Facile Electrochemical Sensor for Nanomolar Rutin Detection Based on Magnetite Nanoparticles and Reduced Graphene Oxide Decorated Electrode

 ,
, 
Abstract
1. Introduction
2. Experimental
2.1. Materials and Reagents
2.2. Characterization Apparatus
2.3. Preparation of NH2-Fe3O4 NPs and GO Composites
2.4. Fabrication of the Decorated Electrodes
2.5. Analytical Procedure
3. Results and Discussion
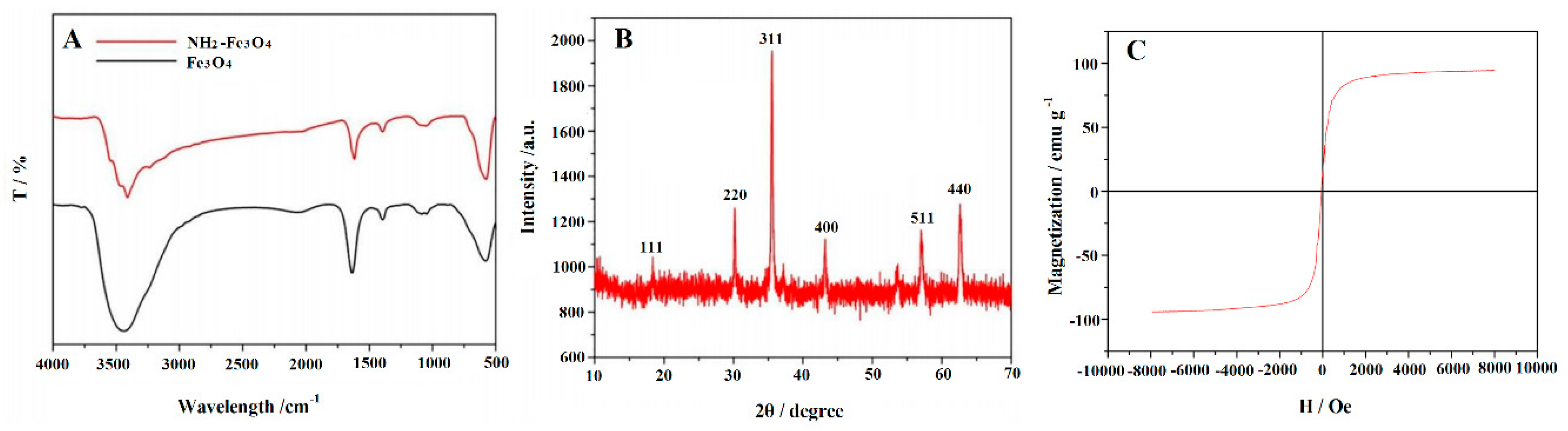
3.1. Characterization of Morphology and Structure of the Synthetic Materials
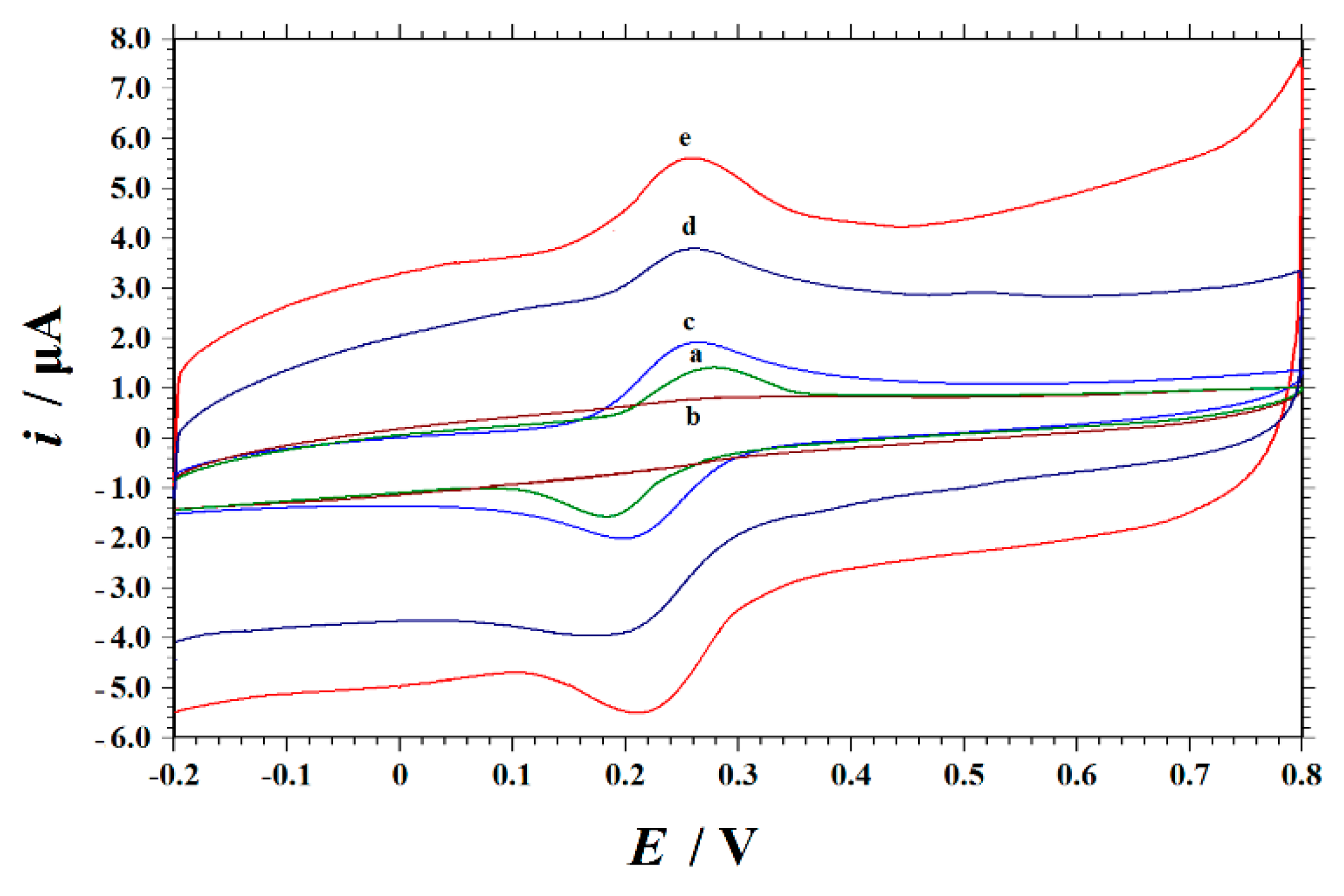
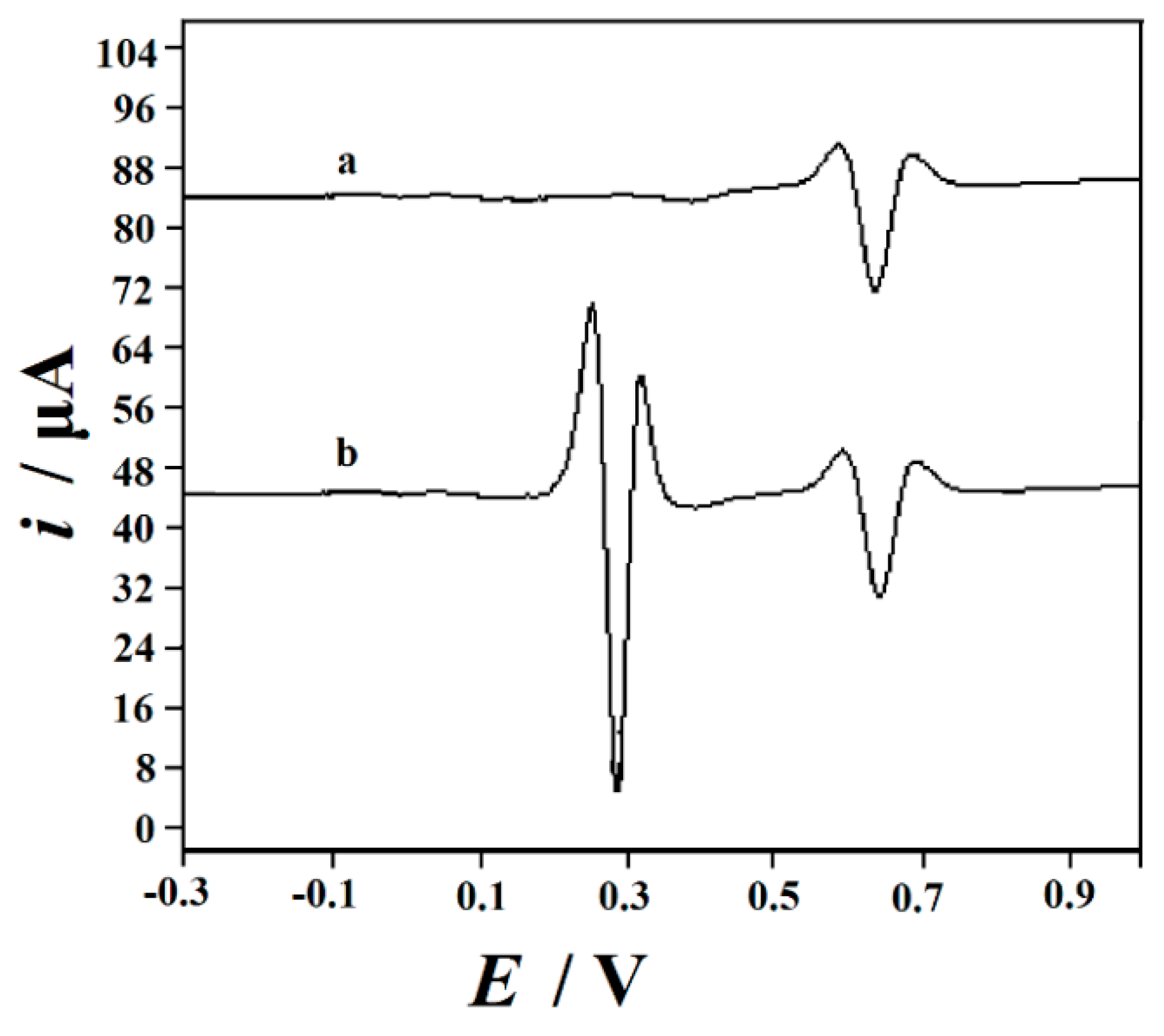
3.2. Electrochemical Properties of Modified Electrodes
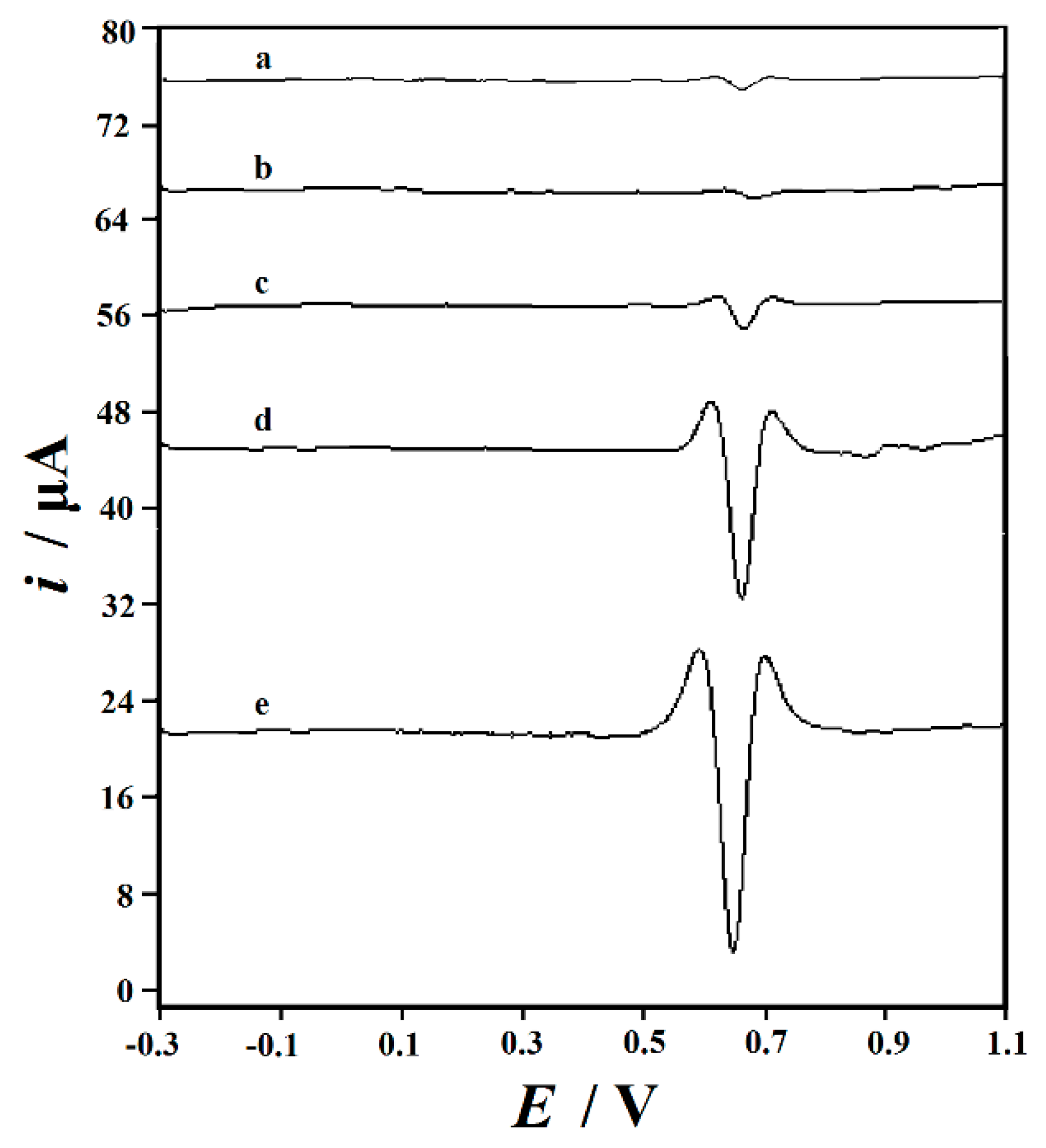
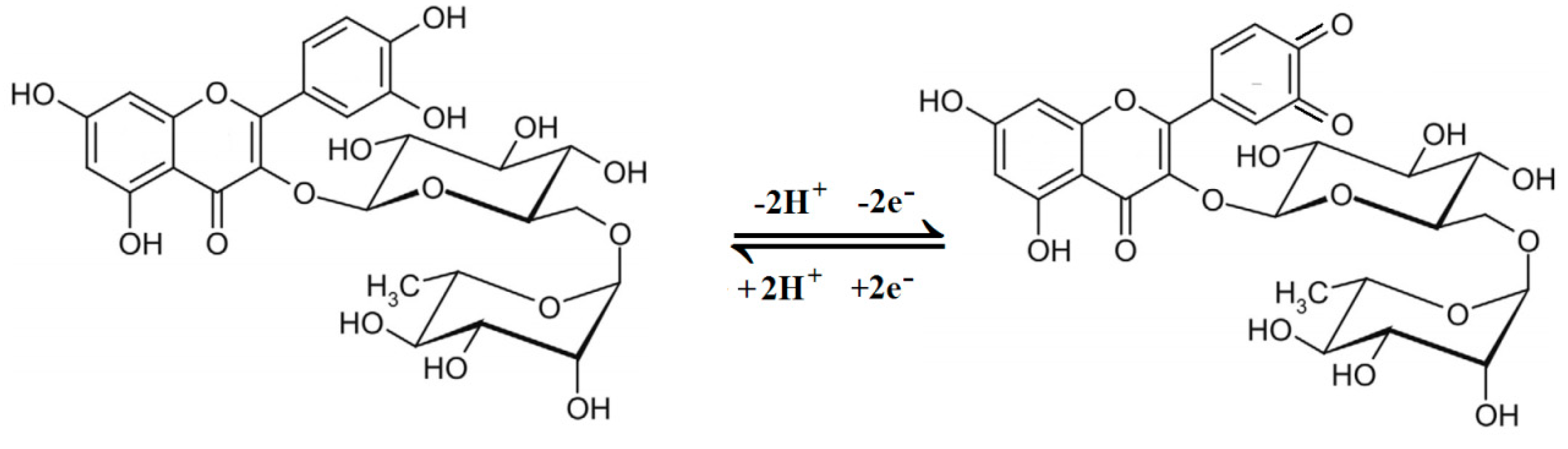
3.3. Rutin Cyclic Voltammograms on the Decorated Electrodes
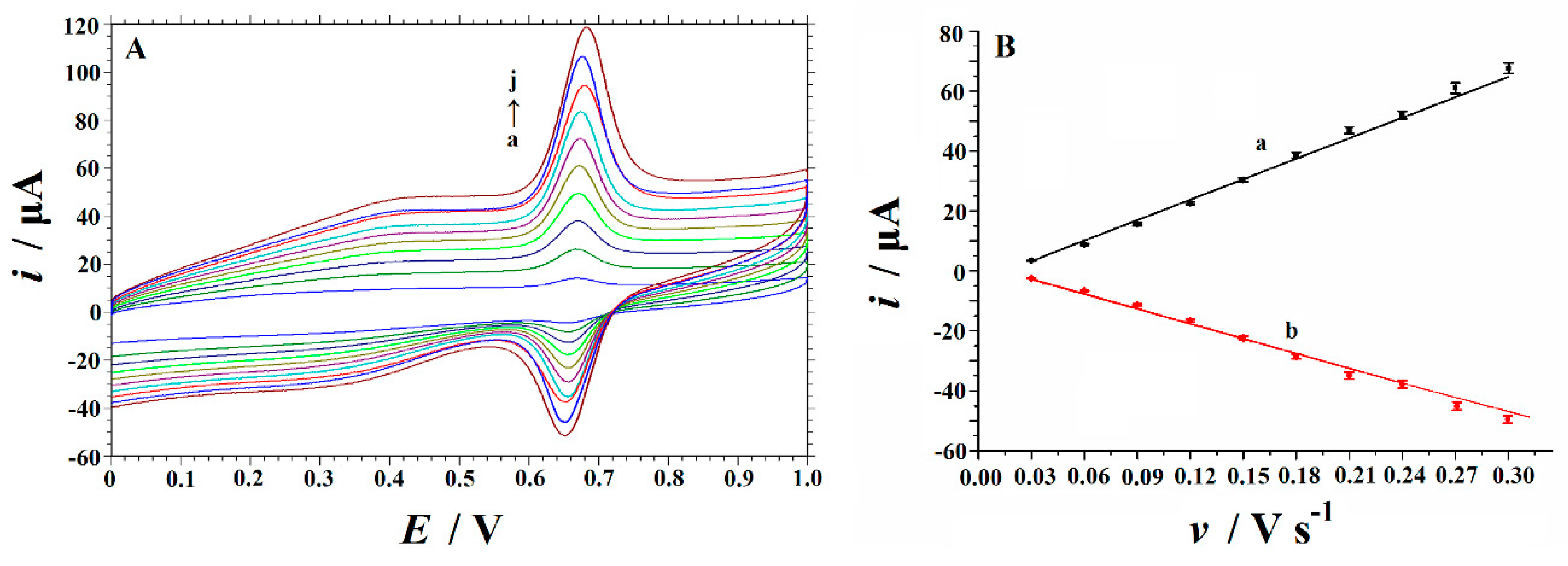
3.4. Effect of Scan Rate
3.5. Effects of Supporting Electrolyte and Solution pH Variation
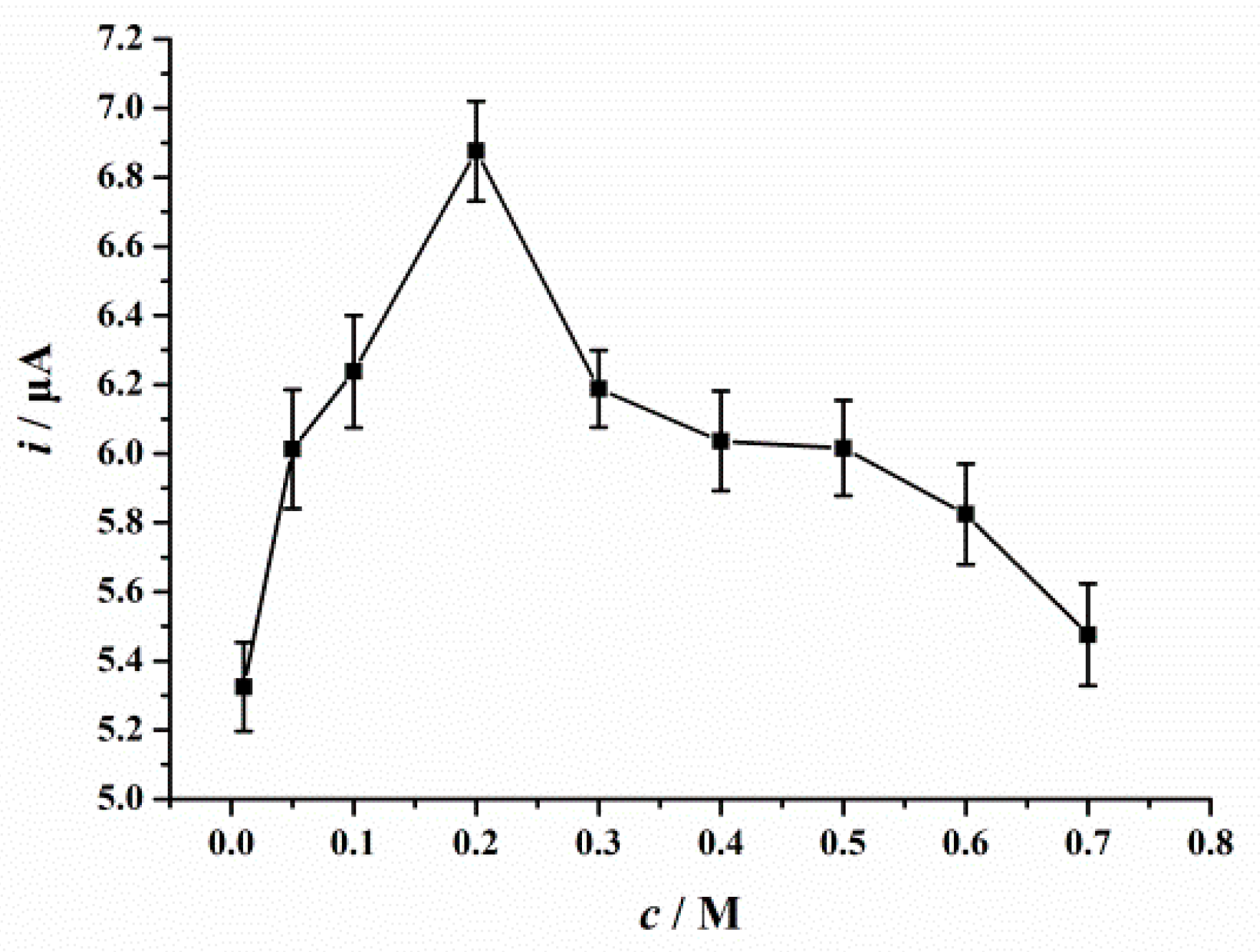
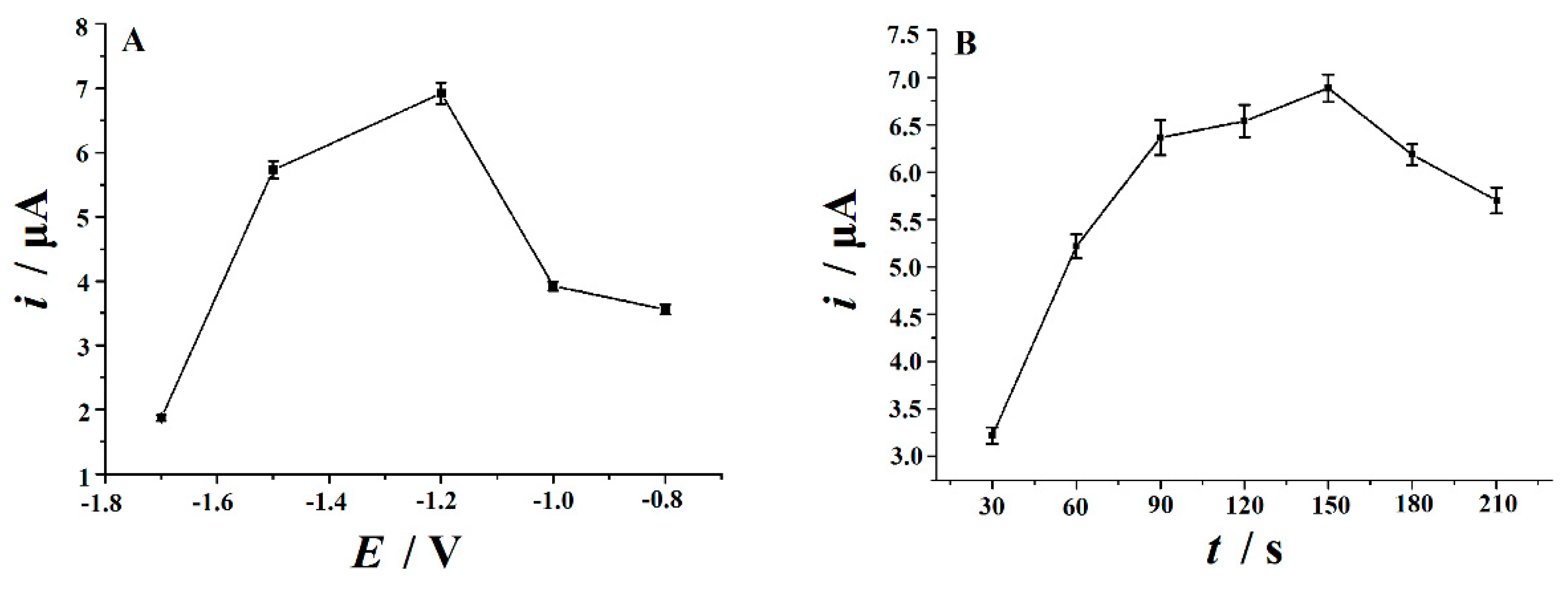
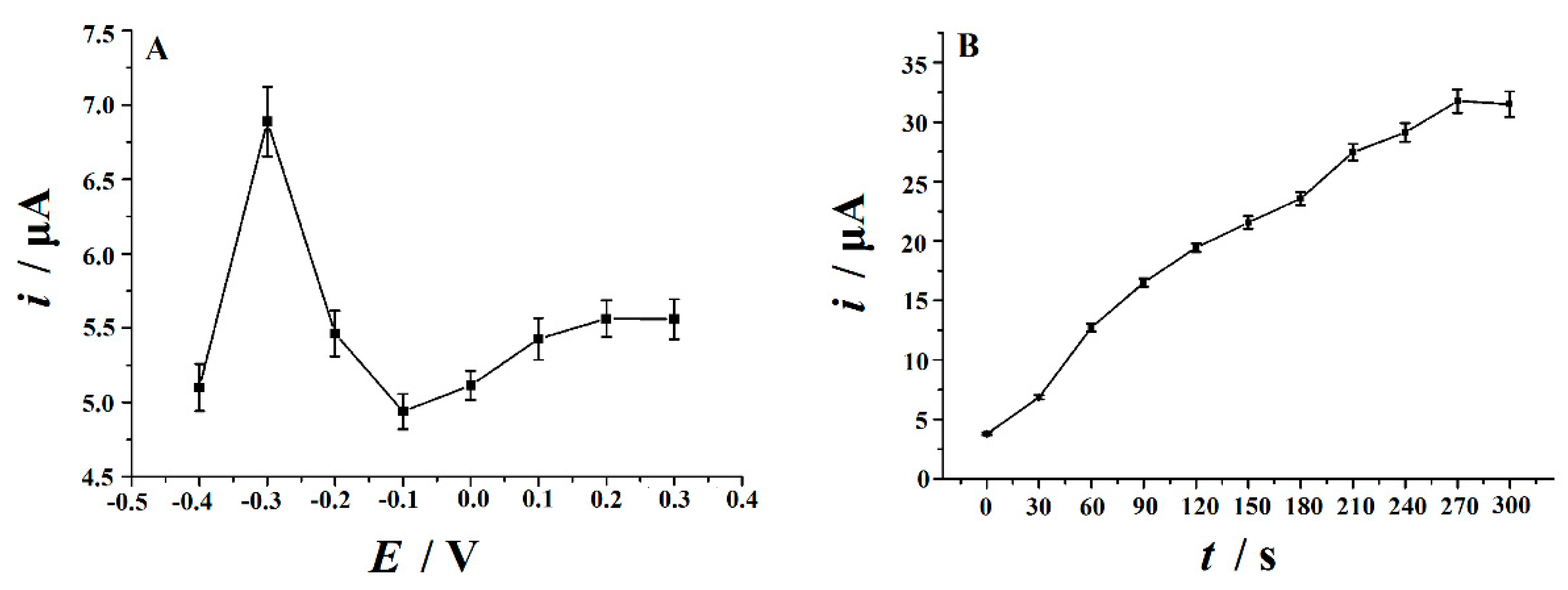
3.6. Optimization of Parameters
3.6.1. Electrochemical Reduction Conditions
3.6.2. Accumulation Conditions
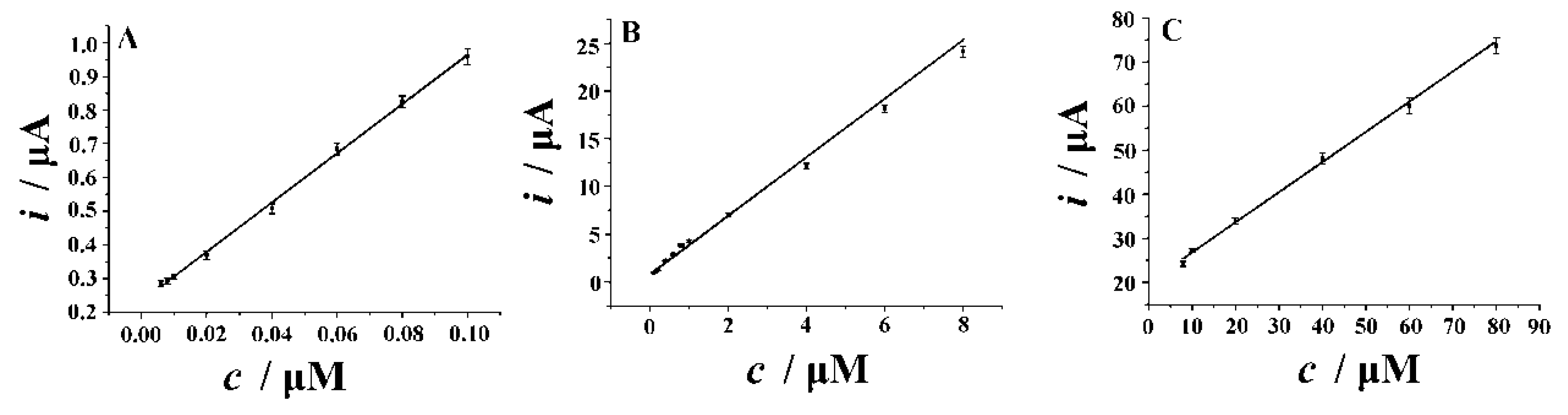
3.7. Figures of Merit
3.7.1. The Study of Interferences
3.7.2. Calibration and Detection Limit
3.7.3. Repeatability and Stability of the Decorated Electrode
3.8. Analytical Application
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tian, Q.; Li, D.; Patil, B.S. Identification and determination of flavonoids in buckwheat (Fagopyrum esculentum Moench, Polygonaceae) by high-performance liquid chromatography with electrospray ionisation mass spectrometry and photodiode array ultraviolet detection. Phytochem. Anal. 2002, 13, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Koval’skii, I.V.; Krasnyuk, I.I.; Nikulina, O.I.; Belyatskaya, A.V.; Kharitonov, Y.Y.; Feldman, N.B.; Lutsenko, S.V. Mechanisms of rutin pharmacological action (review). Pharm. Chem. J. 2014, 48, 73–76. [Google Scholar] [CrossRef]
- Sharma, S.; Ali, A.; Ali, J.; Sahni, J.K.; Baboota, S. Rutin: Therapeutic potential and recent advances in drug delivery. Expert Opin. Investig. Drugs 2013, 22, 1063–1079. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, M.; Fan, Y.; Elsebaei, F.; Zhu, Y. Determination of rutin and quercetin in Chinese herbal medicine by ionic liquid-based pressurized liquid extraction-liquid chromatography-chemiluminescence detection. Talanta 2012, 88, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Yıldırım, S.; Kadıoğlu, A.; Sağlam, A.; Yaşar, A. Determination of phenolic acids and rutin in Heliotropium thermophilum by high-performance liquid chromatography with photodiode array detection. Instrum. Sci. Technol. 2017, 45, 35–48. [Google Scholar] [CrossRef]
- Song, Z.; Hou, S. Sensitive determination of sub-nanogram amounts of rutin by its inhibition on chemiluminescence with immobilized reagents. Talanta 2002, 57, 59–67. [Google Scholar] [CrossRef]
- Du, J.; Li, Y.; Lu, J. Flow injection chemiluminescence determination of rutin based on its enhancing effect on the luminol-ferricyanide/ferrocyanide system. Anal. Lett. 2001, 34, 1741–1748. [Google Scholar] [CrossRef]
- Memon, A.F.; Solangi, A.R.; Memon, S.Q.; Mallah, A.; Memon, N.; Memon, A.A. Simultaneous determination of quercetin, rutin, naringin, and naringenin in different fruits by capillary zone electrophoresis. Food Anal. Methods 2017, 10, 83–91. [Google Scholar] [CrossRef]
- Xu, H.; Li, Y.; Tang, H.W.; Liu, C.M.; Wu, Q.S. Determination of rutin with UV-vis spectrophotometric and laser-induced fluorimetric detections using a non-scanning spectrometer. Anal. Lett. 2010, 43, 893–904. [Google Scholar] [CrossRef]
- Lu, Y.; Gao, B.; Chen, P.; Charles, D.; Yu, L. Characterisation of organic and conventional sweet basil leaves using chromatographic and flow-injection mass spectrometric (FIMS) fingerprints combined with principal component analysis. Food Chem. 2014, 154, 262–268. [Google Scholar] [CrossRef]
- Duan, L.; Yang, L.; Xiong, H.; Zhang, X.; Wang, S. Studies on the electrochemistry of rutin and its interaction with bovine serum albumin using a glassy carbon electrode modified with carbon-coated nickel nanoparticles. Microchim. Acta 2013, 180, 355–361. [Google Scholar] [CrossRef]
- Apetrei, I.M.; Apetrei, C. A modified nanostructured graphene-gold nanoparticle carbon screenprinted electrode for the sensitive voltammetric detection of rutin. Measurement 2018, 114, 37–43. [Google Scholar] [CrossRef]
- Gao, F.; Qi, X.; Cai, X.; Wang, Q.; Gao, F.; Sun, W. Electrochemically reduced graphene modified carbon ionic liquid electrode for the sensitive sensing of rutin. Thin Solid Films 2012, 520, 5064–5069. [Google Scholar] [CrossRef]
- Gholivand, M.B.; Mohammadi-Behzad, L.; Hosseinkhani, H. Application of a Cu–chitosan/multiwalled carbon nanotube film-modified electrode for the sensitive determination of rutin. Anal. Biochem. 2016, 493, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Weng, W.; Yin, C.; Niu, Y.; Li, G.; Dong, R.; Men, Y.; Sun, W. Black phosphorene modified glassy carbon electrode for the sensitive voltammetric detection of rutin. J. Electroanal. Chem. 2018, 811, 78–83. [Google Scholar] [CrossRef]
- Sun, W.; Wang, X.; Zhu, H.; Sun, X.; Shi, F.; Li, G.; Sun, Z. Graphene-MnO2 nanocomposite modified carbon ionic liquid electrode for the sensitive electrochemical detection of rutin. Sens. Actuators B 2013, 178, 443–449. [Google Scholar] [CrossRef]
- Yang, S.; Li, G.; Zhao, J.; Zhu, H.; Qu, L. Electrochemical preparation of Ag nanoparticles/poly(methylene blue) functionalized graphene nanocomposite film modified electrode for sensitive determination of rutin. J. Electroanal. Chem. 2014, 717–718, 225–230. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, J. Sensitive voltammetric determination of rutin at an ionic liquid modified carbon paste electrode. Talanta 2008, 77, 325–330. [Google Scholar] [CrossRef]
- Deng, P.; Xu, Z.; Li, J. Simultaneous determination of ascorbic acid and rutin in pharmaceutical preparations with electrochemical method based on multi-walled carbon nanotubes-chitosan composite film modified electrode. J. Pharm. Biomed. Anal. 2013, 76, 234–242. [Google Scholar] [CrossRef]
- Deng, P.; Xu, Z.; Feng, Y. Highly sensitive and simultaneous determination of ascorbic acid and rutin at an acetylene black paste electrode coated with cetyltrimethyl ammonium bromide film. J. Electroanal. Chem. 2012, 683, 47–54. [Google Scholar] [CrossRef]
- Park, S.; Ruoff, R.S. Chemical methods for the production of graphenes. Nat. Nanotechnol. 2009, 4, 217–224. [Google Scholar] [CrossRef]
- He, Q.; Liu, J.; Liu, X.; Li, G.; Deng, P.; Liang, J. Manganese dioxide Nanorods/electrochemically reduced graphene oxide nanocomposites modified electrodes for cost-effective and ultrasensitive detection of Amaranth. Colloids Surf. B Biointerfaces 2018, 172, 565–572. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liu, X.; Li, G.; Chen, D.; Deng, P.; Liang, J. A promising sensing platform toward dopamine using MnO2 nanowires/electro-reduced graphene oxide composites. Electrochim. Acta 2019, 296, 683–692. [Google Scholar] [CrossRef]
- He, Q.; Liu, J.; Liang, J.; Liu, X.; Li, W.; Liu, Z.; Ding, Z.; Tuo, D. Towards Improvements for Penetrating the Blood–Brain Barrier—Recent Progress from a Material and Pharmaceutical Perspective. Cells 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liu, X.; Li, G.; Deng, P.; Liang, J.; Chen, D. Sensitive and Selective Detection of Tartrazine Based on TiO2-Electrochemically Reduced Graphene Oxide Composite-Modified Electrodes. Sensors 2018, 18, 1911. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liu, X.; Xia, Y.; Li, G.; Deng, P.; Chen, D. Novel Electrochemical Sensors Based on Cuprous Oxide-Electrochemically Reduced Graphene Oxide Nanocomposites Modified Electrode toward Sensitive Detection of Sunset Yellow. Molecules 2018, 23, 2130. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liu, X.; Li, G.; Chen, D.; Deng, P.; Liang, J. Fabrication of Amine-Modified Magnetite-Electrochemically Reduced Graphene Oxide Nanocomposite Modified Glassy Carbon Electrode for Sensitive Dopamine Determination. Nanomaterials 2018, 8, 194. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; He, Q.; Luo, X.; Wang, M.; Liu, D.; Wang, J.; Li, G.; Liu, J. Determination of Uric Acid in Co-Presence of Dopamine and Ascorbic Acid Using Cuprous Oxide Nanoparticle-Functionalized Graphene Decorated Glassy Carbon Electrode. Catalysts 2018, 8, 407. [Google Scholar] [CrossRef]
- Ning, J.; He, Q.; Luo, X.; Wang, M.; Liu, D.; Wang, J.; Liu, J.; Li, G. Rapid and Sensitive Determination of Vanillin Based on a Glassy Carbon Electrode Modified with Cu2O-Electrochemically Reduced Graphene Oxide Nanocomposite Film. Sensors 2018, 18, 2762. [Google Scholar] [CrossRef]
- He, Q.; Liu, J.; Liu, X.; Li, G.; Deng, P.; Liang, J. Preparation of Cu2O-reduced graphene nanocomposite modified electrodes towards ultrasensitive dopamine detection. Sensors 2018, 18, 199. [Google Scholar] [CrossRef]
- He, Q.; Liu, J.; Liang, J.; Liu, X.; Tuo, D.; Li, W. Chemically surface tunable solubility parameter for controllable drug delivery-an example and perspective from hollow PAA-coated magnetite nanoparticles with R6G model drug. Materials 2018, 11, 247. [Google Scholar] [CrossRef]
- He, Q.; Liu, J.; Liang, J.; Liu, X.; Ding, Z.; Tuo, D.; Li, W. Sodium acetate orientated hollow/mesoporous magnetite nanoparticles: Facile synthesis, characterization and formation mechanism. Appl. Sci. 2018, 8, 292. [Google Scholar] [CrossRef]
- Liu, J.; Huang, C.; He, Q. Pharmaceutical application of magnetic iron oxide nanoparticles. Sci. Adv. Mater. 2015, 7, 672–685. [Google Scholar] [CrossRef]
- Ye, Y.; Kong, T.; Yu, X.; Wu, Y.; Zhang, K.; Wang, X. Enhanced nonenzymatic hydrogen peroxide sensing with reduced graphene oxide/ferroferric oxide nanocomposites. Talanta 2012, 89, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zheng, J.; Shi, J.; Lin, Z.; Huang, Q.; Zhang, H.; Wei, C.; Chen, J.; Hu, S.; Hao, A. Nafion covered core–shell structured Fe3O4@graphene nanospheres modified electrode for highly selective detection of dopamine. Anal. Chim. Acta 2015, 853, 285–290. [Google Scholar] [CrossRef]
- Yin, H.; Zhou, Y.; Ma, Q.; Ai, S.; Chen, Q.; Zhu, L. Electrocatalytic oxidation behavior of guanosine at graphene, chitosan and Fe3O4 nanoparticles modified glassy carbon electrode and its determination. Talanta 2010, 82, 1193–1199. [Google Scholar] [CrossRef]
- Teymourian, H.; Salimi, A.; Khezrian, S. Fe3O4 magnetic nanoparticles/reduced graphene oxide nanosheets as a novel electrochemical and bioeletrochemical sensing platform. Biosens. Bioelectron. 2013, 49, 1–8. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, W.; Yu, H.; Hou, C.; Li, D.; Zhang, Y.; Liu, Y. Controlled synthesis various shapes Fe3O4 decorated reduced graphene oxide applied in the electrochemical detection. J. Alloy Compd. 2015, 638, 182–187. [Google Scholar] [CrossRef]
- He, Q.; Lang, J.; Li, G.; Deng, P.; Liu, J.; Liu, X. Electrochemical Detection of Dopamine Based on MnO2 Nanowires/Reduced Graphene Oxide Composites Modified Glassy Carbon Electrode. Chin. J. Anal. Chem. 2018, 46, 438–445. [Google Scholar]
- Wu, D.; Li, Y.; Zhang, Y.; Wang, P.; Wei, Q.; Du, B. Sensitive electrochemical sensor for simultaneous determination of dopamine, ascorbic Acid, and uric acid enhanced by amino-group functionalized mesoporous Fe3O4@graphene sheets. Electrochim. Acta 2014, 116, 244–249. [Google Scholar] [CrossRef]
- Guo, S.; Li, D.; Zhang, L.; Li, J.; Wang, E. Monodisperse mesoporous superparamagnetic single-crystal magnetite nanoparticles for drug delivery. Biomaterials 2009, 30, 1881–1889. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, G.; Li, G.; Du, J.; Qu, L. Decoration of graphene modified carbon paste electrode with flower-globular terbium hexacyanoferrate for nanomolar detection of rutin. Electrochim. Acta 2014, 144, 268–274. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals Applications, 2nd ed.; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Guo, H.L.; Wang, X.F.; Qian, Q.Y.; Wang, F.B.; Xia, X.H. A green approach to the synthesis of graphene nanosheets. ACS Nano 2009, 3, 2653–2659. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Jin, B. The electrochemical behavior of AA and DA on graphene oxide modified electrodes containing various content of oxygen functional groups. J. Electroanal. Chem. 2011, 661, 77–83. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Working Electrodes | ipa/µA | ipc/µA | Epa/V | Epc/V | ipa/ipc | ΔEp |
|---|---|---|---|---|---|---|
| a GCE | 1.431 | 1.093 | 0.674 | 0.653 | 1.309 | 0.021 |
| b GO/GCE | 0.5843 | 0.309 | 0.676 | 0.651 | 1.890 | 0.025 |
| c NH2-Fe3O4 NPs-GO/GCE | 2.396 | 1.929 | 0.678 | 0.657 | 1.242 | 0.021 |
| d ErGO/GCE | 14.35 | 13.58 | 0.678 | 0.661 | 1.057 | 0.017 |
| e NH2-Fe3O4 NPs-ErGO/GCE | 21.16 | 17.00 | 0.663 | 0.648 | 1.245 | 0.015 |
| Electrolyte | Peak Current (μA) | Peak Potential (V) |
|---|---|---|
| HAc-NaAc buffer (pH 4.0) | 4.275 | 0.540 |
| HAc-NH4Ac (pH 4.0) | 4.645 | 0.564 |
| (CH2)6N4-HCl (pH 5.0) | 3.688 | 0.528 |
| phosphate buffer (pH 6.0) | 3.951 | 0.480 |
| NH3-NH4Cl (pH 9.0) | 1.264 | 0.432 |
| HCl | 6.200 | 0.648 |
| HNO3 | 6.418 | 0.660 |
| H2SO4 | 6.863 | 0.664 |
| NaOH | 0.645 | 0.328 |
| Coexisting Substance | Concentration/mM | Change of Peak Current/µA | RE/% | Coexisting Substance | Concentration/mM | Change of Peak Current/µA | RE/% |
|---|---|---|---|---|---|---|---|
| glucose | 1 | 0.0546 | 1.28 | uric acid | 0.1 | 0.1761 | 4.13 |
| citric acid | 1 | 0.0674 | 1.58 | Mn2+ | 1 | −0.0942 | −2.21 |
| glutamate | 1 | 0.1006 | 2.36 | Zn2+ | 1 | 0.1530 | 3.59 |
| arginine | 1 | 0.1351 | 3.17 | Ca2+ | 1 | −0.1343 | −3.15 |
| tryptophan | 0.1 | 0.1842 | 4.32 | Mg2+ | 1 | −0.0840 | −1.97 |
| tyrosine | 0.1 | 0.1543 | 3.62 | Pb2+ | 1 | 0.1100 | 2.58 |
| glycine | 1 | 0.0503 | 1.18 | Cu2+ | 1 | 0.1778 | 4.17 |
| lactose | 1 | 0.0678 | 1.59 | Al3+ | 1 | −0.0972 | −2.28 |
| lactic acid | 1 | 0.1006 | 2.36 | K+ | 1 | −0.0495 | 1.16 |
| starch | 1 | −0.1215 | −2.85 | Na+ | 1 | 0.0622 | 1.46 |
| sucrose | 1 | 0.1177 | 2.76 | PO43− | 1 | 0.0925 | 2.17 |
| dopamine | 1 | 0.2063 | 4.84 | NO3− | 1 | 0.0674 | 1.58 |
| ascorbic acid | 1 | −0.1253 | −2.94 | Cl− | 1 | −0.0657 | −1.54 |
| Electrochemical Sensors | Technique | Supporting Electrolyte | Linear Range/µM | Correlation Coefficient | Detection Limit/µM | References |
|---|---|---|---|---|---|---|
| a GR–AuNPs/CSPE | j SWV | HAc-NaAc buffer (pH 5.0) | 0.1–15 | 0.9988 | 0.011 | [12] |
| b GR/CILE | k DPV | PBS buffer (pH 2.5) | 0.07–10; 10–100 | 0.997, 0.991 | 0.024 | [13] |
| c Cu–CS/MWCNT/GCE | DPV | B-R buffer (pH 3) | 0.05–100 | 0.9994 | 0.01 | [14] |
| d BP–PEDOT:PSS/GCE | DPV | PBS buffer (pH 2.5) | 0.02–15.0; 15.0–80.0 | 0.998, 0.996 | 0.007 | [15] |
| e GR-MnO2/CILE | DPV | PBS buffer (pH 2.5) | 0.01–500.0 | 0.992 | 0.00273 | [16] |
| f AgNPs/PMB–GR/Au electrode | DPV | PBS buffer (pH 3.0) | 0.01–0.1; 0.1–2.0; 2.0–10.0 | 0.9947, 0.9959, 0.9958 | 0.01 | [17] |
| g IL/CPE | SWV | B-R buffer (pH 3.29) | 0.04–10 | 0.9984 | 0.01 | [18] |
| h MWCNTs–CHIT/ABPE | SDLSV | 0.1 M HCl | 0.02–10 | 0.9994 | 0.01 | [19] |
| i CTAB/ABPE | SDLSV | 0.1 M HCl | 0.006–0.2; 0.2–10 | 0.9985, 0.9992 | 0.004 | [20] |
| NH2-Fe3O4 NPs-ErGO/GCE | SDLSV | 0.2 M H2SO4 | 0.006–0.1; 0.1–8.0; 8.0–80 | 0.004 | This work |
| Sample | Labelled/µM | d Detected by UV Method/µM | d Detected by this method/µM | Added/µM | d Total Found by this Method/µM | Recovery/% |
|---|---|---|---|---|---|---|
| a Tablet 1 | 32.76 | 32.95 ± 0.94 | 31.48 ± 0.86 | 30.00 | 61.13 ± 2.14 | 98.8 |
| b Tablet 2 | 3.28 | 3.08 ± 0.11 | 3.37 ± 0.15 | 3.00 | 6.48 ± 0.20 | 103.7 |
| Urine 1 c | - | e ND | ND | 5.00 | 5.07 ± 0.18 | 101.4 |
| Urine 2 c | - | ND | ND | 1.00 | 1.03 ± 0.03 | 103.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Q.; Wu, Y.; Tian, Y.; Li, G.; Liu, J.; Deng, P.; Chen, D. Facile Electrochemical Sensor for Nanomolar Rutin Detection Based on Magnetite Nanoparticles and Reduced Graphene Oxide Decorated Electrode. Nanomaterials 2019, 9, 115. https://doi.org/10.3390/nano9010115
He Q, Wu Y, Tian Y, Li G, Liu J, Deng P, Chen D. Facile Electrochemical Sensor for Nanomolar Rutin Detection Based on Magnetite Nanoparticles and Reduced Graphene Oxide Decorated Electrode. Nanomaterials. 2019; 9(1):115. https://doi.org/10.3390/nano9010115
Chicago/Turabian StyleHe, Quanguo, Yiyong Wu, Yaling Tian, Guangli Li, Jun Liu, Peihong Deng, and Dongchu Chen. 2019. "Facile Electrochemical Sensor for Nanomolar Rutin Detection Based on Magnetite Nanoparticles and Reduced Graphene Oxide Decorated Electrode" Nanomaterials 9, no. 1: 115. https://doi.org/10.3390/nano9010115
APA StyleHe, Q., Wu, Y., Tian, Y., Li, G., Liu, J., Deng, P., & Chen, D. (2019). Facile Electrochemical Sensor for Nanomolar Rutin Detection Based on Magnetite Nanoparticles and Reduced Graphene Oxide Decorated Electrode. Nanomaterials, 9(1), 115. https://doi.org/10.3390/nano9010115