Electrochemical Analysis for Demonstrating CO Tolerance of Catalysts in Polymer Electrolyte Membrane Fuel Cells


Abstract
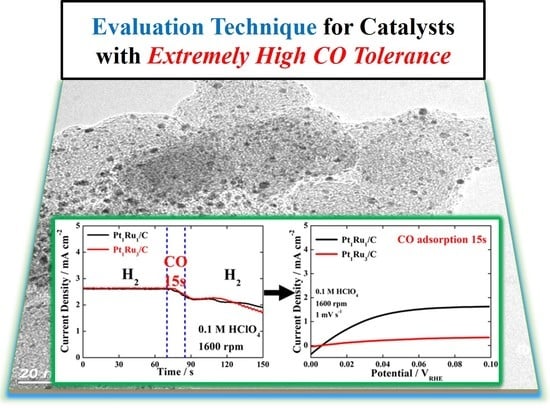
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Pt1Ru3/C catalyst
2.3. Characterizations
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Qingfeng, L.; Ronghuan, H.; Jens, O.J.; Niels, J.B. Approaches and recent development of polymer electrolyte membranes for fuel cells operating above 100 °C. Chem. Mater. 2003, 15, 4896–4915. [Google Scholar]
- Sharma, M.; Jang, J.H.; Shin, D.Y.; Kwon, J.A.; Lim, D.H.; Choi, D.Y.; Sung, H.K.; Jang, J.H.; Lee, S.Y.; Lee, L.Y.; et al. Work function-tailored graphene via transition metal encapsulation as highly active and durable catalyst for oxygen reduction reaction. Energy Environ. Sci. 2019, 12, 2200–2211. [Google Scholar] [CrossRef]
- Jang, J.H.; Sharma, M.; Choi, D.I.; Kang, Y.S.; Kim, Y.J.; Min, J.H.; Sung, H.K.; Jung, N.G.; Yoo, S.J. Boosting fuel cell durability under shut-down/start-up conditions using hydrogen oxidation-selective metal-carbon hybrid core-shell catalyst. ACS Appl. Mater. Interfaces 2019, 11, 27735–27742. [Google Scholar] [CrossRef] [PubMed]
- Lulianelli, A.; Ribeirinha, P.; Mendes, A.; Basile, A. Methanol steam reforming for hydrogen generation via conventional and membrane reactors: A review. J. Renew. Sustain. Ener. 2014, 29, 355–368. [Google Scholar] [CrossRef]
- Boyano, A.; Blanco-Marigorta, A.M.; Morosuk, T.; Tsatsaronis, G. Exergoenvironmental analysis of a steam methane reforming process for hydrogen production. Energy 2011, 36, 2202–2214. [Google Scholar] [CrossRef]
- Philippe, T.; Robert, D.; Bernard, C.; Christophe, C.; Severine, R.; Claude, L. Poisoning of Pt/C catalysts by CO and its consequences over the kinetics of hydrogen chemisorption. Appl. Catal. B Environ. 2009, 92, 280–284. [Google Scholar]
- Xuan, C.; Zheng, S.; Nancy, G.; Zhang, L.; Zhang, J.; Datong, S.; Zhong, S.L.; Wang, H.; Shen, J. A review of PEM hydrogen fuel cell contamination: Impacts, mechanisms, and mitigation. J. Power Sources 2007, 165, 739–756. [Google Scholar]
- Hsieh, Y.C.; Zhang, Y.; Su, D.; Volkov, V.; Si, R.; Wu, L.; Zhu, Y.; An, W.; Liu, P.; He, P.; et al. Ordered bilayer ruthenium–platinum core-shell nanoparticles as carbon monoxide-tolerant fuel cell catalysts. Nat. Commun. 2013, 4, 2466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kim, J.; Zhang, J.; Nan, F.; Gauquelin, N.; Botton, G.A.; He, P.; Bashyam, R.; Knights, S. Ti4O7 supported Ru@Pt core–shell catalyst for CO-tolerance in PEM fuel cell hydrogen oxidation reaction. Appl. Energ. 2013, 103, 507–513. [Google Scholar] [CrossRef]
- Jackson, A.; Strickler, A.; Higgins, D.; Jaramillo, T. Engineering Ru@Pt Core-Shell Catalysts for Enhanced Electrochemical Oxygen Reduction Mass Activity and Stability. Nanomaterials 2018, 8, 38. [Google Scholar] [CrossRef]
- Rigdon, W.A.; Huang, X. Carbon monoxide tolerant platinum electrocatalysts on niobium doped titania and carbon nanotube composite supports. J. Power Sources 2014, 272, 845–859. [Google Scholar] [CrossRef]
- Lee, M.J.; Kang, J.S.; Kang, Y.S.; Chung, D.Y.; Shin, H.J.; Ahn, C.Y.; Park, S.B.; Kim, S.j.; Lee, K.S.; Sung, Y.E. Understanding the bifunctional effect for removal of CO poisoning: Blend of a platinum nanocatalyst and hydrous ruthenium oxide as a model system. ACS Catal. 2016, 6, 2398–2407. [Google Scholar] [CrossRef]
- Liu, P.; Logadottir, A.; Nørskov, J.K. Modeling the electro-oxidation of CO and H2/CO on Pt, Ru, PtRu and Pt3Sn. Electrochim. Acta. 2003, 48, 3731–3742. [Google Scholar] [CrossRef]
- Lopes, P.P.; Ticianelli, E.A. The CO tolerance pathways on the Pt–Ru electrocatalytic system. J. Electroanal. Chem. 2010, 644, 110–116. [Google Scholar] [CrossRef]
- Roth, C.; Papworth, A.J.; Hussain, I.; Nichols, R.J.; Schiffrin, D.J. A Pt/Ru nanoparticulate system to study the bifunctional mechanism of electrocatalysis. J. Electroanal. Chem. 2005, 581, 79–85. [Google Scholar] [CrossRef]
- Sugimoto, W.; Aoyama, K.; Kawaguchi, T.; Murakami, Y.; Takasu, Y. Kinetics of CH3OH oxidation on PtRu/C studied by impedance and CO stripping voltammetry. J. Electroanal. Chem. 2005, 576, 215–221. [Google Scholar] [CrossRef]
- Dickinson, E.J.F.; Streeter, I.; Compton, R.G. Theory of chronoamperometry at cylindrical microelectrodes and their arrays. J. Phys. Chem. C 2008, 31, 11637–11644. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, L.; Amemiya, S. Chronoamperometry at micropipet electrodes for determination of diffusion coefficients and transferred charges at liquid/liquid interfaces. Anal. Chem. 2004, 76, 5570–5578. [Google Scholar] [CrossRef] [PubMed]
- Ham, D.J.; Kim, Y.K.; Han, S.H.; Lee, J.S. Pt/WC as an anode catalyst for PEMFC: Activity and CO tolerance. Catal. Today 2008, 132, 117–122. [Google Scholar] [CrossRef]
- Xi, J.; Wang, J.; Yu, L.; Qiu, X.; Chen, L. Facile approach to enhance the Pt utilization and CO-tolerance of Pt/C catalysts by physically mixing with transition-metal oxide nanoparticles. Chem. Commun. 2007, 16, 1656–1658. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Guo, D.J.; Wang, J.S.; Qiu, X.P.; Chen, L.Q.; Zhu, W.T. Using phosphomolybdic acid (H3PMo12O40) to efficiently enhance the electrocatalytic activity and CO-tolerance of platinum nanoparticles supported on multi-walled carbon nanotubes catalyst in acidic medium. J. Electroanal. Chem. 2010, 638, 167–172. [Google Scholar] [CrossRef]
- Li, M.; Pan, Y.; Guo, X.; Liang, Y.; Wu, Y.; Wen, Y.; Yang, H. Pt/single-stranded DNA/graphene nanocomposite with improved catalytic activity and CO tolerance. J. Mater. Chem. A 2015, 19, 10353–10359. [Google Scholar] [CrossRef]
- Martinez, H.V.; Rodriguez, J.L.; Tsiouvaras, N.; Pena, M.A.; Fierro, J.L.G.; Pastor, E. Novel synthesis method of CO-tolerant PtRu−MoOx nanoparticles: Structural characteristics and performance for methanol electrooxidation. Chem. Mater. 2008, 20, 4249–4259. [Google Scholar] [CrossRef]
- González-Hernández, M.; Antolini, E.; Perez, J. Synthesis, Characterization and CO Tolerance Evaluation in PEMFCs of Pt2RuMo Electrocatalysts. Catalysis 2019, 9, 61. [Google Scholar] [CrossRef]
- Ianniello, R.; Schmidt, V.M.; Stimming, U.; Stumper, J.; Wallau, A. CO adsorption and oxidation on Pt and PtRu alloys: Dependence on substrate composition. Electrochim. Acta 1994, 39, 1863–1869. [Google Scholar] [CrossRef]
- Jeon, T.Y.; Lee, K.S.; Yoo, S.J.; Cho, Y.H.; Kang, S.H.; Sung, Y.E. Effect of Surface Segregation on the Methanol Oxidation Reaction in Carbon-Supported Pt−Ru Alloy Nanoparticles. Langmuir 2010, 26, 9123–9129. [Google Scholar] [CrossRef]
- Jang, Y.; Choi, K.-H.; Chung, D.Y.; Lee, J.E.; Jung, N.; Sung, Y.-E. Self-assembled dendritic Pt nanostructure with high-index facets as highly active and durable electrocatalyst for oxygen reduction. ChemSusChem 2017, 10, 3063–3068. [Google Scholar] [CrossRef]
- Cuesta, A.; Couto, A.; Rincón, A.; Pérez, M.C.; López-Cudero, A.; Gutiérrez, C. Potential dependence of the saturation CO coverage of Pt electrodes: The origin of the pre-peak in CO-stripping voltammograms. Part 3: Pt(poly). J. Electroanal. Chem. 2006, 586, 184–195. [Google Scholar] [CrossRef]
- López-Cudero, A.; Cuesta, A.; Gutiérrez, C. Potential dependence of the saturation CO coverage of Pt electrodes: The origin of the pre-peak in CO-stripping voltammograms. Part 1: Pt(111). J. Electroanal. Chem. 2005, 579, 1–12. [Google Scholar]
- Li, Q.; He, R.; Gao, J.A.; Jensen, J.O.; Bjerrum, N.J. The CO poisoning effect in PEMFCs operational at temperatures up to 200 °C. J. Electrochem. Soc. 2003, 150, A1599–A1605. [Google Scholar] [CrossRef]
- Lopes, P.P.; Freitas, K.S.; Ticianelli, E.A. CO Tolerance of PEMFC Anodes: Mechanisms and Electrode Designs. Electrocatalysis 2010, 1, 200–212. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, J.; Jeffery, A.A.; Kim, Y.; Jung, N. Electrochemical Analysis for Demonstrating CO Tolerance of Catalysts in Polymer Electrolyte Membrane Fuel Cells. Nanomaterials 2019, 9, 1425. https://doi.org/10.3390/nano9101425
Min J, Jeffery AA, Kim Y, Jung N. Electrochemical Analysis for Demonstrating CO Tolerance of Catalysts in Polymer Electrolyte Membrane Fuel Cells. Nanomaterials. 2019; 9(10):1425. https://doi.org/10.3390/nano9101425
Chicago/Turabian StyleMin, Jiho, A. Anto Jeffery, Youngjin Kim, and Namgee Jung. 2019. "Electrochemical Analysis for Demonstrating CO Tolerance of Catalysts in Polymer Electrolyte Membrane Fuel Cells" Nanomaterials 9, no. 10: 1425. https://doi.org/10.3390/nano9101425
APA StyleMin, J., Jeffery, A. A., Kim, Y., & Jung, N. (2019). Electrochemical Analysis for Demonstrating CO Tolerance of Catalysts in Polymer Electrolyte Membrane Fuel Cells. Nanomaterials, 9(10), 1425. https://doi.org/10.3390/nano9101425