Optimization of the Production Process and Product Quality of Titanate Nanotube–Drug Composites

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
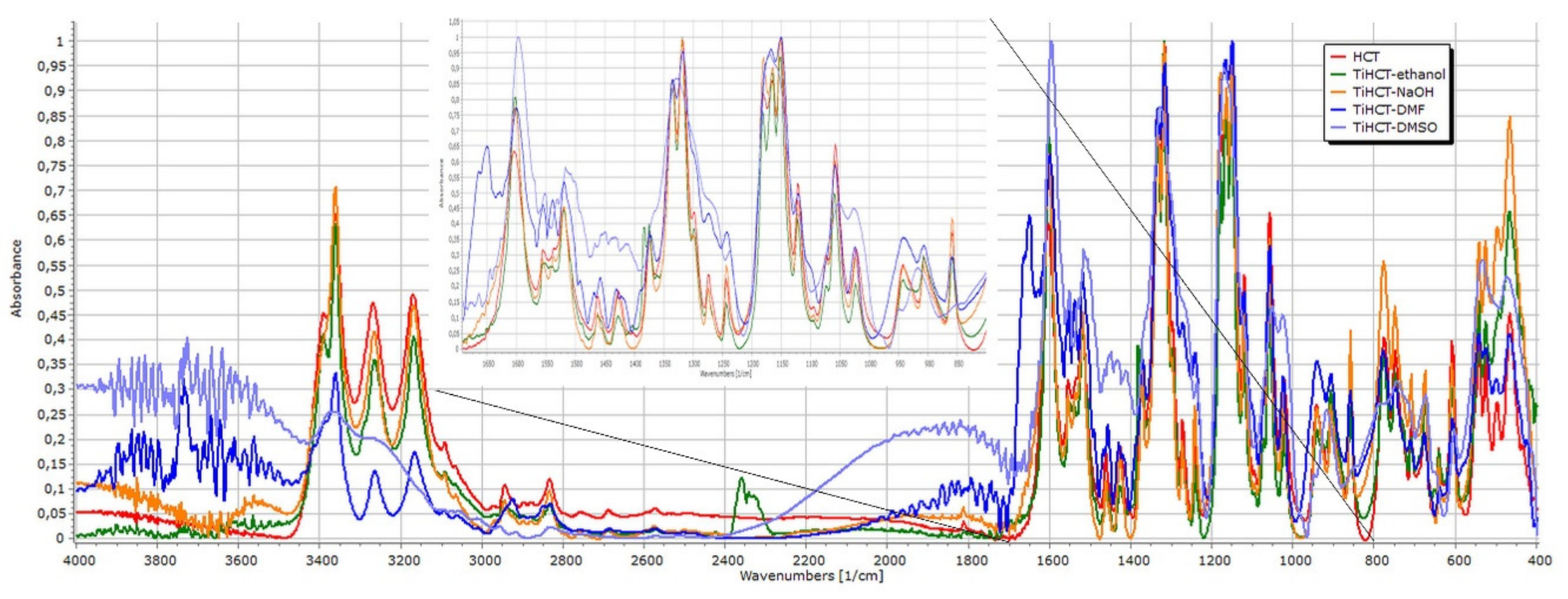
3. Results
3.1. Properties of the TNTs
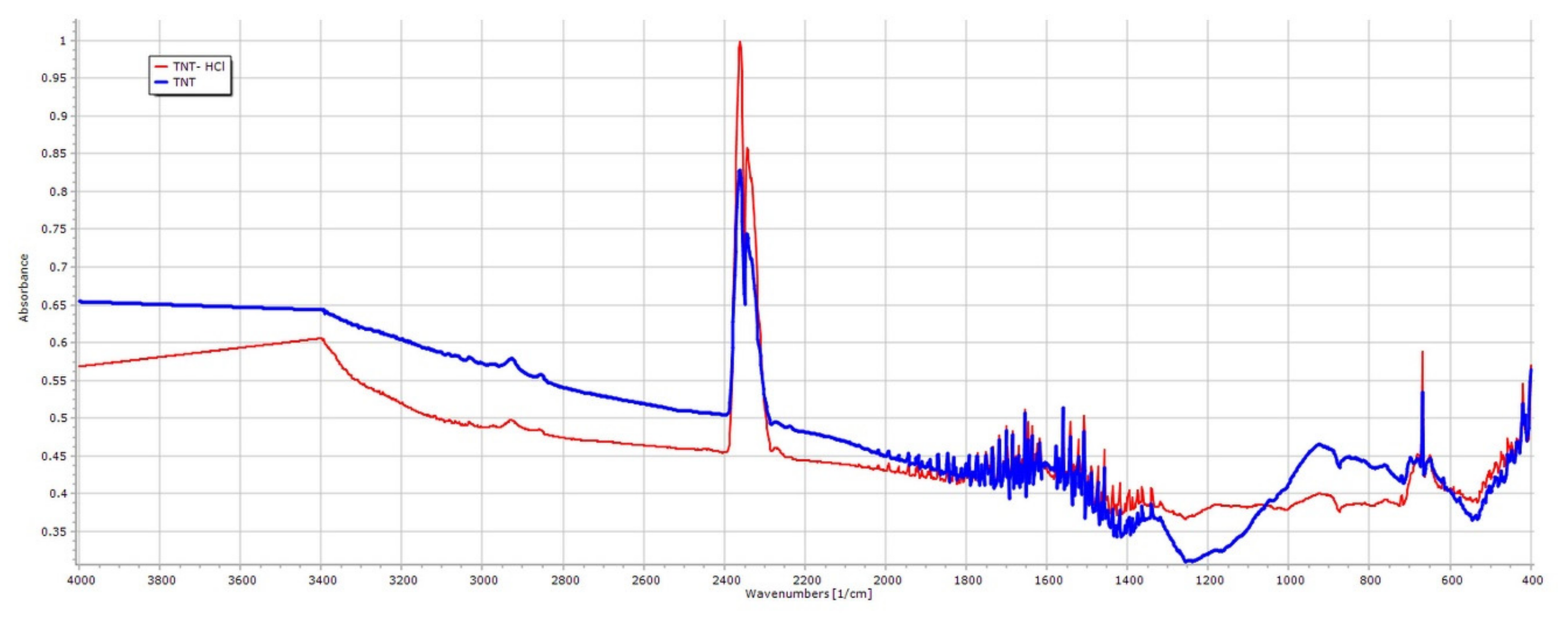
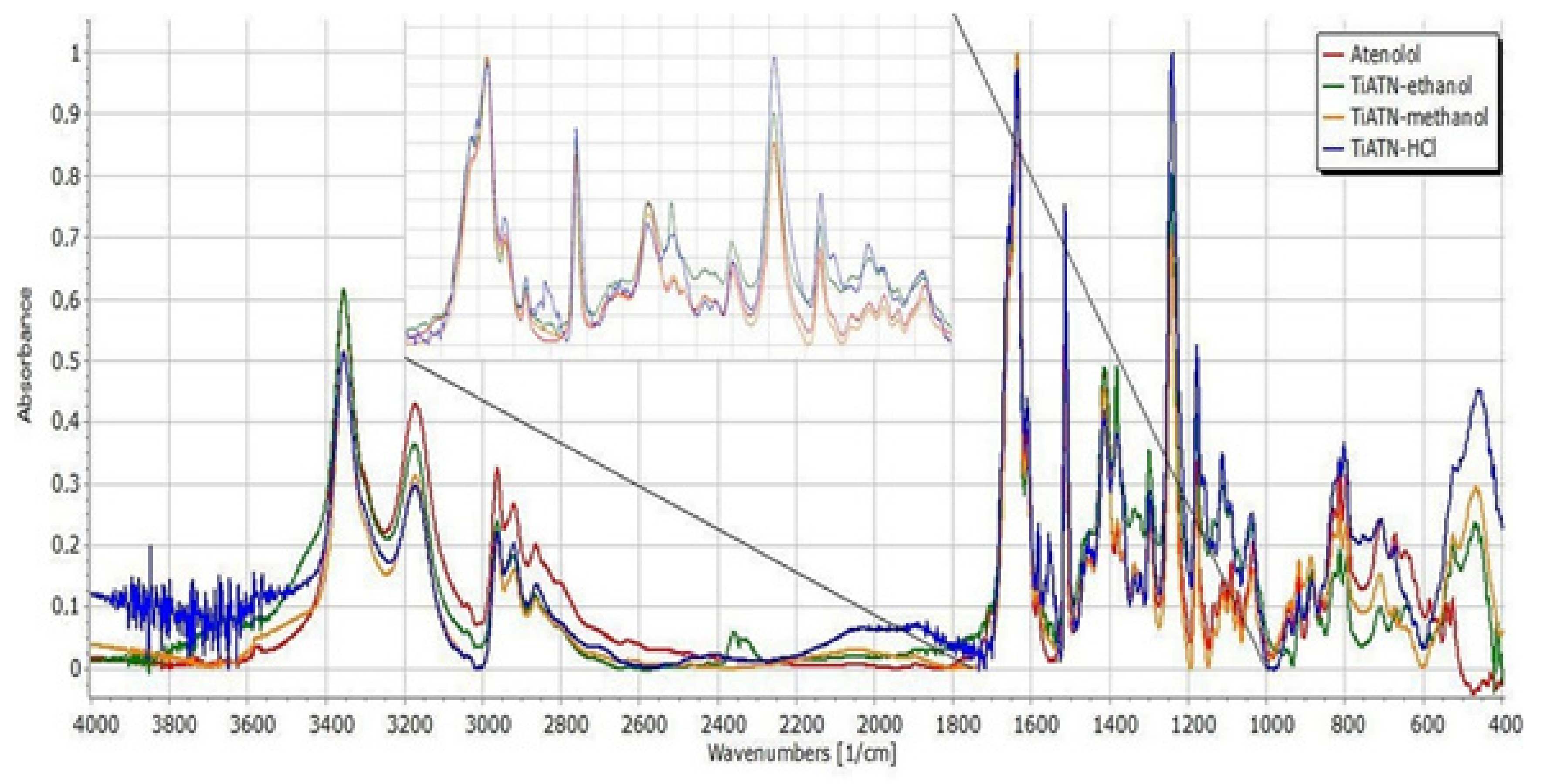
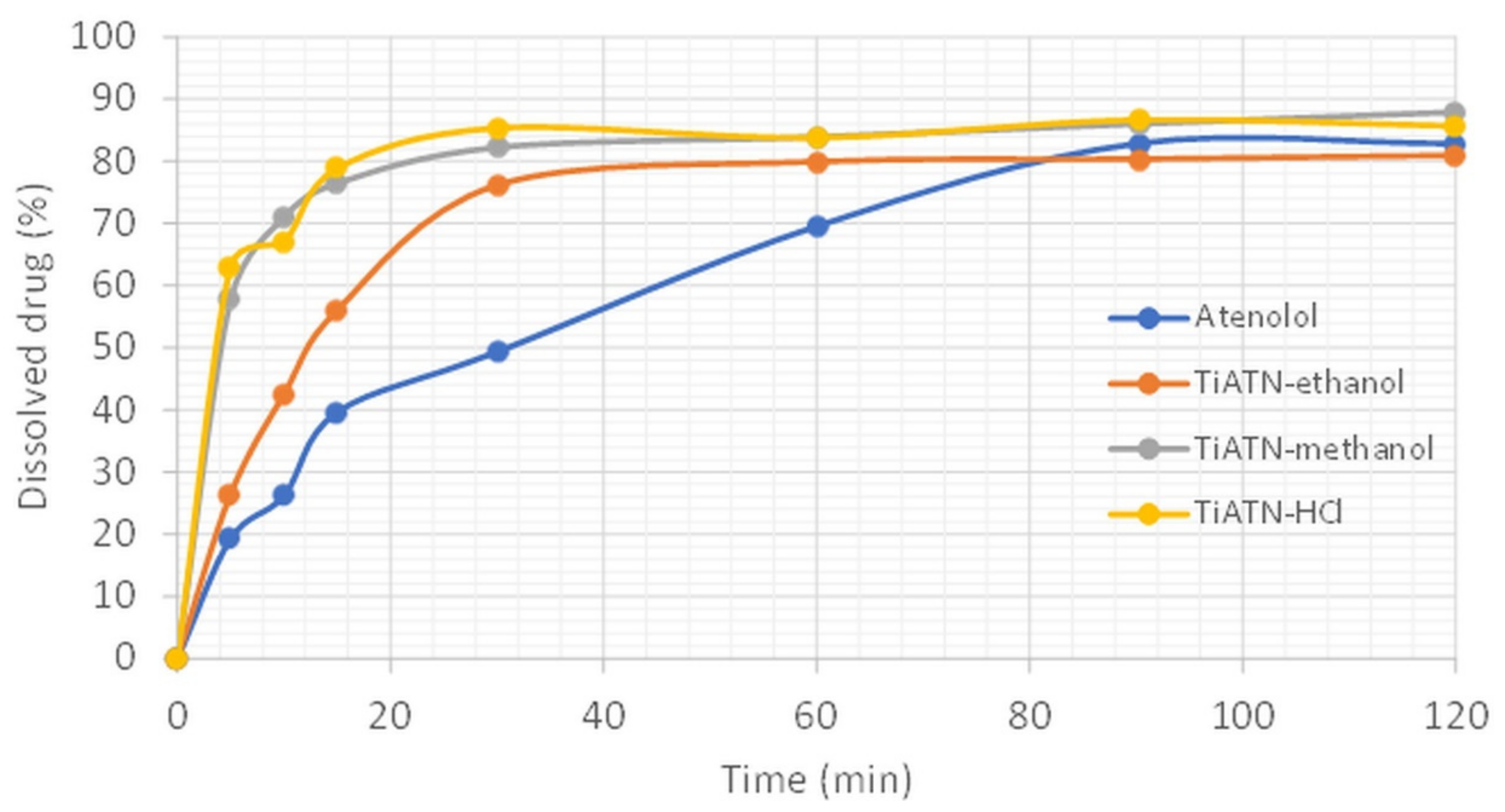
3.2. Effect of Various Solvents on Composite Formation with ATN
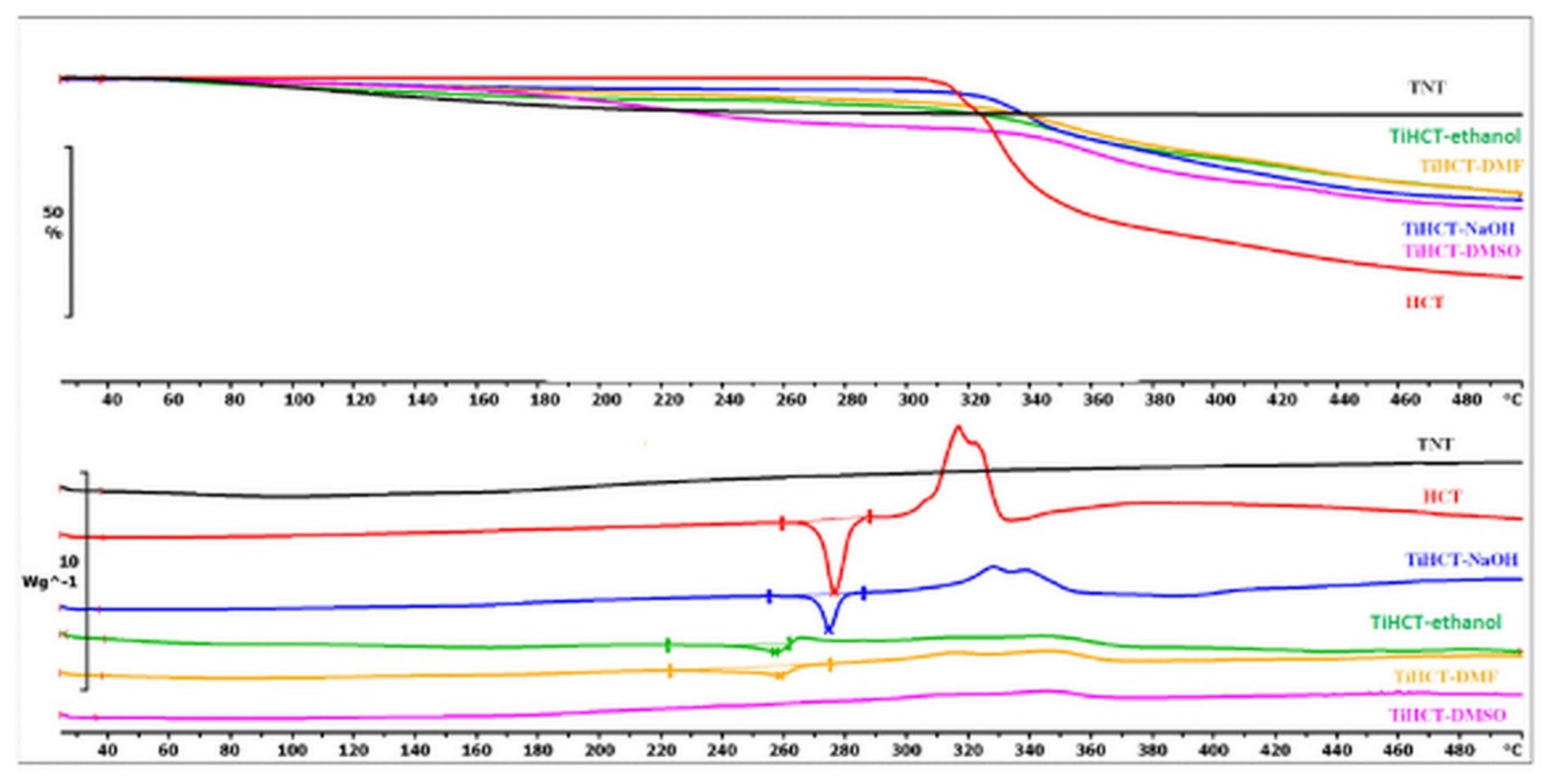
3.3. Effect of Various Solvents on the Composite Formation with HCT
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shukla, A.K.; Bishnoi, R.S.; Dev, S.K.; Kumar, M.; Fenin, V. Biopharmaceutical Classification System: Tool based prediction for drug dosage formulation. Adv. Pharm. J. 2017, 2, 204–209. [Google Scholar]
- Kumar, P.; Singh, C. A study on solubility enhancement methods for poorly water soluble drugs. Am. J. Pharmacol. Sci. 2013, 1, 67–73. [Google Scholar] [CrossRef]
- Habib, M.J. Pharmaceutical Solid Dispersion Technology; CRC Press: Washington, DC, USA, 2000. [Google Scholar]
- Joshi, H.N.; Tejwani, R.W.; Davidovich, M.; Sahasrabudhe, V.P.; Jemal, M.; Bathala, M.S.; Varia, S.A.; Serajuddin, A.T. Bioavailability enhancement of a poorly water-soluble drug by solid dispersion in polyethylene glycol–polysorbate 80 mixture. Int. J. Pharmaceut. 2004, 269, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Taylor, L.S. Influence of different polymers on the crystallization tendency of molecularly dispersed amorphous felodipine. J. Pharm. Sci. 2006, 95, 2692–2705. [Google Scholar] [CrossRef] [PubMed]
- Van den Mooter, G.; Wuyts, M.; Blaton, N.; Busson, R.; Grobet, P.; Augustijns, P.; Kinget, R. Physical stabilisation of amorphous ketoconazole in solid dispersions with polyvinylpyrrolidone K25. Eur. J. Pharm. Sci. 2001, 12, 261–269. [Google Scholar] [CrossRef]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Verheyen, S.; Blaton, N.; Kinget, R.; van den Mooter, G. Mechanism of increased dissolution of diazepam and temazepam from polyethylene glycol 6000 solid dispersions. Int. J. Pharmaceut. 2002, 249, 45–58. [Google Scholar] [CrossRef]
- Martınez-Oharriz, M.; Martın, C.; Goni, M.; Rodrıguez-Espinosa, C.; Tros-Ilarduya, M.; Zornoza, A. Influence of polyethylene glycol 4000 on the polymorphic forms of diflunisal. Eur. J. Pharm. Sci. 1999, 8, 127–132. [Google Scholar] [CrossRef]
- Chiou, W.L.; Riegelman, S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef]
- Nelson, E.; Knoechel, E.; Hamlin, W.; Wagner, J. Influence of the absorption rate of tolbutamide on the rate of decline of blood sugar levels in normal humans. J. Pharm. Sci. 1962, 51, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Lachman, L.; Swartz, C.; Huebner, C. Preformulation investigation I: Relation of salt forms and biological activity of an experimental antihypertensive. J. Pharm. Sci. 1972, 61, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Losic, D.; Simovic, S. Self-ordered nanopore and nanotube platforms for drug delivery applications. Exp. Op. Drug Deliv. 2009, 6, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Aw, M.S.; Kurian, M.; Losic, D. Non-eroding drug-releasing implants with ordered nanoporous and nanotubular structures: Concepts for controlling drug release. Biomaterial. Sci. 2014, 2, 10–34. [Google Scholar]
- Mainardes, R.M.; Silva, L.P. Drug delivery systems: Past, present, and future. Curr. Drug Target. 2004, 5, 449–455. [Google Scholar] [CrossRef]
- Fahr, A.; Liu, X. Drug delivery strategies for poorly water-soluble drugs. Exp. Op. Drug Deliv. 2007, 4, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.B.; Colson, Y.L.; Grinstaff, M.W. Local drug delivery strategies for cancer treatment: Gels, nanoparticles, polymeric films, rods, and wafers. J. Control. Rel. 2012, 159, 14–26. [Google Scholar] [CrossRef]
- Pison, U.; Welte, T.; Giersig, M.; Groneberg, D.A. Nanomedicine for respiratory diseases. Eur. J. Pharmacol. 2006, 533, 341–350. [Google Scholar] [CrossRef]
- Wang, S.; Su, R.; Nie, S.; Sun, M.; Zhang, J.; Wu, D.; Moustaid-Moussa, N. Application of nanotechnology in improving bioavailability and bioactivity of diet-derived phytochemicals. J. Nutr. Biochem. 2014, 25, 363–376. [Google Scholar] [CrossRef]
- Kulkarni, H.P. Synthesis and Applications of Titania Nanotubes: Drug Delivery and Ionomer Composites. Ph.D. Thesis, University of North Carolina, Chapel Hill, NC, USA, 2008. [Google Scholar]
- Gulati, K.; Ramakrishnan, S.; Aw, M.S.; Atkins, G.J.; Findlay, D.M.; Losic, D. Biocompatible polymer coating of titania nanotube arrays for improved drug elution and osteoblast adhesion. Acta Biomater. 2012, 8, 449–456. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, J.-Y.; Li, H.-Q.; Zhao, A.Z.-J.; Wang, Y.; Zhang, K.-Q.; Sun, H.-T.; Lai, Y.-K. Recent advances on smart TiO2 nanotube platforms for sustainable drug delivery applications. Int. J. Nanomed. 2017, 12, 151. [Google Scholar]
- Lai, S.; Zhang, W.; Liu, F.; Wu, C.; Zeng, D.; Sun, Y.; Xu, Y.; Fang, Y.; Zhou, W. TiO2 nanotubes as animal drug delivery system and in vitro controlled release. J. Nanosci Nanotechnol. 2013, 13, 91–97. [Google Scholar]
- Sipos, B.; Pintye-Hódi, K.; Kónya, Z.; Kelemen, A.; Regdon, G., Jr.; Sovány, T. Physicochemical characterisation and investigation of the bonding mechanisms of API-titanate nanotube composites as new drug carrier systems. Int. J. Pharmaceut. 2017, 518, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Saei, A.A.; Jabbaribar, F.; Fakhree, M.A.A.; Acree, W.E., Jr.; Jouyban, A. Solubility of sodium diclofenac in binary water + alcohol solvent mixtures at 25 °C. J. Drug Del. Sci. Tech. 2008, 18, 149–151. [Google Scholar] [CrossRef]
- Hamidi, S.; Jouyban, A. Solubility of atenolol in ethanol + water mixtures at various temperatures. J. Serb. Chem. Soc. 2015, 80, 695–704. [Google Scholar] [CrossRef]
- Wang, S.; Xi, S.; Qu, Y.; Wang, J. Measurement and Correlation of Solubility of Hydrochlorothiazide in Monosolvents and Binary Solvent Mixtures from 283.15 to 323.15 K. J. Chem. Eng. Data 2019, 64, 3128–3138. [Google Scholar] [CrossRef]
- Singh, M.; Lara, S.; Tlali, S. Effects of size and shape on the specific heat, melting entropy and enthalpy of nanomaterials. J. Taibah Univ. Sci. 2017, 11, 922–929. [Google Scholar] [CrossRef]
- Meot-Ner, M. The ionic hydrogen bond 4. Intramolecular and multiple bonds. Protonation and complexes of amides and amino acid derivatives. J. Am. Chem. Soc. 1984, 106, 278–283. [Google Scholar] [CrossRef]
- Johnston, A.; Florence, A.J.; Shankland, N.; Kennedy, A.R.; Shankland, K.; Price, S.L. Crystallization and Crystal Energy Landscape of Hydrochlorothiazide. Cryst. Growth Des. 2007, 7, 705–712. [Google Scholar] [CrossRef]
- Saini, A.; Chadha, R.; Gupta, A.; Singh, P.; Bhandari, S.; Khullar, S.; Mandal, S.; Jain, D.D. New conformational polymorph of hydrochlorothiazide with improved solubility. Pharm. Dev. Techn. 2016, 21, 611–618. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | API Tablets | TNT-API Tablets |
|---|---|---|
| API | 16.7% | - |
| TNT–API | - | 33.3% |
| Avicel PH 112 | 50.0% | 39.5% |
| Tablettose | 29.3% | 23.2% |
| Talc | 3.0% | 3.0% |
| Mg stearate | 1.0% | 1.0% |
| Material | Уs (mJ/m2) | SD | УsDisp (mJ/m2) | SD | УsPol (mJ/m2) | SD | Polarity % |
|---|---|---|---|---|---|---|---|
| TNTs (previous) | 80.72 | ±0.64 | 43.78 | ±0.54 | 36.94 | ±0.35 | 45.76 |
| TNTs (current) | 80.85 | ±1.18 | 44.55 | ±0.53 | 36.31 | ±1.04 | 44.90 |
| TNT–HCl | 78.63 | ±2.07 | 43.10 | ±0.27 | 35.53 | ±2.05 | 45.19 |
| ATN | 59.48 | ±3.99 | 36.70 | ±2.96 | 22.77 | ±2.68 | 38.20 |
| TiATN–ethanol | 60.14 | ±4.25 | 40.45 | ±1.48 | 19.68 | ±3.87 | 32.72 |
| TiATN–methanol | 58.04 | ±2.01 | 37.12 | ±1.19 | 20.92 | ±1.47 | 36.04 |
| TiATN–HCl | 68.37 | ±2.26 | 34.83 | ±0.05 | 33.54 | ±2.26 | 49.06 |
| HCT | 69.51 | ±2.71 | 43.33 | ±0.79 | 26.18 | ±2.59 | 37.60 |
| TiHCT–ethanol | 78.25 | ±0.86 | 44.65 | ±0.57 | 33.60 | ±0.64 | 42.93 |
| TiHCT–NaOH 1M | 77.54 | ±1.89 | 44.52 | ±0.80 | 33.02 | ±1.71 | 42.59 |
| TiHCT–DMF | 71.47 | ±2.63 | 42.53 | ±0.29 | 28.94 | ±2.63 | 40.49 |
| TiHCT–DMS | 73.92 | ±1.42 | 45.29 | ±0.08 | 28.63 | ±1.42 | 38.72 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranjous, Y.; Regdon, G., Jr.; Pintye-Hódi, K.; Varga, T.; Szenti, I.; Kónya, Z.; Sovány, T. Optimization of the Production Process and Product Quality of Titanate Nanotube–Drug Composites. Nanomaterials 2019, 9, 1406. https://doi.org/10.3390/nano9101406
Ranjous Y, Regdon G Jr., Pintye-Hódi K, Varga T, Szenti I, Kónya Z, Sovány T. Optimization of the Production Process and Product Quality of Titanate Nanotube–Drug Composites. Nanomaterials. 2019; 9(10):1406. https://doi.org/10.3390/nano9101406
Chicago/Turabian StyleRanjous, Yasmin, Géza Regdon, Jr., Klára Pintye-Hódi, Tamás Varga, Imre Szenti, Zoltán Kónya, and Tamás Sovány. 2019. "Optimization of the Production Process and Product Quality of Titanate Nanotube–Drug Composites" Nanomaterials 9, no. 10: 1406. https://doi.org/10.3390/nano9101406
APA StyleRanjous, Y., Regdon, G., Jr., Pintye-Hódi, K., Varga, T., Szenti, I., Kónya, Z., & Sovány, T. (2019). Optimization of the Production Process and Product Quality of Titanate Nanotube–Drug Composites. Nanomaterials, 9(10), 1406. https://doi.org/10.3390/nano9101406