Applicability of Goethite/Reduced Graphene Oxide Nanocomposites to Remove Lead from Wastewater

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Graphite Cleaning
2.3. Graphene Oxide Synthesis
2.4. The α-FeOOH/rGO Composites Synthesis
2.5. Characterization
2.6. Optimum pH Value Determination
2.7. Adsorption Kinetics
2.8. Adsorption Isotherms
3. Results and Discussion
3.1. Graphite Cleaning
3.2. Graphene Oxide Synthesis
3.3. The α-FeOOH/rGO Composites Synthesis
3.4. Characterizations
3.4.1. SEM Analysis
3.4.2. STEM Analysis
3.4.3. Raman Spectroscopy
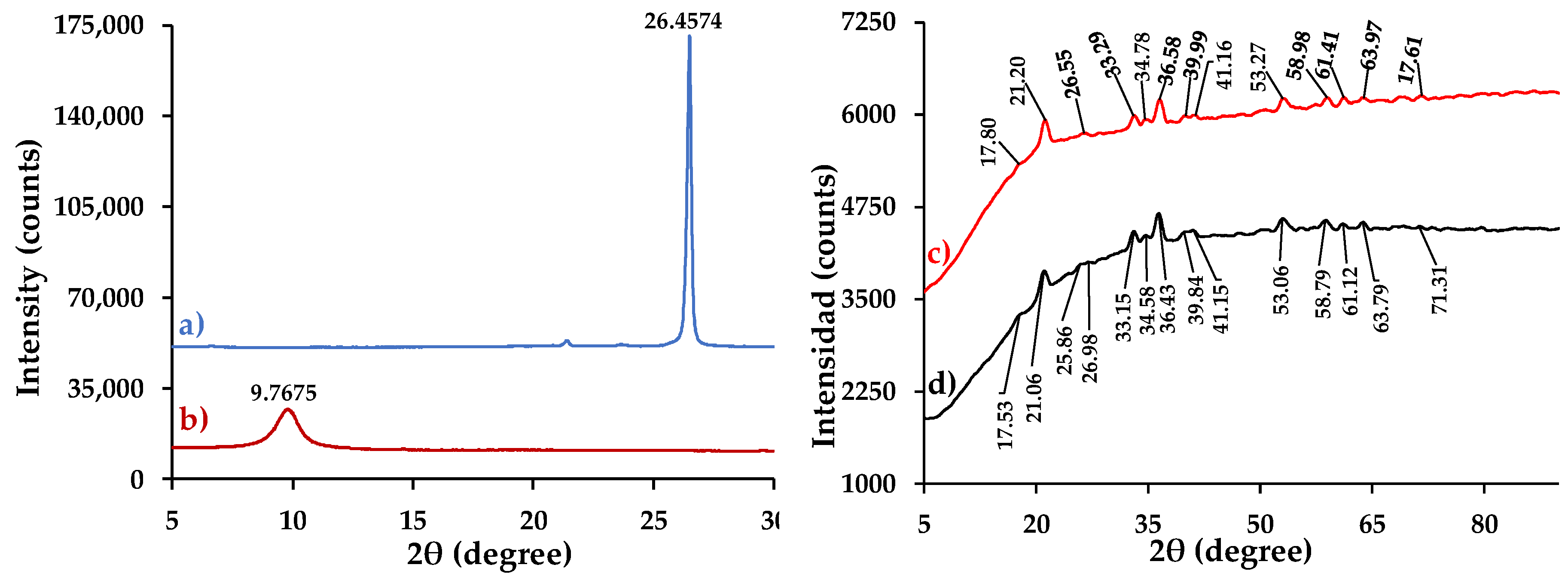
3.4.4. XRD Analysis
3.4.5. FTIR Analysis
3.4.6. Z-Potential Determination through DLS Analysis
3.5. Optimum pH Value Determination
3.6. Adsorption Kinetics
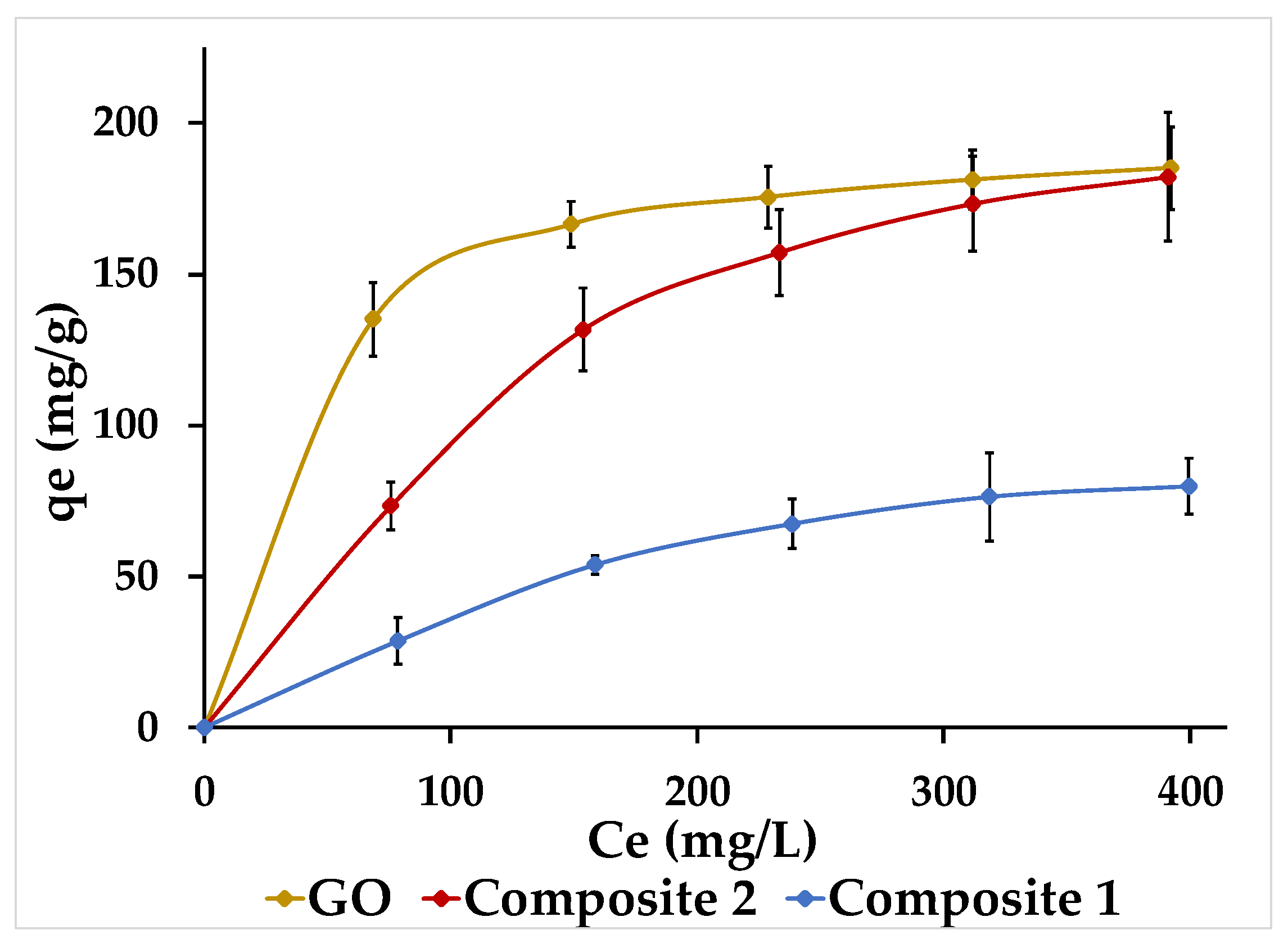
3.7. Adsorption Isotherms
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fu, F.; Wang, Q. Removal of heavy metal ions from wastewaters: A review. J. Environ. Manag. 2011, 92, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Valdivia, M. Intoxicación por plomo. Rev. Soc. Per. Med. Inter. 2005, 18, 22–27. [Google Scholar]
- Rouquerol, J.; Rouquerol, F.; Llewellyn, P.; Maurin, G.; Sing, K.S. Adsorption by Powders and Porous Solids: Principles, Methodology and Applications; Academic Press: Cambridge, MA, USA, 2013; pp. 2–22. [Google Scholar]
- Babel, S.; Kurniawan, T.A. Low-cost adsorbents for heavy metals uptake from contaminated water: A review. J. Hazard. Mater. 2003, 97, 219–243. [Google Scholar] [CrossRef]
- Yang, S.T.; Chang, Y.; Wang, H.; Liu, G.; Chen, S.; Wang, Y.; Liu, Y.; Cao, A. Folding/aggregation of graphene oxide and its application in Cu2+ removal. J. Colloid Interface Sci. 2010, 351, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, C.; Hu, J.; Wu, X.; Wang, X. Synthesis of magnetite/graphene oxide composite and application for cobalt (II) removal. J. Phys. Chem. C. 2011, 115, 25234–25240. [Google Scholar] [CrossRef]
- Bigham, J.M.; Fitzpatrick, R.W.; Schulze, D.G. Iron Oxides. In Soil Mineralogy with Environmental Applications; Dixon, J.B., Schulze, D.G., Eds.; Soil Science Society of America Inc.: Madison, WI, USA, 2002; pp. 323–366. [Google Scholar]
- Lehmann, M.; Zouboulis, A.I.; Matis, K.A. Removal of metal ions from dilute aqueous solutions: A comparative study of inorganic sorbent materials. Chemosphere 1999, 39, 881–892. [Google Scholar] [CrossRef]
- Chen, Y.H.; Li, F.A. Kinetic study on removal of copper (II) using goethite and hematite nano-photocatalysts. J. Colloid Interface Sci. 2010, 347, 277–281. [Google Scholar] [CrossRef]
- Fan, Y.; Fu, D.; Zhou, S.; Lu, Y.; Zhao, X.; Jin, W.; Zhao, Y. Facile synthesis of goethite anchored regenerated graphene oxide nanocomposite and its application in the removal of fluoride from drinking water. Desalin. Water Treat. 2016, 57, 28393–28404. [Google Scholar] [CrossRef]
- Xu, H.; Hu, Z.; Lu, A.; Hu, Y.; Li, L.; Yang, Y.; Zhang, Z.; Wu, H. Synthesis and super capacitance of goethite/reduced graphene oxide for supercapacitors. Mater. Chem. Phys. 2013, 141, 310–317. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved synthesis of graphene oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]
- Foo, K.Y.; Hameed, B.H. Insights into the modeling of adsorption isotherm systems. Chem. Eng. J. 2010, 156, 2–10. [Google Scholar] [CrossRef]
- Mushtaq, M.; Bhatti, H.N.; Iqbal, M.; Noreen, S. Eriobotrya japonica seed biocomposite efficiency for copper adsorption: Isotherms, kinetics, thermodynamic and desorption studies. J. Environ. Manag. 2016, 176, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Kudin, K.N.; Ozbas, B.; Schniepp, H.C.; Prud’Homme, R.K.; Aksay, I.A.; Car, R. Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett. 2008, 8, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 147, 47–57. [Google Scholar] [CrossRef]
- Hummers, W., Jr.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Park, S.; Bielawski, W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef]
- Saini, A.; Kumar, A.; Kumar, V.; Chander, S. Synthesis of graphene oxide using modified Hummer’s method and its reduction using hydrazine hydrate. Int. J. Eng. Trends Technol. 2016, 40, 67–71. [Google Scholar] [CrossRef]
- Dimiev, A.; Kosynkin, D.V.; Alemany, L.B.; Chaguine, P.; Tour, J.M. Pristine graphite oxide. J. Am. Chem. Soc. 2012, 134, 2815–2822. [Google Scholar] [CrossRef]
- Higginbotham, A.L.; Kosynkin, D.V.; Sinitskii, A.; Sun, Z.; Tour, J.M. Lower-defect graphene oxide nanoribbons from multiwalled carbon nanotubes. ACS Nano 2010, 4, 2059–2069. [Google Scholar] [CrossRef]
- Choi, E.K.; Jeon, I.Y.; Oh, S.J.; Baek, J.B. “Direct” grafting of linear macromolecular “wedges” to the edge of pristine graphite to prepare edge-functionalized graphene-based polymer composites. J. Mater. Chem. 2010, 20, 10936–10942. [Google Scholar] [CrossRef]
- Deng, J.H.; Zhang, X.R.; Zeng, G.M.; Gong, J.L.; Niu, Q.Y.; Liang, J. Simultaneous removal of Cd(II) and ionic dyes from aqueous solution using magnetic graphene oxide nanocomposite as an adsorbent. Chem. Eng. J. 2013, 226, 189–200. [Google Scholar] [CrossRef]
- Lian, P.; Zhu, X.; Liang, S.; Li, Z.; Yang, W.; Wang, H. Large reversible capacity of high quality graphene sheets as an anode material for lithium-ion batteries. Electrochim. Acta 2010, 55, 3909–3914. [Google Scholar] [CrossRef]
- Li, C.; Feng, C.; Peng, Z.; Gong, W.; Kong, L. Ammonium-assisted green fabrication of graphene/natural rubber latex composite. Polym. Compos. 2013, 34, 88–95. [Google Scholar] [CrossRef]
- Chen, J.; Yao, B.; Li, C.; Shi, G. An improved Hummers method for eco-friendly synthesis of graphene oxide. Carbon 2013, 64, 225–229. [Google Scholar] [CrossRef]
- Shahriary, L.; Athawale, A.A. Graphene oxide synthesized by using modified Hummers approach. Int. J. Renew. Energy Environ. Eng. 2014, 2, 58–63. [Google Scholar]
- Wang, H.; Yuan, X.; Wu, Y.; Huang, H.; Zeng, G.; Liu, Y.; Wang, X.; Lin, N.; Qi, Y. Adsorption characteristics and behaviors of graphene oxide for Zn(II) removal from aqueous solution. Appl. Surf. Sci. 2013, 279, 432–440. [Google Scholar] [CrossRef]
- Bradder, P.; Ling, S.K.; Wang, S.; Liu, S. Dye adsorption on layered graphite oxide. J. Chem. Eng. Data 2011, 56, 138–141. [Google Scholar] [CrossRef]
- Villacís-García, M.; Ugalde-Arzate, M.; Vaca-Escobar, K.; Villalobos, M.; Zanella, R.; Martínez-Villegas, N. Laboratory synthesis of goethite and ferrihydrite of controlled particle sizes. Bol. La Soc. Geol. Mex. 2015, 67, 433–446. [Google Scholar] [CrossRef]
- El-Khodary, S.A.; El-Enany, G.M.; El-Okr, M.; Ibrahim, M. Preparation and characterization of microwave reduced graphite oxide for high-performance supercapacitors. Electrochim. Acta 2014, 150, 269–278. [Google Scholar] [CrossRef]
- Chaiyakun, S.; Witit-Anun, N.; Nuntawong, N.; Chindaudom, P.; Oaew, S.; Kedkeaw, C.; Limsuwan, P. Preparation and characterization of graphene oxide nanosheets. Procedia Eng. 2012, 32, 759–764. [Google Scholar] [CrossRef]
- Hu, C.; Liu, Y.; Yang, Y.; Cui, J.; Huang, Z.; Wang, Y.; Yang, L.; Wang, H.; Xiao, Y.; Rong, J. One-step preparation of nitrogen-doped graphene quantum dots from oxidized debris of graphene oxide. J. Mater. Chem. B. 2013, 1, 39–42. [Google Scholar] [CrossRef]
- Lakshmipathiraj, P.; Narasimhan, B.R.V.; Prabhakar, S.; Bhaskar Raju, G. Adsorption of arsenate on synthetic goethite from aqueous solutions. J. Hazard. Mater. 2006, 136, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Dimiev, A.M.; Alemany, L.B.; Tour, J.M. Graphene oxide. Origin of acidity, its instability in water, and a new dynamic structural model. ACS Nano 2012, 7, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Aquino, A.J.; Tunega, D.; Haberhauer, G.; Gerzabek, M.H.; Lischka, H. Acid–base properties of a goethite surface model: A theoretical view. Geochim. Cosmochim. Acta 2008, 72, 3587–3602. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, C.; Zhou, W.; Lei, A.; Zhang, Q.; Wan, Q.; Zou, B. Fast and considerable adsorption of methylene blue dye onto graphene oxide. Bull. Environ. Contam. Toxicol. 2011, 87, 86–90. [Google Scholar] [CrossRef]
- Mallakpour, S.; Abdolmaleki, A.; Borandeh, S. Covalently functionalized graphene sheets with biocompatible natural amino acids. Appl. Surf. Sci. 2014, 307, 533–542. [Google Scholar] [CrossRef]
- Wang, K.; Xing, B. Adsorption and desorption of cadmium by goethite pretreated with phosphate. Chemosphere 2002, 48, 665–670. [Google Scholar] [CrossRef]
- Essington, M.E. Soil and Water Chemistry, 1st ed.; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2007; pp. 408–447. [Google Scholar]
- Sposito, G. The Chemistry of Soils; Oxford University Press: Oxford, UK, 2008; pp. 219–239. [Google Scholar]
- Wu, Z.; Gu, Z.; Wang, X.; Evans, L.; Guo, H. Effects of organic acids on adsorption of lead onto montmorillonite, goethite and humic acid. Environ. Pollut. 2003, 121, 469–475. [Google Scholar] [CrossRef]
- Naseem, R.; Tahir, S.S. Removal of Pb(II) from aqueous/acidic solutions by using bentonite as an adsorbent. Water Res. 2001, 35, 3982–3986. [Google Scholar] [CrossRef]
- Li, Y.H.; Wang, S.; Wei, J.; Zhang, X.; Xu, C.; Luan, Z.; Wu, D.; Wei, B. Lead adsorption on carbon nanotubes. Chem. Phys. Lett. 2002, 357, 263–266. [Google Scholar] [CrossRef]
- Cechinel, M.A.P.; de Souza, A.A.U. Study of lead (II) adsorption onto activated carbon originating from cow bone. J. Clean. Prod. 2014, 65, 342–349. [Google Scholar] [CrossRef]
- Farajzadeh, M.A.; Monji, A.B. Adsorption characteristics of wheat bran towards heavy metal cations. Sep. Purif. Technol. 2004, 38, 197–207. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Norm. Mass Percent (%) | C | O | Al | Si | S | Cl | K | Fe |
|---|---|---|---|---|---|---|---|---|
| Graphite | 70.67 | 19.64 | 4.68 | 3.86 | 0.04 | 0.01 | 0.32 | 0.77 |
| GO | 45.02 | 38.87 | 4.83 | 3.94 | 3.72 | 2.03 | 0.50 | 1.09 |
| Composite 1 | 21.16 | 33.56 | 0.56 | 0.52 | 0.76 | 0.03 | 0.03 | 43.37 |
| Composite 2 | 16.55 | 38.17 | 0.42 | 0.29 | 0.84 | 0.01 | 0.02 | 43.70 |
| Langmuir Isotherm | Freundlich Isotherm | |||||
|---|---|---|---|---|---|---|
| Material | qmax (mg/g) | K (L/mg) | R2 | N | K (L/mg) | R2 |
| GO | 200 | 0.3 | 0.99 | 5.57 | 65.07 | 0.94 |
| Composite 1 | 138.89 | 3.65 × 10−3 | 0.96 | 1.56 | 1.90 | 0.95 |
| Composite 2 | 277.78 | 5.28 × 10−3 | 0.98 | 1.80 | 7.17 | 0.94 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordon-Nuñez, F.; Vaca-Escobar, K.; Villacís-García, M.; Fernández, L.; Debut, A.; Aldás-Sandoval, M.B.; Espinoza-Montero, P.J. Applicability of Goethite/Reduced Graphene Oxide Nanocomposites to Remove Lead from Wastewater. Nanomaterials 2019, 9, 1580. https://doi.org/10.3390/nano9111580
Gordon-Nuñez F, Vaca-Escobar K, Villacís-García M, Fernández L, Debut A, Aldás-Sandoval MB, Espinoza-Montero PJ. Applicability of Goethite/Reduced Graphene Oxide Nanocomposites to Remove Lead from Wastewater. Nanomaterials. 2019; 9(11):1580. https://doi.org/10.3390/nano9111580
Chicago/Turabian StyleGordon-Nuñez, Franklin, Katherine Vaca-Escobar, Milton Villacís-García, Lenys Fernández, Alexis Debut, María Belén Aldás-Sandoval, and Patricio J. Espinoza-Montero. 2019. "Applicability of Goethite/Reduced Graphene Oxide Nanocomposites to Remove Lead from Wastewater" Nanomaterials 9, no. 11: 1580. https://doi.org/10.3390/nano9111580
APA StyleGordon-Nuñez, F., Vaca-Escobar, K., Villacís-García, M., Fernández, L., Debut, A., Aldás-Sandoval, M. B., & Espinoza-Montero, P. J. (2019). Applicability of Goethite/Reduced Graphene Oxide Nanocomposites to Remove Lead from Wastewater. Nanomaterials, 9(11), 1580. https://doi.org/10.3390/nano9111580