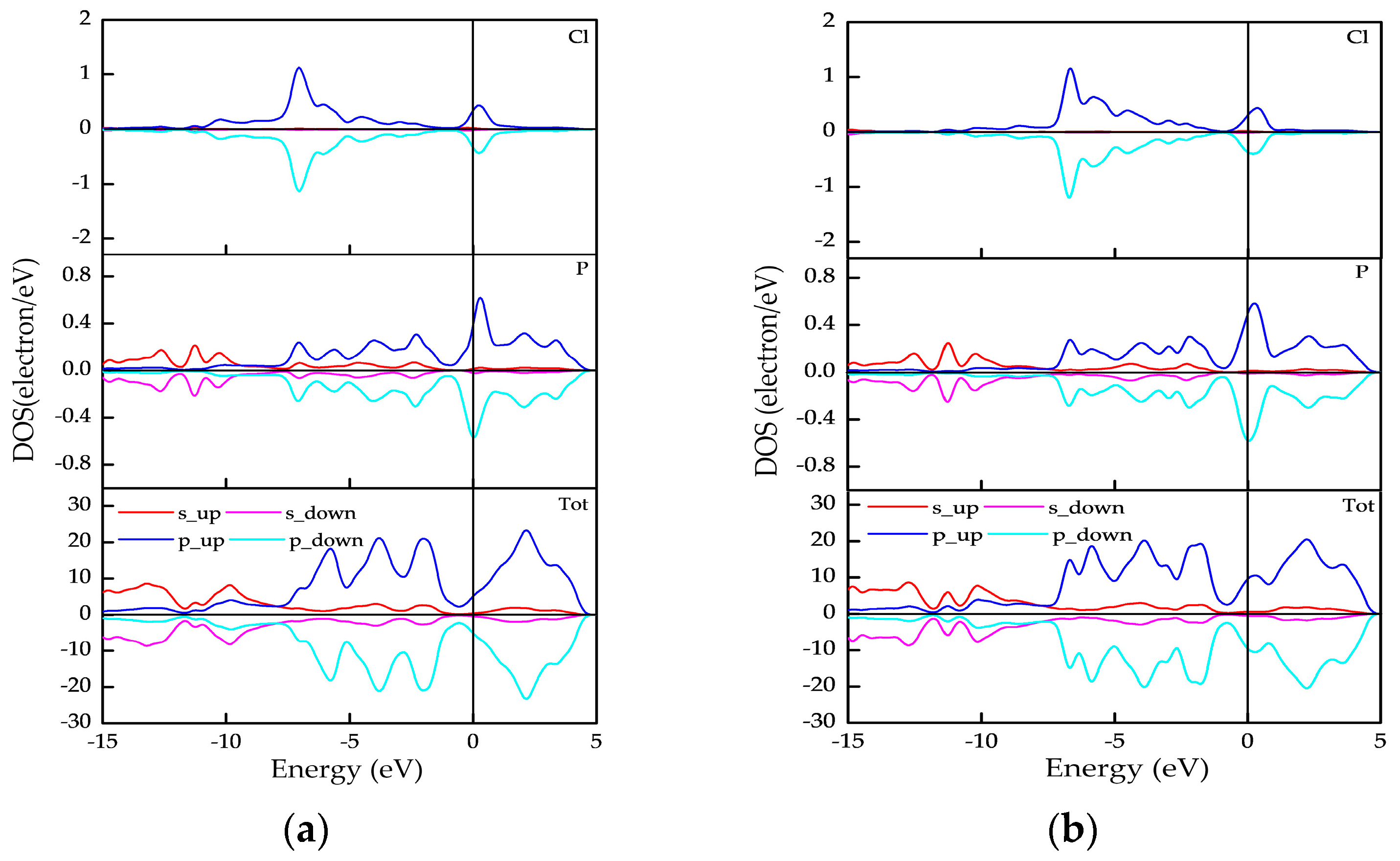
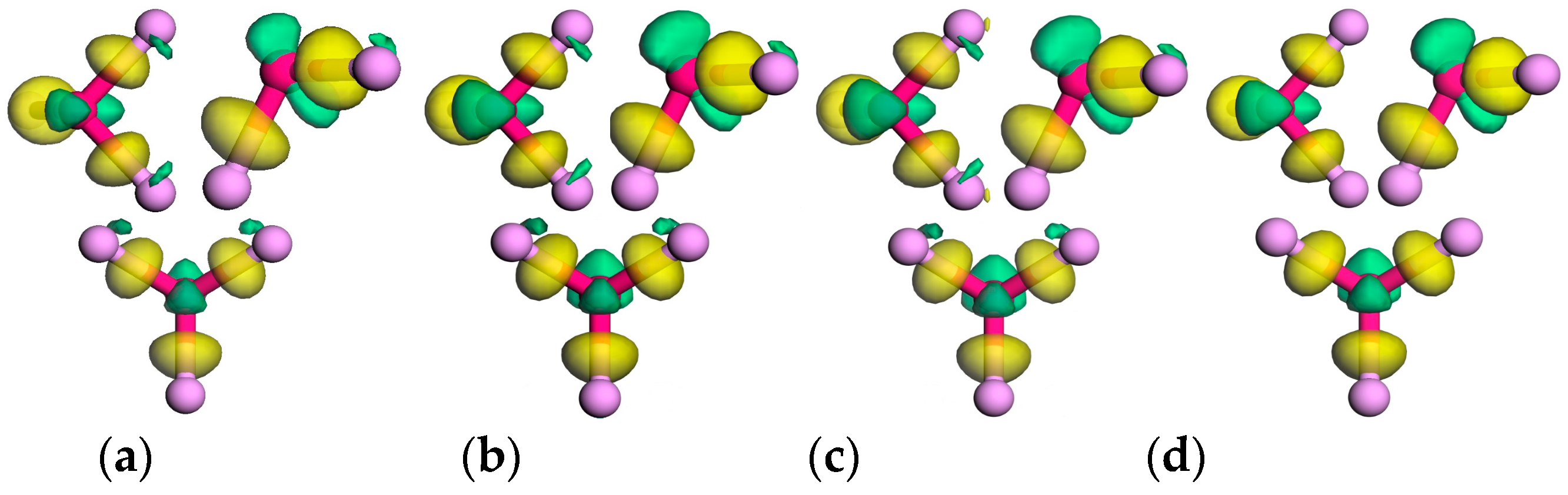
3.2.2. Spin Distribution and Spin Splitting
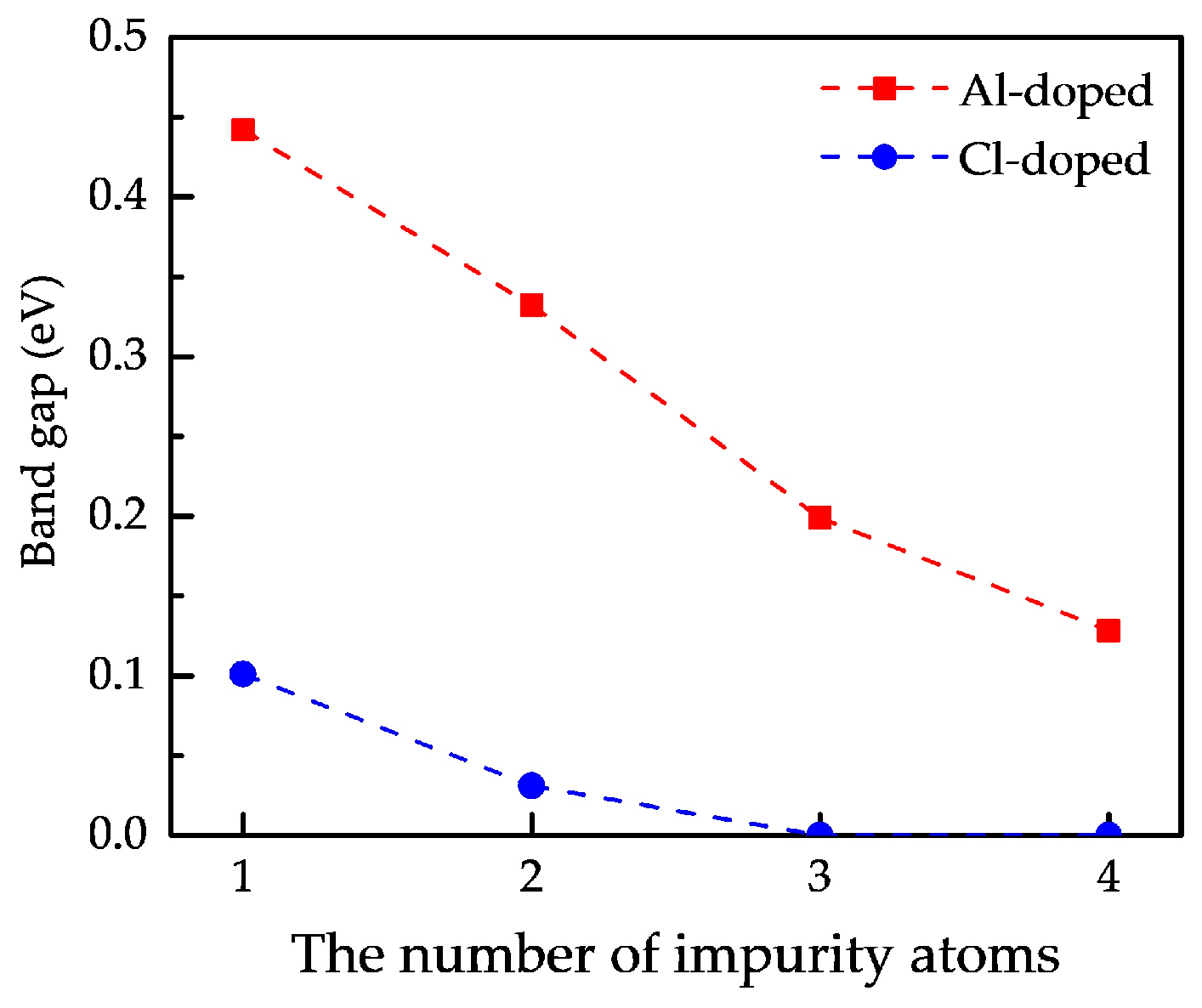
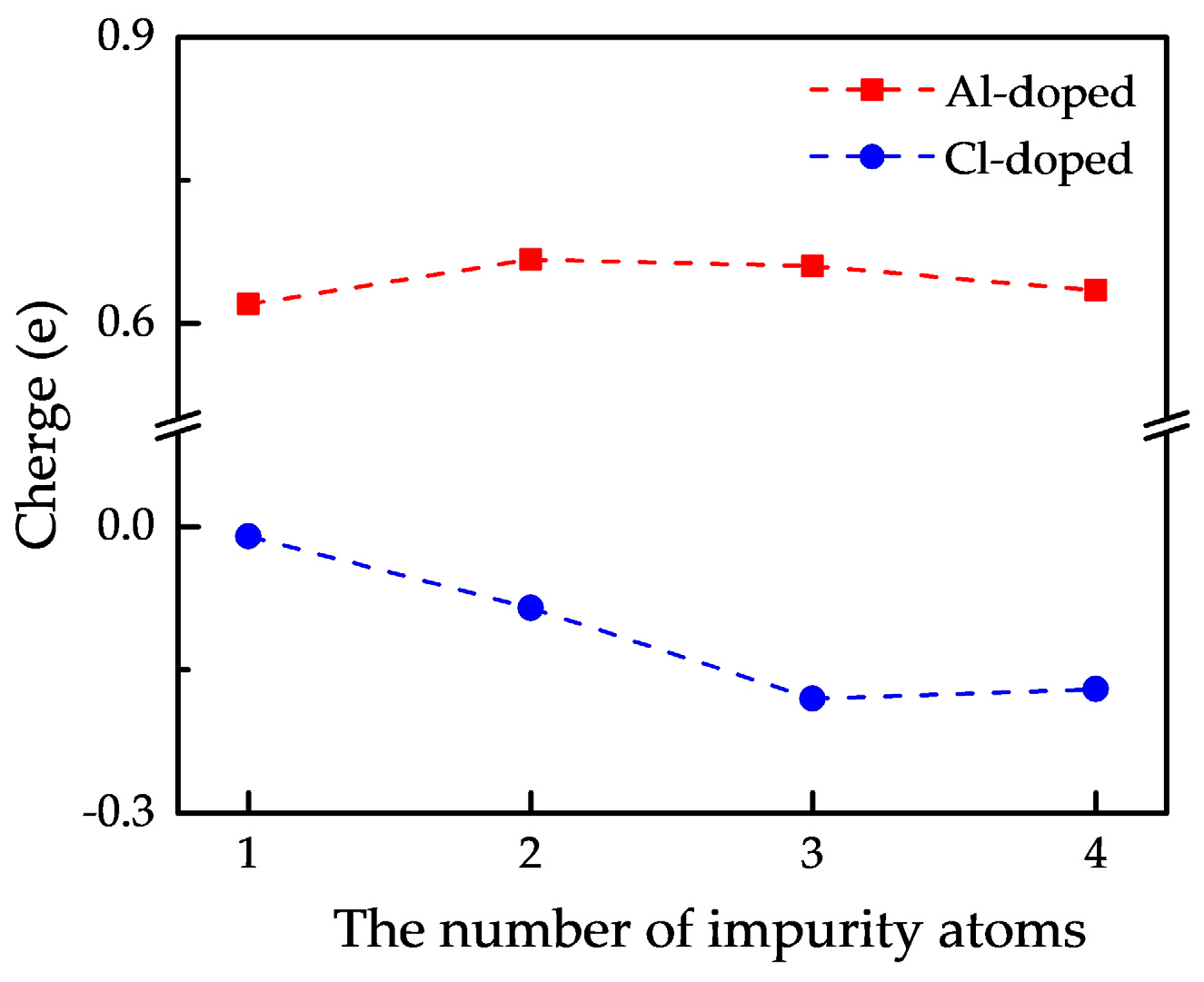
3.3. Electronic Properties
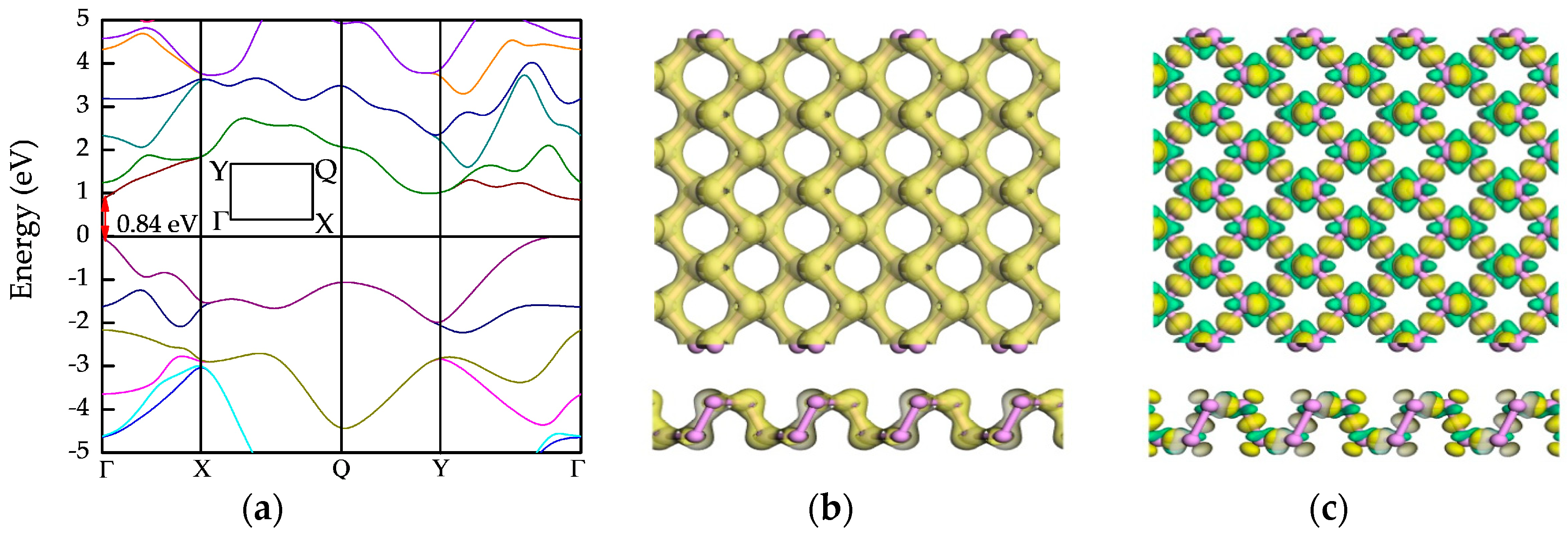
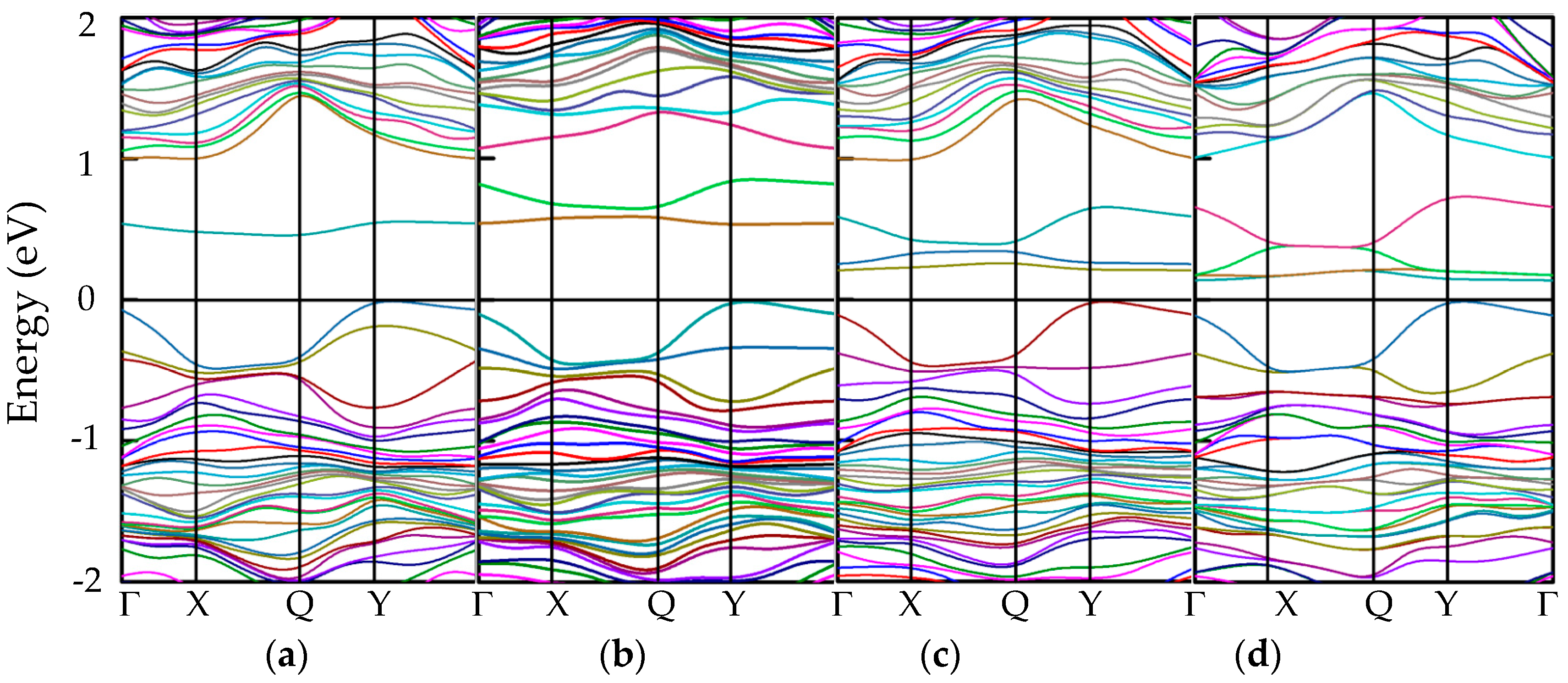
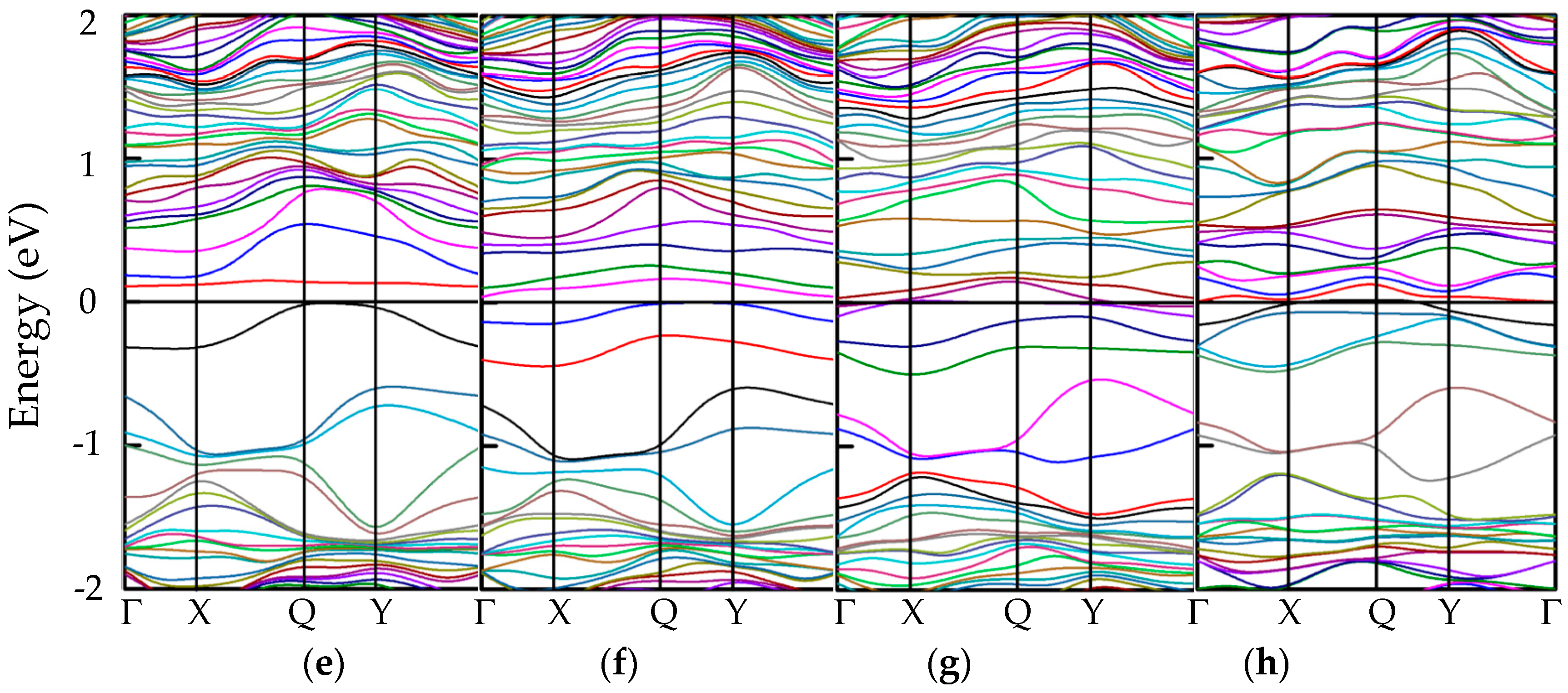
3.3.1. Band Structures
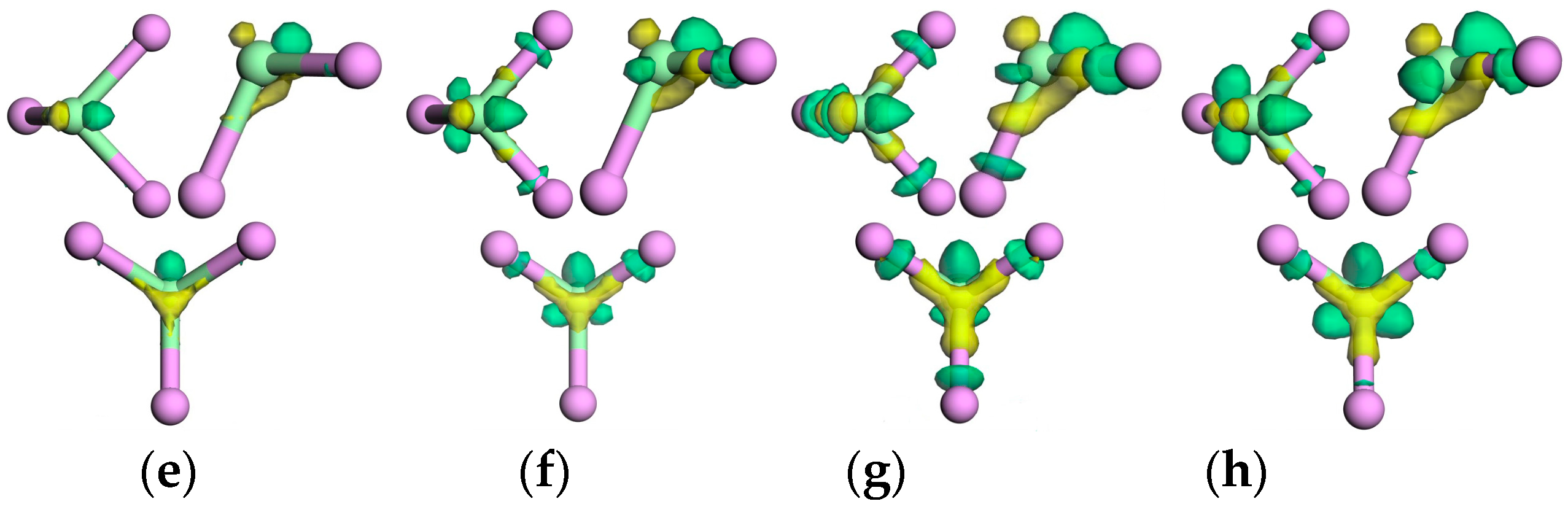
3.3.2. Electron Density and Charge Density Difference
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rodin, A.S.; Carvalho, A.; Castro Neto, A.H. Strain-induced gap modification in black phosphorus. Phys. Rev. Lett. 2014, 112, 176801. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.Q.; Ke, Q.Q.; Zhang, G.; Feng, Y.P.; Shenoy, V.B.; Zhang, Y.W. Giant phononic anisotropy and unusual anharmonicity of phosphorene: Interlayer coupling and strain engineering. Adv. Funct. Mater. 2016, 25, 2230–2236. [Google Scholar] [CrossRef]
- Liu, B.; Bai, L.C.; Korznikova, E.A.; Dmitriev, S.V.; Wing-Keung Law, A.; Zhou, K. Thermal conductivity and tensile response of phosphorene nanosheets with vacancy defects. J. Phys. Chem. C 2017, 121, 13876–13887. [Google Scholar] [CrossRef]
- Liu, H.; Neal, A.T.; Zhu, Z.; Luo, Z.; Xu, X.F.; Tomanek, D.; Ye, P.D. Phosphorene: An unexplored 2D semiconductor with a high hole mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef] [PubMed]
- Li, L.K.; Yu, Y.J.; Ye, G.J.; Ge, Q.Q.; Ou, X.D.; Hua, W.; Feng, D.L.; Chen, X.H.; Zhang, Y.B. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K. Graphene: Status and prospects. Science 2009, 324, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Tosun, M.; Seol, G.; Chang, T.C.; Takei, K.; Javey, A. Degenerate n-doping of few-layer transition metal dichalcogenides by potassium. Nano Lett. 2013, 13, 1991–1995. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.Q.; Zhang, G.; Zhang, Y.W. Layer-dependent band alignment and work function of few-layer phosphorene. Sci. Rep. 2014, 4, 6677. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.Y.; Wang, H.; Huang, L.; Sun, Y.Y.; Zhang, S.B. Stacking fault enriching the electronic and transport properties of few-layer phosphorenes and black phosphorus. Nano Lett. 2016, 16, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.Q.; He, J.T.; Yang, S.L.; Jia, H.Y.; Liu, Y.Y.; Liu, W.; Liu, Y.; Li, T.Z. Investigation of black phosphorus field-effect transistors and its stability. Opt. Quant. Electron. 2016, 48, 344. [Google Scholar] [CrossRef]
- Lim, S.K.; Kang, S.C.; Yoo, T.J.; Lee, S.K.; Hwang, H.J.; Lee, B.H. Operation mechanism of a MoS2/BP heterojunction FET. Nanomaterials 2018, 8, 797. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.N.; Yogeesh, M.N.; Yang, S.X.; Aldave, S.H.; Kim, J.S.; Sonde, S.; Tao, L.; Lu, N.S.; Akinwande, D. Flexible black phosphorus ambipolar transistors, circuits and AM demodulator. Nano Lett. 2015, 15, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Dong, B.W.; Guo, X.; Chang, Y.H.; Chen, N.; Huang, X.; Liao, W.G.; Zhu, C.X.; Wang, H.; Lee, C.; et al. Waveguide-integrated black phosphorus photodetector for mid-infrared applications. ACS Nano 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Liu, Y.; Fang, C.Z.; Shao, Y.; Han, G.Q.; Zhang, J.C.; Hao, Y. Theoretical study of strained black phosphorus photodetector integrated with silicon waveguide. Superlattice Microst. 2018, 122, 501–509. [Google Scholar] [CrossRef]
- Gong, F.; Wu, F.; Long, M.S.; Chen, F.S.; Su, M.; Yang, Z.Y.; Shi, J. Black phosphorus infrared photodetectors with fast response and high photoresponsivity. Phys. Status Solidi-R 2018, 12, 1800310. [Google Scholar] [CrossRef]
- Zheng, H.L.; Zhang, J.M.; Yang, B.S.; Dua, X.B.; Yan, Y. A first-principles study on the magnetic properties of nonmetal atom doped phosphorene monolayers. Phys. Chem. Chem. Phys. 2015, 17, 16341. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.Y.; Li, N. Manipulating the magnetic moment in phosphorene by lanthanide atom doping: A first-principles study. RSC Adv. 2016, 6, 92048–92056. [Google Scholar] [CrossRef]
- Yu, Q.H.; Jiang, Y.; Zhang, W.; Wu, B.Z.; Yin, J.R.; Zhang, P.; Ding, Y.H. Noble metal atoms doped phosphorene: Electronic properties and gas adsorption ability. Mater. Res. Express 2017, 4, 045703. [Google Scholar] [CrossRef]
- Feng, X.W.; Kulish, V.V.; Wu, P.; Liu, X.K.; Ang, K.W. Anomalously enhanced thermal stability of phosphorene via metal adatom doping: An experimental and first-principles study. Nano Res. 2016, 9, 2687–2695. [Google Scholar] [CrossRef]
- Hashmi, A.; Hong, J.S. Transition metal doped phosphorene: First-principles study. J. Phys. Chem. C 2015, 119, 9198–9204. [Google Scholar] [CrossRef]
- Yu, W.Y.; Zhu, Z.L.; Niu, C.Y.; Li, C.; Cho, J.H.; Jia, Y. Dilute magnetic semiconductor and half-metal behaviors in 3d transition-metal doped black and blue phosphorenes: A first-principles study. Nanoscale Res. Lett. 2016, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.D.; Bai, L.W.; Yang, G.G.; Fan, K.Q.; Xie, Y.; Li, M.L. The electronic properties of O-doped pure and sulfur vacancy-defect monolayer WS2: A first-principles study. Materials 2018, 11, 218. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zhu, Z.; Niu, C.Y.; Li, C.; Cho, J.H.; Jia, Y. Anomalous doping effect in black phosphorene from first-principles calculations. Phys. Chem. Chem. Phys. 2015, 17, 16351–16358. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Pederson, M.; Perdew, J. Fully self-consistent Fermi-orbital self-interaction correction in density-functional theory. Bull. Am. Phys. Soc. 2017, 62, 052505–052514. [Google Scholar]
- Boukhvalov, D.W. The atomic and electronic structure of nitrogen and boron-doped phosphorene. Phys. Chem. Chem. Phys. 2015, 17, 27210–27216. [Google Scholar] [CrossRef] [PubMed]
- Segall, M.D.; Pickard, C.J.; Shah, R.; Payne, M.C. Population analysis in plane wave electronic structure calculations. Mol. Phys. 1996, 89, 571–577. [Google Scholar] [CrossRef]
- Segall, M.D.; Shah, R.; Pickard, C.J.; Payne, M.C. Population analysis of plane-wave electronic structure calculations of bulk materials. Phys. Rev. B 1996, 54, 16317–16320. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Impurity Number | Al-Doped | Cl-Doped | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 1.423 | - | - | - | 3.474 | - | - | - |
| 2 | 3.963 | −1 | 2.0 × 10−13 | 7.5 × 10−3 | 8.523 | −33 | 4.5 × 10−13 | 2.0 |
| 3 | 4.916 | 803 | - | - | 13.995 | 944 | - | - |
| 4 | 6.783 | −6 | 3.0 × 10−13 | 2.0 × 10−3 | 20.454 | −14 | 5.0 × 10−13 | 3.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Wang, H.; Zhang, M.; Zhao, W.; Liu, Y.; Qin, H. The Electronic and Magnetic Properties of Multi-Atom Doped Black Phosphorene. Nanomaterials 2019, 9, 311. https://doi.org/10.3390/nano9020311
Wang K, Wang H, Zhang M, Zhao W, Liu Y, Qin H. The Electronic and Magnetic Properties of Multi-Atom Doped Black Phosphorene. Nanomaterials. 2019; 9(2):311. https://doi.org/10.3390/nano9020311
Chicago/Turabian StyleWang, Ke, Hai Wang, Min Zhang, Wei Zhao, Yan Liu, and Hongbo Qin. 2019. "The Electronic and Magnetic Properties of Multi-Atom Doped Black Phosphorene" Nanomaterials 9, no. 2: 311. https://doi.org/10.3390/nano9020311
APA StyleWang, K., Wang, H., Zhang, M., Zhao, W., Liu, Y., & Qin, H. (2019). The Electronic and Magnetic Properties of Multi-Atom Doped Black Phosphorene. Nanomaterials, 9(2), 311. https://doi.org/10.3390/nano9020311