Morphology, Optical Properties and Photocatalytic Activity of Photo- and Plasma-Deposited Au and Au/Ag Core/Shell Nanoparticles on Titania Layers

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
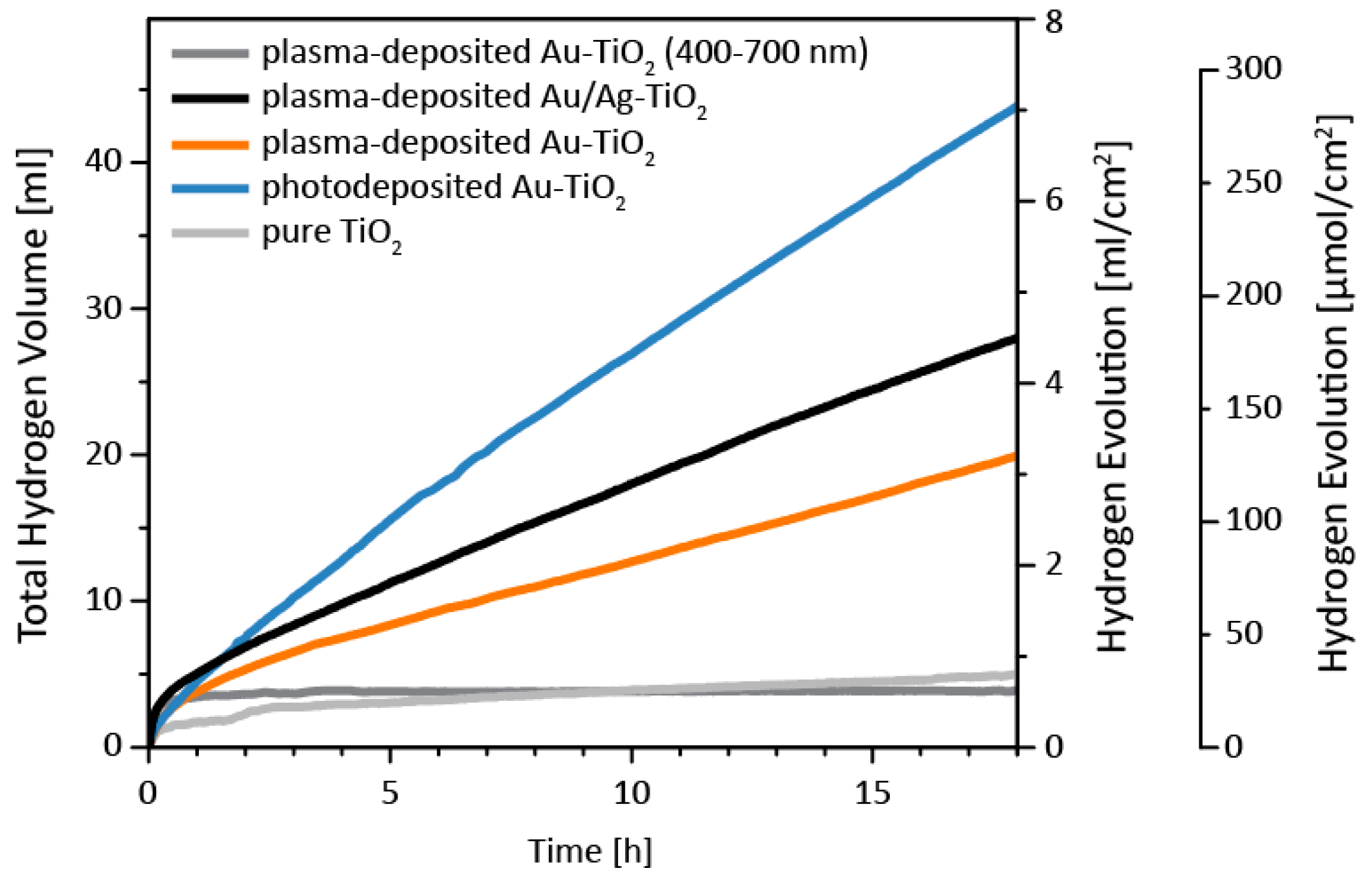
3. Results and Discussion
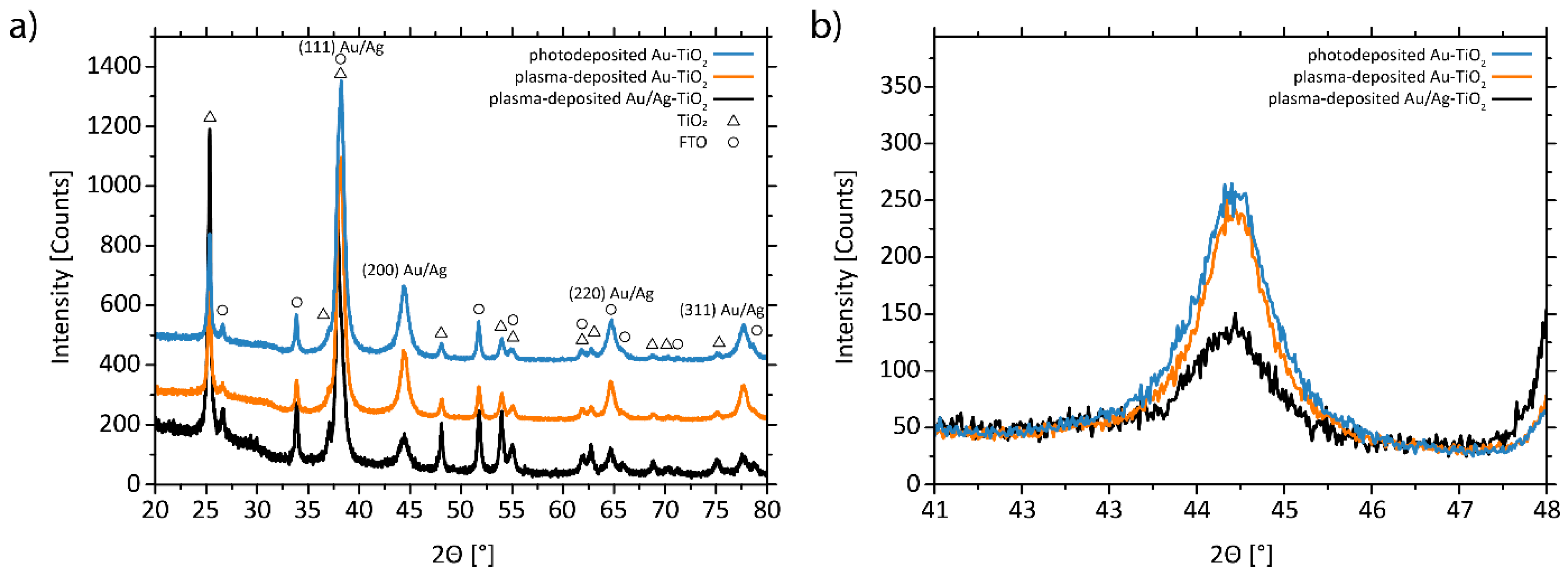
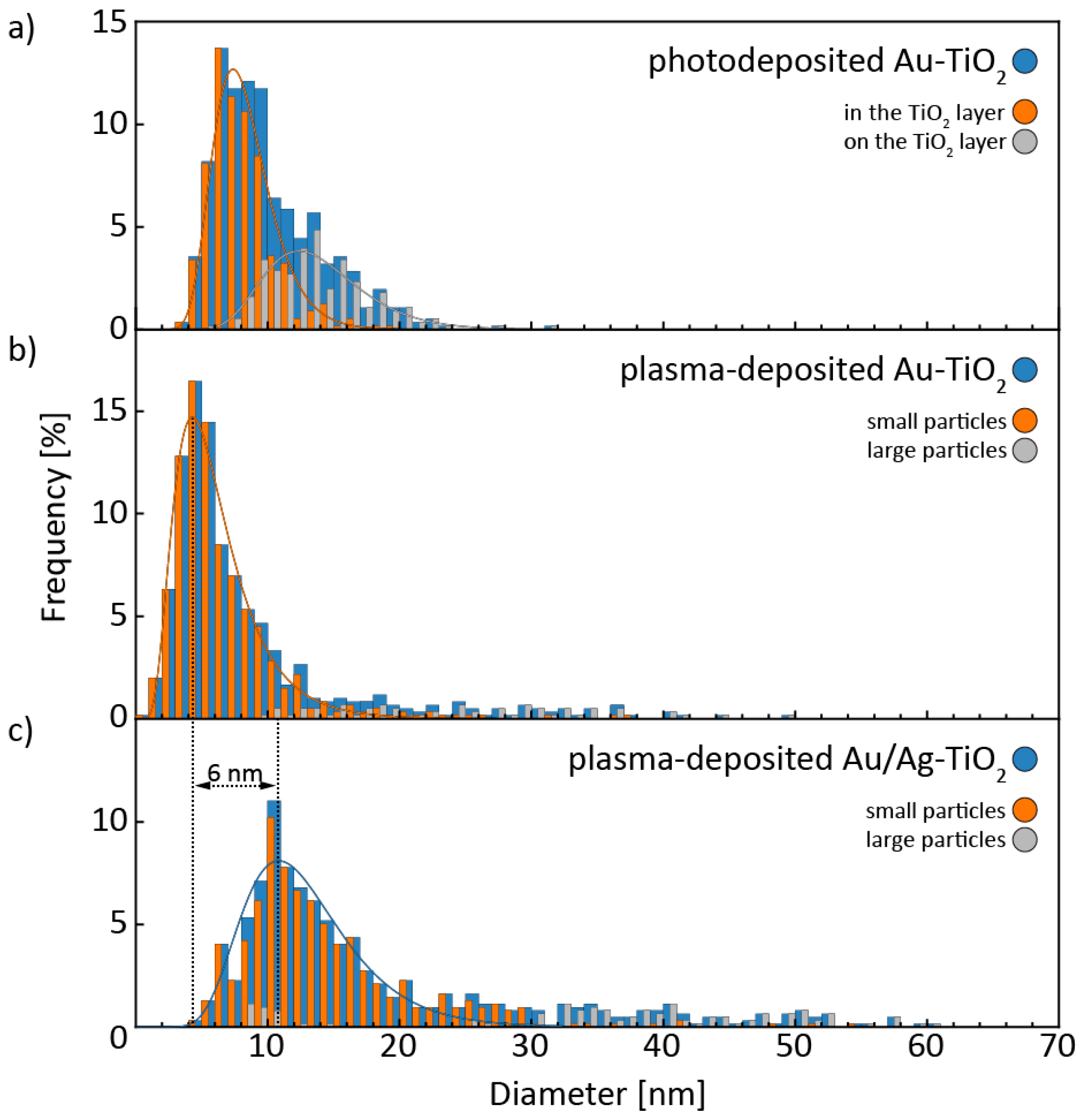
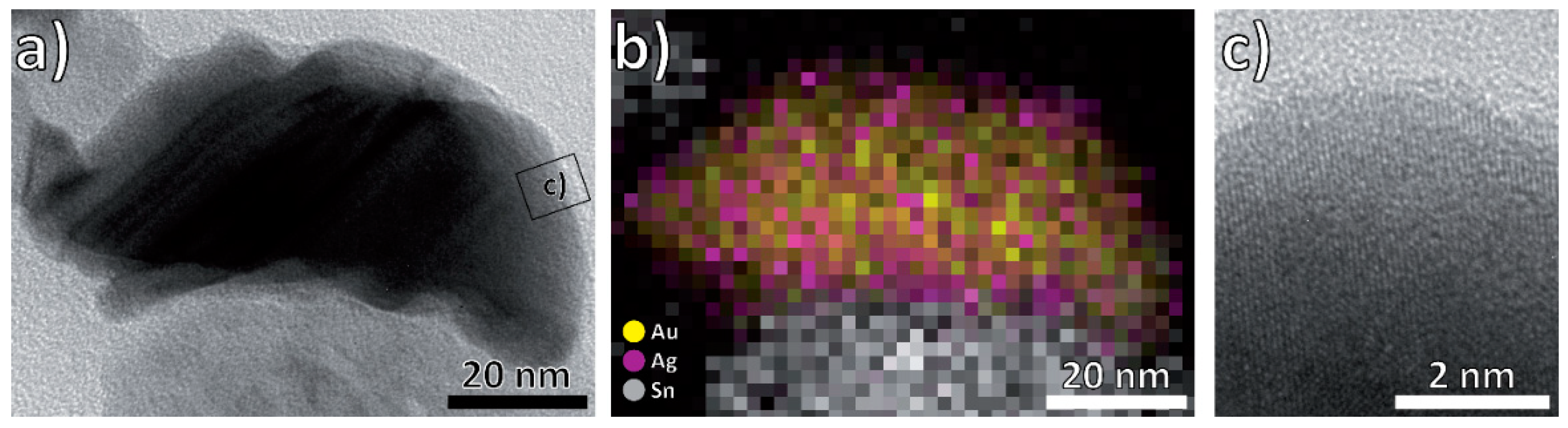
3.1. Structural and Morphological Characterization
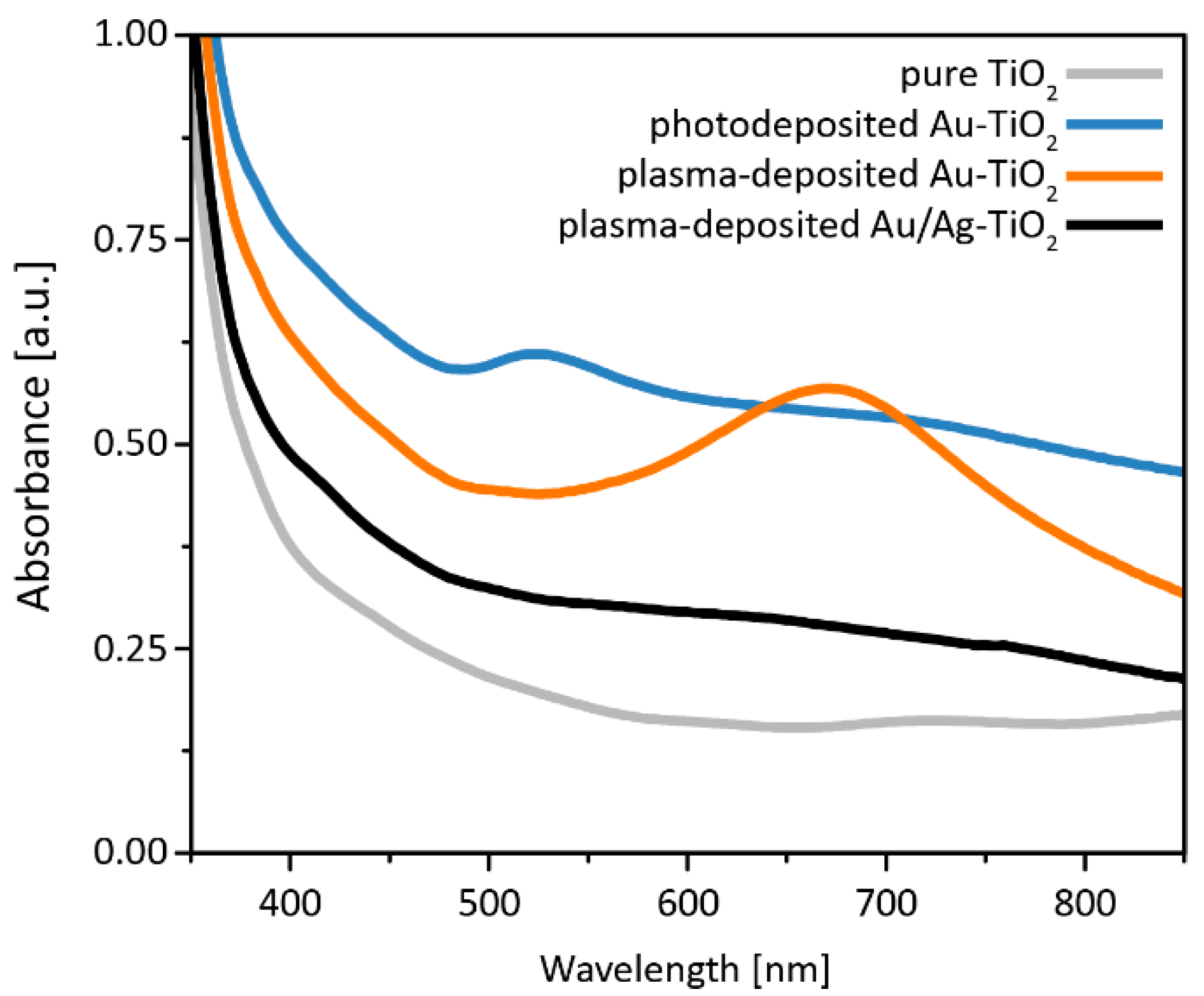
3.2. Optical Properties
3.3. Catalytic Properties
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Armaroli, N.; Balzani, V. The future of energy supply: Challenges and opportunities. Angew. Chem. Int. Ed. 2007, 46, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Schiermeier, Q.; Tollefson, J.; Scully, T.; Witze, A.; Morton, O. Electricity without carbon. Nature 2008, 5, 10–15. [Google Scholar] [CrossRef]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Styring, S. Artificial photosynthesis for solar fuels. Faraday Discuss. 2012, 155, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, J.J.; House, R.L.; Papanikolas, J.M.; Meyer, T.J. Chemical approaches to artificial photosynthesis. Proc. Natl. Acad. Sci. USA 2012, 109, 15560–15564. [Google Scholar] [CrossRef] [PubMed]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Amaranatha Reddy, D.; Han, N.S.; Jeong, S.; Hong, S.; Praveen Kumar, D.; Song, J.K.; Kim, T.K. Modulation of charge carrier pathways in CdS nanospheres by integrating MoS2 and Ni2P for improved migration and separation toward enhanced photocatalytic hydrogen evolution. Catal. Sci. Technol. 2017, 7, 641–649. [Google Scholar] [CrossRef]
- Reddy, D.A.; Park, H.; Gopannagari, M.; Kim, E.H.; Lee, S.; Kumar, D.P.; Kim, T.K. Designing CdS Mesoporous Networks on Co-C@Co9S8 Double-Shelled Nanocages as Redox-Mediator-Free Z-Scheme Photocatalyst. ChemSusChem 2018, 11, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.A.; Park, H.; Ma, R.; Kumar, D.P.; Lim, M.; Kim, T.K. Heterostructured WS2-MoS2 Ultrathin Nanosheets Integrated on CdS Nanorods to Promote Charge Separation and Migration and Improve Solar-Driven Photocatalytic Hydrogen Evolution. ChemSusChem 2017, 10, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.A.; Choi, J.; Lee, S.; Kim, Y.; Hong, S.; Kumar, D.P.; Kim, T.K. Hierarchical dandelion-flower-like cobalt-phosphide modified CdS/reduced graphene oxide-MoS2 nanocomposites as a noble-metal-free catalyst for efficient hydrogen evolution from water. Catal. Sci. Technol. 2016, 6, 6197–6206. [Google Scholar] [CrossRef]
- Reddy, D.A.; Park, H.; Hong, S.; Kumar, D.P.; Kim, T.K. Hydrazine-assisted formation of ultrathin MoS2 nanosheets for enhancing their co-catalytic activity in photocatalytic hydrogen evolution. J. Mater. Chem. A 2017, 5, 6981–6991. [Google Scholar] [CrossRef]
- Reddy, D.A.; Kim, H.K.; Kim, Y.; Lee, S.; Choi, J.; Islam, M.J.; Kumar, D.P.; Kim, T.K. Multicomponent transition metal phosphides derived from layered double hydroxide double-shelled nanocages as an efficient non-precious co-catalyst for hydrogen production. J. Mater. Chem. A 2016, 4, 13890–13898. [Google Scholar] [CrossRef]
- Scanlon, D.O.; Dunnill, C.W.; Buckeridge, J.; Shevlin, S.A.; Logsdail, A.J.; Woodley, S.M.; Catlow, C.R.A.; Powell, M.J.; Palgrave, R.G.; Parkin, I.P.; et al. Band alignment of rutile and anatase TiO2. Nat. Mater. 2013, 12, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.M.; Manorama, S.V.; Reddy, A.R. Bandgap studies on anatase titanium dioxide nanoparticles. Mater. Chem. Phys. 2003, 78, 239–245. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, P.; Liu, J.; Yu, J. New understanding of the difference of photocatalytic activity among anatase, rutile and brookite TiO2. Phys. Chem. Chem. Phys. 2014, 16, 20382–20386. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, S.; Guo, L.; Mao, S.S. Semiconductor-based Photocatalytic Hydrogen Generation. Chem. Rev. 2010, 110, 6503–6570. [Google Scholar] [CrossRef] [PubMed]
- Primo, A.; Corma, A.; García, H. Titania supported gold nanoparticles as photocatalyst. Phys. Chem. Chem. Phys. 2011, 13, 886–910. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Rasul, M.G.; Brown, R.; Hashib, M.A. Influence of parameters on the heterogeneous photocatalytic degradation of pesticides and phenolic contaminants in wastewater: A short review. J. Environ. Manag. 2011, 92, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Lu, Y.; Ren, Z. Mini Review on photocatalysis of titanium dioxide nanoparticles and their solar applications. Nano Energy 2013, 2, 1031–1045. [Google Scholar] [CrossRef]
- Pu, Y.C.; Wang, G.; Chang, K.D.; Ling, Y.; Lin, Y.K.; Fitzmorris, B.C.; Liu, C.M.; Lu, X.; Tong, Y.; Zhang, J.Z.; et al. Au nanostructure-decorated TiO2 nanowires exhibiting photoactivity across entire UV-visible region for photoelectrochemical water splitting. Nano Lett. 2013, 13, 3817–3823. [Google Scholar] [CrossRef] [PubMed]
- Linic, S.; Christopher, P.; Ingram, D.B. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 2011, 10, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Astruc, D. Nanogold plasmonic photocatalysis for organic synthesis and clean energy conversion. Chem. Soc. Rev. 2014, 43, 7188–7216. [Google Scholar] [CrossRef] [PubMed]
- Furube, A.; Du, L.; Hara, K.; Katoh, R.; Tachiya, M. Ultrafast plasmon-induced electron transfer from gold nanodots into TiO2 nanoparticles. J. Am. Chem. Soc. 2007, 129, 14852–14853. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, W.J.; Lee, S.A.; Maeda, K.; Mallouk, T.E. Visible Light Water Splitting Using Dye- Sensitized Oxide Semiconductors. Acc. Chem. Res. 2009, 42, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Yum, J.-H.; Chen, P.; Grätzel, M.; Nazeeruddin, M.K. Recent developments in solid-state dye-sensitized solar cells. ChemSusChem 2008, 1, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Kruth, A.; Quade, A.; Brüser, V.; Weltmann, K.-D. Plasma-Enhanced Synthesis of Poly (allylamine)-Encapsulated Ruthenium Dye-Sensitized Titania Photocatalysts. J. Phys. Chem. C 2013, 117, 3804–3811. [Google Scholar] [CrossRef]
- Kruth, A.; Peglow, S.; Rockstroh, N.; Junge, H.; Brüser, V.; Weltmann, K.D. Enhancement of photocatalyic activity of dye sensitised anatase layers by application of a plasma-polymerized allylamine encapsulation. J. Photochem. Photobiol. A Chem. 2014, 290, 31–37. [Google Scholar] [CrossRef]
- Kruth, A.; Hansen, S.; Beweries, T.; Brüser, V.; Weltmann, K.D. Plasma synthesis of polymer-capped dye-sensitised anatase nanopowders for visible-light-driven hydrogen evolution. ChemSusChem 2013, 6, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Karnahl, M.; Mejía, E.; Rockstroh, N.; Tschierlei, S.; Luo, S.P.; Grabow, K.; Kruth, A.; Brüser, V.; Junge, H.; Lochbrunner, S.; et al. Photocatalytic hydrogen production with copper photosensitizer-titanium dioxide composites. ChemCatChem 2014, 6, 82–86. [Google Scholar] [CrossRef]
- Warren, S.C.; Thimsen, E. Plasmonic solar water splitting. Energy Environ. Sci. 2012, 5, 5133–5146. [Google Scholar] [CrossRef]
- Petryayeva, E.; Krull, U.J. Localized surface plasmon resonance: Nanostructures, bioassays and biosensing—A review. Anal. Chim. Acta 2011, 706, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Noguez, C. Surface Plasmons on Metal Nanoparticles: The Influence of Shape and Physical Environment. J. Phys. Chem. C 2007, 111, 3806–3819. [Google Scholar] [CrossRef]
- Link, S.; El-Sayed, M.A. Shape and size dependence of radiative, non-radiative and photothermal properties of gold nanocrystals. Int. Rev. Phys. Chem. 2000, 19, 409–453. [Google Scholar] [CrossRef]
- Jain, P.K.; Lee, K.S.; El-Sayed, I.H.; El-Sayed, M.A. Calculated absorption and scattering properties of gold nanoparticles of different size, shape, and composition: Applications in biological imaging and biomedicine. J. Phys. Chem. B 2006, 110, 7238–7248. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Zhang, J.Z. Mechanisms Behind Plasmonic Enhancement of Photocurrent in Metal Oxides. Austin J. Nanomed. Nanotechnol. 2014, 2, 1–4. [Google Scholar]
- Cushing, S.K.; Li, J.; Meng, F.; Senty, T.R.; Suri, S.; Zhi, M.; Li, M.; Bristow, A.D.; Wu, N. Photocatalytic Activity Enhanced by Plasmonic Resonant Energy Transfer Transfer from Metal to Semiconductor. J. Am. Chem. Soc. 2012, 134, 105033–105041. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.C.; Dai, Y.; Yu, L.; Huang, B.B. Energy transfer in plasmonic photocatalytic composites. Light Sci. Appl. 2016, 5, 1–13. [Google Scholar] [CrossRef]
- Garcia, M.A. Surface plasmons in metallic nanoparticles: Fundamentals and applications. J. Phys. D Appl. Phys. 2012, 45, 389501. [Google Scholar] [CrossRef]
- Gärtner, F.; Losse, S.; Boddien, A.; Pohl, M.-M.; Denurra, S.; Junge, H.; Beller, M. Hydrogen evolution from water/Alcohol mixtures: Effective in situ generation of an active Au/TiO2 catalyst. ChemSusChem 2012, 5, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, D.; Han, H.; Li, C. Roles of cocatalysts in photocatalysis and photoelectrocatalysis. Acc. Chem. Res. 2013, 46, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Priebe, J.B.; Karnahl, M.; Junge, H.; Beller, M.; Hollmann, D.; Brückner, A. Water Reduction with Visible Light: Synergy between Optical Transitions and Electron Transfer in Au-TiO2 Catalysts Visualized by In situ EPR Spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 11420–11424. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Tatsuma, T. Plasmon-induced photoelectrochemistry at metal nanoparticles supported on nanoporous TiO2. Chem. Commun. 2004, 1810–1811. [Google Scholar] [CrossRef] [PubMed]
- Priebe, J.B.; Radnik, J.; Lennox, A.J.J.; Pohl, M.-M.; Karnahl, M.; Hollmann, D.; Grabow, K.; Bentrup, U.; Junge, H.; Beller, M.; et al. Solar Hydrogen Production by Plasmonic Au-TiO2 Catalysts: Impact of Synthesis Protocol and TiO2 Phase on Charge Transfer Efficiency and H2 Evolution Rates. ACS Catal. 2015, 5, 2137–2148. [Google Scholar] [CrossRef]
- Silva, C.G.; Juárez, R.; Marino, T.; Molinari, R.; García, H. Influence of Excitation Wavelength (UV or Visible Light) on the Photocatalytic Activity of Titania Containing Gold Nanoparticles for the Generation of Hydrogen or Oxygen from Water. J. Am. Chem. Soc. 2011, 133, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, E.; Rau, S.; Ohtani, B. Plasmonic titania photocatalysts active under UV and visible-light irradiation: Influence of gold amount, size, and shape. J. Nanotechnol. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Yan, J.; Wu, G.; Guan, N.; Li, L. Synergetic promotion of the photocatalytic activity of TiO2 by gold deposition under UV-visible light irradiation. Chem. Commun. 2013, 49, 11767–11769. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Tiruvalam, R.; Logsdail, A.J.; He, Q.; Downing, C.A.; Jensen, M.T.; Dimitratos, N.; Kesavan, L.; Wells, P.P.; Bechstein, R.; et al. Designer titania-supported Au-Pd nanoparticles for efficient photocatalytic hydrogen production. ACS Nano 2014, 8, 3490–3497. [Google Scholar] [CrossRef] [PubMed]
- Osterloh, F.E. Inorganic nanostructures for photoelectrochemical and photocatalytic water splitting. Chem. Soc. Rev. 2013, 42, 2294–2320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Gao, L.; Song, X.; Sun, J. Micro- and Nanostructures of Photoelectrodes for Solar-Driven Water Splitting. Adv. Mater. 2015, 27, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Wenderich, K.; Mul, G. Methods, Mechanism, and Applications of Photodeposition in Photocatalysis: A Review. Chem. Rev. 2016, 116, 14587–14619. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Haba, M.; Okumura, M.; Akita, T.; Hashimoto, S.; Toshima, N. Novel formation of Ag/Au bimetallic nanoparticles by physical mixture of monometallic nanoparticles in dispersions and their application to catalysts for aerobic glucose oxidation. Langmuir 2013, 29, 10330–10339. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Akita, T.; Ishida, T. Synergistic Catalysis of Au@Ag Core−Shell Nanoparticles Stabilized on Metal−Organic Framework. J. Am. Chem. Soc. 2011, 133, 1304–1306. [Google Scholar] [CrossRef] [PubMed]
- Kruth, A.; Peglow, S.; Quade, A.; Pohl, M.-M.; Weltmann, K.-D. Structural and Photoelectrochemical Properties of DC Magnetron- Sputtered TiO2 Layers on FTO. J. Phys. Chem. C 2014, 118, 25234–25244. [Google Scholar] [CrossRef]
- Peglow, S.; Pohl, M.-M.; Kruth, A.; Brüser, V. Plasma Based Synthesis, Electron Microscopy, and Optical Characterization of Au-, Ag-, and Ag/Au-Core−Shell Nanoparticles. J. Phys. Chem. C 2015, 119, 563–572. [Google Scholar] [CrossRef]
- Schneider, J.; Bahnemann, D.W. Undesired Role of Sacrificial Reagents in Photocatalysis. J. Phys. Chem. Lett. 2013, 4, 3479–3483. [Google Scholar] [CrossRef]
- Nada, A.A.; Barakat, M.H.; Hamed, H.A.; Mohamed, N.R.; Veziroglu, T.N. Studies on the photocatalytic hydrogen production using suspended modified TiO2 photocatalysts. Int. J. Hydrogen Energy 2005, 30, 687–691. [Google Scholar] [CrossRef]
- Kawai, T.; Sakata, T. Conversion of carbohydrate into hydrogen fuel by a photocatalytic process. Nature 1980, 286, 474–476. [Google Scholar] [CrossRef]
- Wulff, H. Introduction to Complex Plasmas; Bonitz, M., Horing, N., Ludwig, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Strecker, A.; Salzberger, U.; Mayer, J. Specimen Preparation for Transmission Electron Microscopy: Reliable Method for Cross-Sections and Brittle Materials. Prakt. Metallogr. 1993, 30, 482–495. [Google Scholar]
- Gottwald, W.; Heinrich, K.H. UV/VIS-Spektroskopie für Anwender; Wiley VCH: Weinheim, Germany, 1999. [Google Scholar]
- Djerdj, I.; Tonejc, A.M. Structural investigations of nanocrystalline TiO2 samples. J. Alloys Compd. 2006, 413, 159–174. [Google Scholar] [CrossRef]
- Suh, I.; Ohta, H.; Waseda, Y. High-temperature thermal expansion of six metallic elements measured by dilatation method and X-ray diffraction. J. Mater. Sci. 1988, 23, 757–760. [Google Scholar] [CrossRef]
- Yamanaka, T.; Kurashima, R.; Mimaki, J. X-ray diffraction study of bond character of rutile-type SiO2, GeO2 and SnO2. Z. Kristallogr. 2000, 215, 424–428. [Google Scholar] [CrossRef]
- Petrov, I.; Barna, P.B.; Hultman, L.; Greene, J.E. Microstructural evolution during film growth. J. Vac. Sci. Technol. A 2003, 21, 117–128. [Google Scholar] [CrossRef]
- Thornton, J.A. The microstructure of sputter-deposited coatings. J. Vac. Sci. Technol. A 1986, 4, 3059–3065. [Google Scholar] [CrossRef]
- Williams, D.; Carter, C. Transmission Electron Microscopy—A Textbook for Materials Science; Springer: Boston, MA, USA, 1996; ISBN 9780387765006. [Google Scholar]
- Claus, P.; Brückner, A.; Mohr, C.; Hofmeister, H. Supported Gold Nanoparticles from Quantum Dot to Mesoscopic Size Scale: Effect of Electronic and Structural Properties on Catalytic Hydrogenation of Conjugated Functional Groups. J. Am. Chem. Soc. 2000, 122, 11430–11439. [Google Scholar] [CrossRef]
- Söderlund, J.; Kiss, L.; Niklasson, G.; Granqvist, C. Lognormal Size Distributions in Particle Growth Processes without Coagulation. Phys. Rev. Lett. 1998, 80, 2386–2388. [Google Scholar] [CrossRef]
- Robson, J.D. Modelling the evolution of particle size distribution during nucleation, growth and coarsening. Mater. Sci. Technol. 2004, 20, 441–448. [Google Scholar] [CrossRef]
- Cleveland, C.; Luedtke, W.; Landman, U. Melting of Gold Clusters: Icosahedral Precursors. Phys. Rev. Lett. 1998, 81, 2036–2039. [Google Scholar] [CrossRef]
- Marks, L.D. Experimental studies of small particle structures. Rep. Prog. Phys. 1999, 57, 603–649. [Google Scholar] [CrossRef]
- Nam, H.-S.; Hwang, N.; Yu, B.; Yoon, J.-K. Formation of an Icosahedral Structure during the Freezing of Gold Nanoclusters: Surface-Induced Mechanism. Phys. Rev. Lett. 2002, 89, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ascencio, J.A.; Gutiérrez-Wing, C.; Espinosa, M.E.; Marín, M.; Tehuacanero, S.; Zorrilla, C.; José-Yacamán, M. Structure determination of small particles by HREM imaging: Theory and experiment. Surf. Sci. 1998, 396, 349–368. [Google Scholar] [CrossRef]
- Barnard, A.S. A thermodynamic model for the shape and stability of twinned nanostructures. J. Phys. Chem. B 2006, 110, 24498–24504. [Google Scholar] [CrossRef] [PubMed]
- Hubenthal, F.; Ziegler, T.; Hendrich, C.; Alschinger, M.; Träger, F. Tuning the surface plasmon resonance by preparation of gold-core/silver-shell and alloy nanoparticles. Eur. Phys. J. D 2005, 34, 165–168. [Google Scholar] [CrossRef]
- Douglas, F.; Yañez, R.; Ros, J.; Marín, S.; De La Escosura-Muñiz, A.; Alegret, S.; Merkoçi, A. Silver, gold and the corresponding core shell nanoparticles: Synthesis and characterization. J. Nanopart. Res. 2008, 10, 97–106. [Google Scholar] [CrossRef]
- Shyjumon, I.; Gopinadhan, M.; Ivanova, O.; Quaas, M.; Wulff, H.; Helm, C.A.; Hippler, R. Structural deformation, melting point and lattice parameter studies of size selected silver clusters. Eur. Phys. J. D 2006, 37, 409–415. [Google Scholar] [CrossRef]
- Buffat, P.; Borel, J.-P. Size Effect on the Melting Temperature of Gold Particles. Phys. Rev. A 1976, 13, 2287–2298. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhang, S.; Zhao, M. Size-dependent melting point of noble metals. Mater. Chem. Phys. 2003, 82, 225–227. [Google Scholar] [CrossRef]
- Jette, E.R.; Foote, F. Precision Determination of Lattice Constants. J. Chem. Phys. 1935, 3, 605–616. [Google Scholar] [CrossRef]
- Tsuji, M.; Yamaguchi, D.; Matsunaga, M.; Alam, M.J. Epitaxial growth of Au@Cu core-shell nanocrystals prepared using the PVP-assisted polyol reduction method. Cryst. Growth Des. 2010, 10, 5129–5135. [Google Scholar] [CrossRef]
- Fan, F.R.; Liu, D.Y.; Wu, Y.F.; Duan, S.; Xie, Z.X.; Jiang, Z.Y.; Tian, Z.Q. Epitaxial growth of heterogeneous metal nanocrystals: From gold nano-octahedra to palladium and silver nanocubes. J. Am. Chem. Soc. 2008, 130, 6949–6951. [Google Scholar] [CrossRef] [PubMed]
- Skriver, H.L.; Rosengaard, N.M. Surface energy and work function of elemental metals. Phys. Rev. B 1992, 46, 7157–7168. [Google Scholar] [CrossRef]
- Koo, J.B.; Hong, K.J.; Park, J.S.; Shin, D.C. Effect of grain size on transmittance and mechanical strength of sintered alumina. Mater. Sci. Eng. A 2004, 374, 191–195. [Google Scholar] [CrossRef]
- Apetz, R.; Bruggen, M.P.B. Van Transparent Alumina: A Light-Scattering Model. J. Am. Ceram. Soc. 2003, 86, 480–486. [Google Scholar] [CrossRef]
- Dericioglu, A.F.; Kagawa, Y. Effect of grain boundary microcracking on the light transmittance of sintered transparent MgAl2O4. J. Eur. Ceram. Soc. 2003, 23, 951–959. [Google Scholar] [CrossRef]
- Barbe, C.J.; Arendse, F.; Comte, P.; Jirousek, M.; Lenzmann, F.; Shklover, V.; Gra, M. Nanocrystalline Titanium Oxide Electrodes for Photovoltaic Applications. J. Am. Ceram. Soc. 1997, 71, 3157–3171. [Google Scholar] [CrossRef]
- Nehl, C.L.; Hafner, J.H. Shape-dependent plasmon resonances of gold nanoparticles. J. Mater. Chem. 2008, 2415–2419. [Google Scholar] [CrossRef]
- Rubio, A.; Serra, L. Dielectric screening effects on the photoabsorption cross section of embedded metallic clusters. Phys. Rev. B 1993, 48, 18222–18229. [Google Scholar] [CrossRef]
- Amendola, V.; Meneghetti, M. Size Evaluation of Gold Nanoparticles by UV-vis Spectroscopy. J. Phys. Chem. C 2009, 4277–4285. [Google Scholar] [CrossRef]
- Zook, J.M.; Rastogi, V.; Maccuspie, R.I.; Keene, A.M.; Fagan, J. Measuring Agglomerate Size Distribution and Dependence of Localized Surface Plasmon Resonance Absorbance on Gold Nanoparticle Agglomerate Size Using Analytical Ultracentrifugation. ACS Nano 2011, 5, 8070–8079. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; El-Sayed, M.A. Gold nanoparticles: Optical properties and implementations in cancer diagnosis and photothermal therapy. J. Adv. Res. 2010, 1, 13–28. [Google Scholar] [CrossRef]
- Jain, P.K.; El-Sayed, M.A. Plasmonic coupling in noble metal nanostructures. Chem. Phys. Lett. 2010, 487, 153–164. [Google Scholar] [CrossRef]
- Hayashi, S.; Okamoto, T. Plasmonics: Visit the past to know the future. J. Phys. D. Appl. Phys. 2012, 45, 433001. [Google Scholar] [CrossRef]
- Hartland, G. V Optical Studies of Dynamics in Noble Metal Nanostructures. Chem. Rev. 2011, 111, 3858–3887. [Google Scholar] [CrossRef] [PubMed]
- Stockman, M.I. Nanoplasmonics: The physics behind the applications. Phys. Today 2011, 39–44. [Google Scholar] [CrossRef]
- Romero, I.; Aizpurua, J.; Bryant, G.W.; Abajo, F.J.G. De Plasmons in nearly touching metallic nanoparticles: Singular response in the limit of touching dimers. Opt. Express 2006, 14, 1627–1631. [Google Scholar] [CrossRef]
- Zhu, J. Surface Plasmon Resonance from Bimetallic Interface in Au-Ag Core-Shell Structure Nanowires. Nanoscale Res. Lett. 2009, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, H.; Li, Z.; Wang, P. The Study of Surface Plasmon in Au/Ag Core/Shell Compound Nanoparticles. Plasmonics 2012, 509–513. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, B.-Q.; Li, Z.-Y.; Xia, Y.; Chen, Y.-G. Surface Plasmon Resonance in Bimetallic Core–Shell Nanoparticles. J. Phys. Chem. C 2015, 119, 16836–16845. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Au-Content (μg/cm2) | Ag-Content (μg/cm2) |
|---|---|---|
| photodeposited Au | 17.5 ± 3.7 | - |
| plasma-deposited Au | 19.6 ± 6.5 | - |
| plasma-deposited Au/Ag | 7.8 ± 3.4 | 1.4 ± 0.8 |
| Sample | Total H2 Volume (mL) | H2 evolution Rate a (µL h−1 cm−2) | H2 evolution Rate b (μmol h−1 cm−2) |
|---|---|---|---|
| pure TiO2 | 5.0 | 31 | 1.3 |
| photodeposited Au-TiO2 | 44.1 | 372 | 15.2 |
| plasma-deposited Au-TiO2 | 20.0 | 152 | 6.2 |
| plasma-deposited Ag/Au-TiO2 | 28.2 | 217 | 8.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, A.; Peglow, S.; Karnahl, M.; Kruth, A.; Junge, H.; Brüser, V.; Scheu, C. Morphology, Optical Properties and Photocatalytic Activity of Photo- and Plasma-Deposited Au and Au/Ag Core/Shell Nanoparticles on Titania Layers. Nanomaterials 2018, 8, 502. https://doi.org/10.3390/nano8070502
Müller A, Peglow S, Karnahl M, Kruth A, Junge H, Brüser V, Scheu C. Morphology, Optical Properties and Photocatalytic Activity of Photo- and Plasma-Deposited Au and Au/Ag Core/Shell Nanoparticles on Titania Layers. Nanomaterials. 2018; 8(7):502. https://doi.org/10.3390/nano8070502
Chicago/Turabian StyleMüller, Alexander, Sandra Peglow, Michael Karnahl, Angela Kruth, Henrik Junge, Volker Brüser, and Christina Scheu. 2018. "Morphology, Optical Properties and Photocatalytic Activity of Photo- and Plasma-Deposited Au and Au/Ag Core/Shell Nanoparticles on Titania Layers" Nanomaterials 8, no. 7: 502. https://doi.org/10.3390/nano8070502
APA StyleMüller, A., Peglow, S., Karnahl, M., Kruth, A., Junge, H., Brüser, V., & Scheu, C. (2018). Morphology, Optical Properties and Photocatalytic Activity of Photo- and Plasma-Deposited Au and Au/Ag Core/Shell Nanoparticles on Titania Layers. Nanomaterials, 8(7), 502. https://doi.org/10.3390/nano8070502