Theoretical Study of Aluminum Hydroxide as a Hydrogen-Bonded Layered Material


Abstract
1. Introduction
2. Calculation Methods
3. Results and Discussion
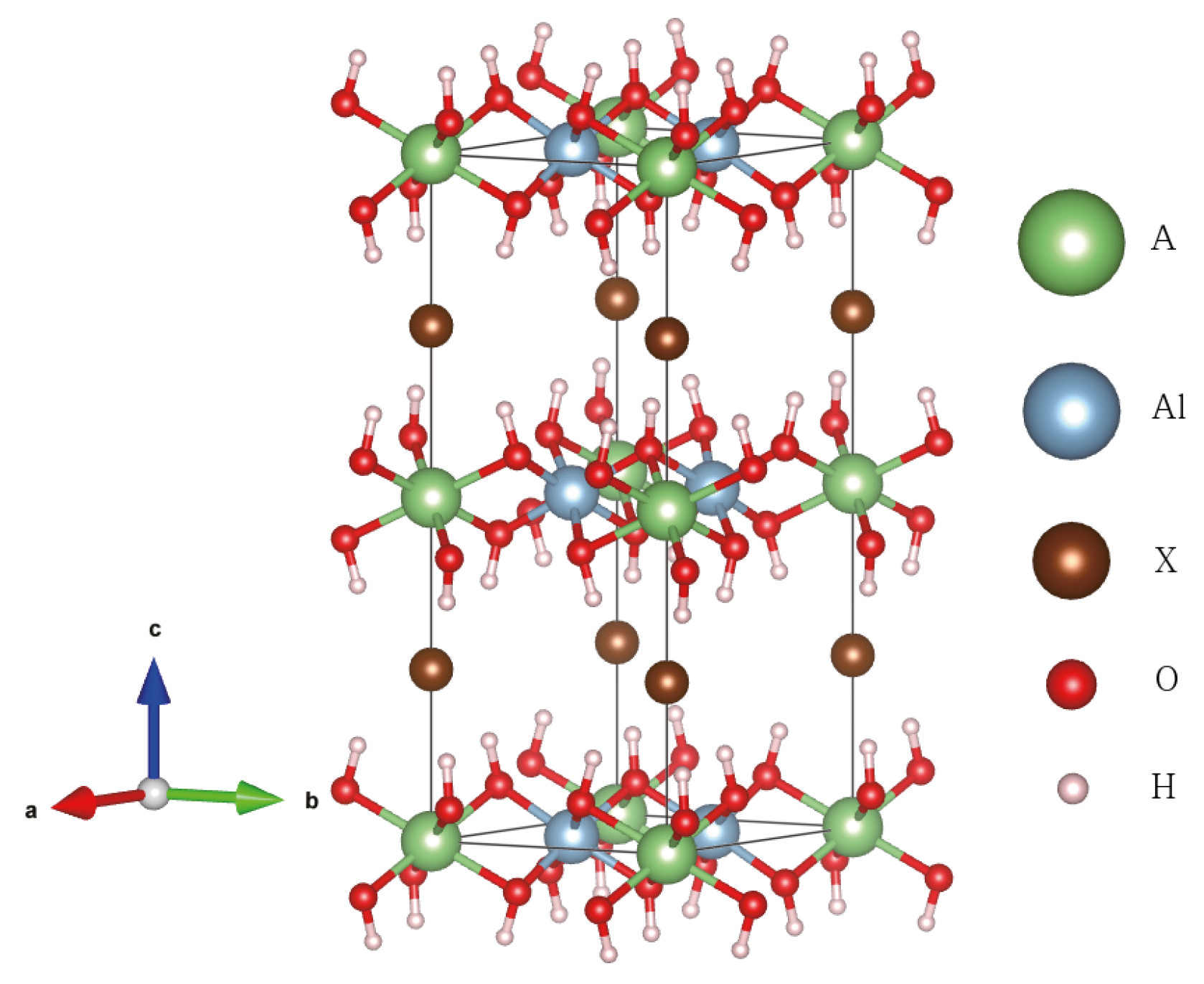
3.1. Crystal Structure of Aluminum Hydroxide
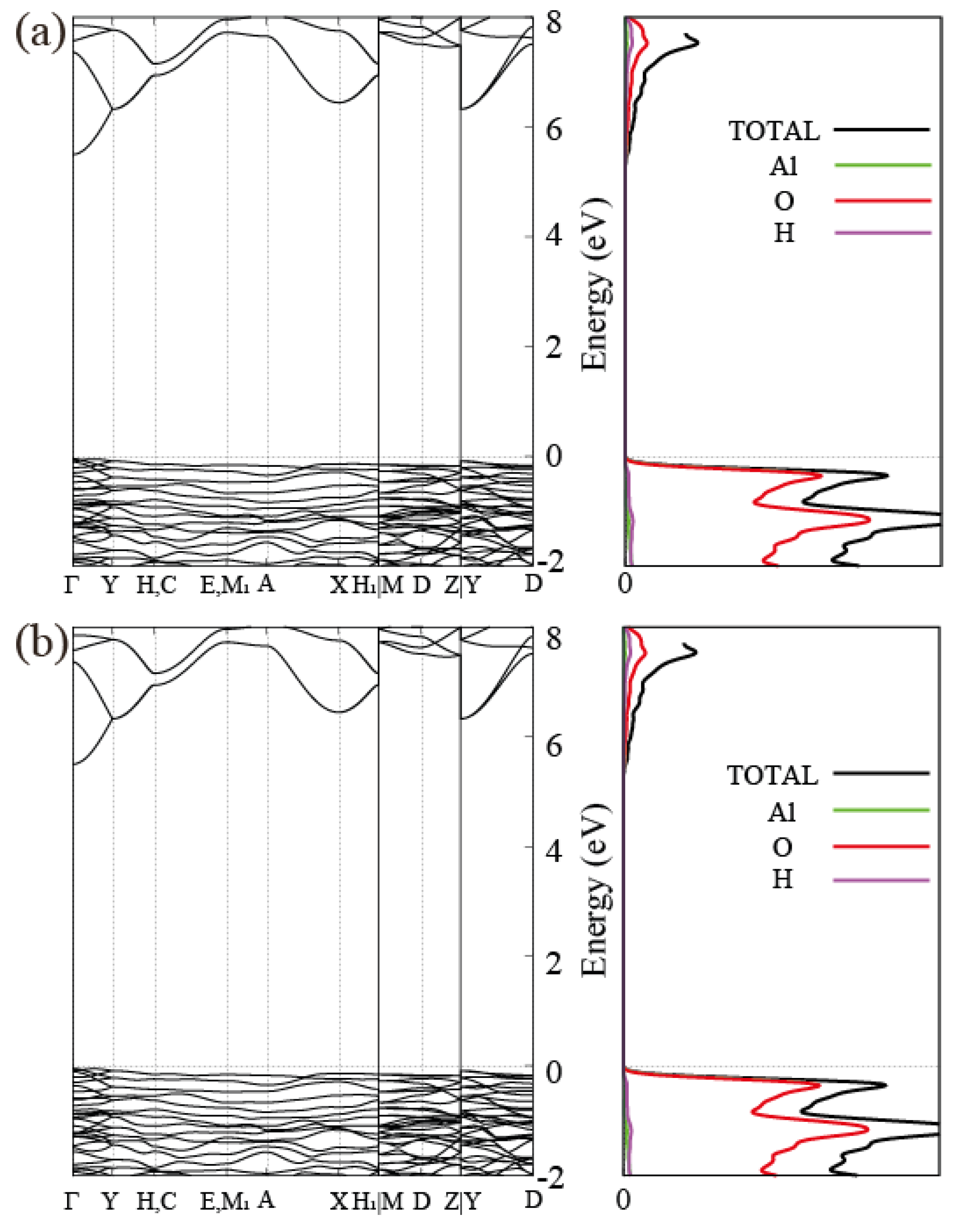
3.2. Electronic Band Structure of Gibbsite, Bayerite and Single Layer
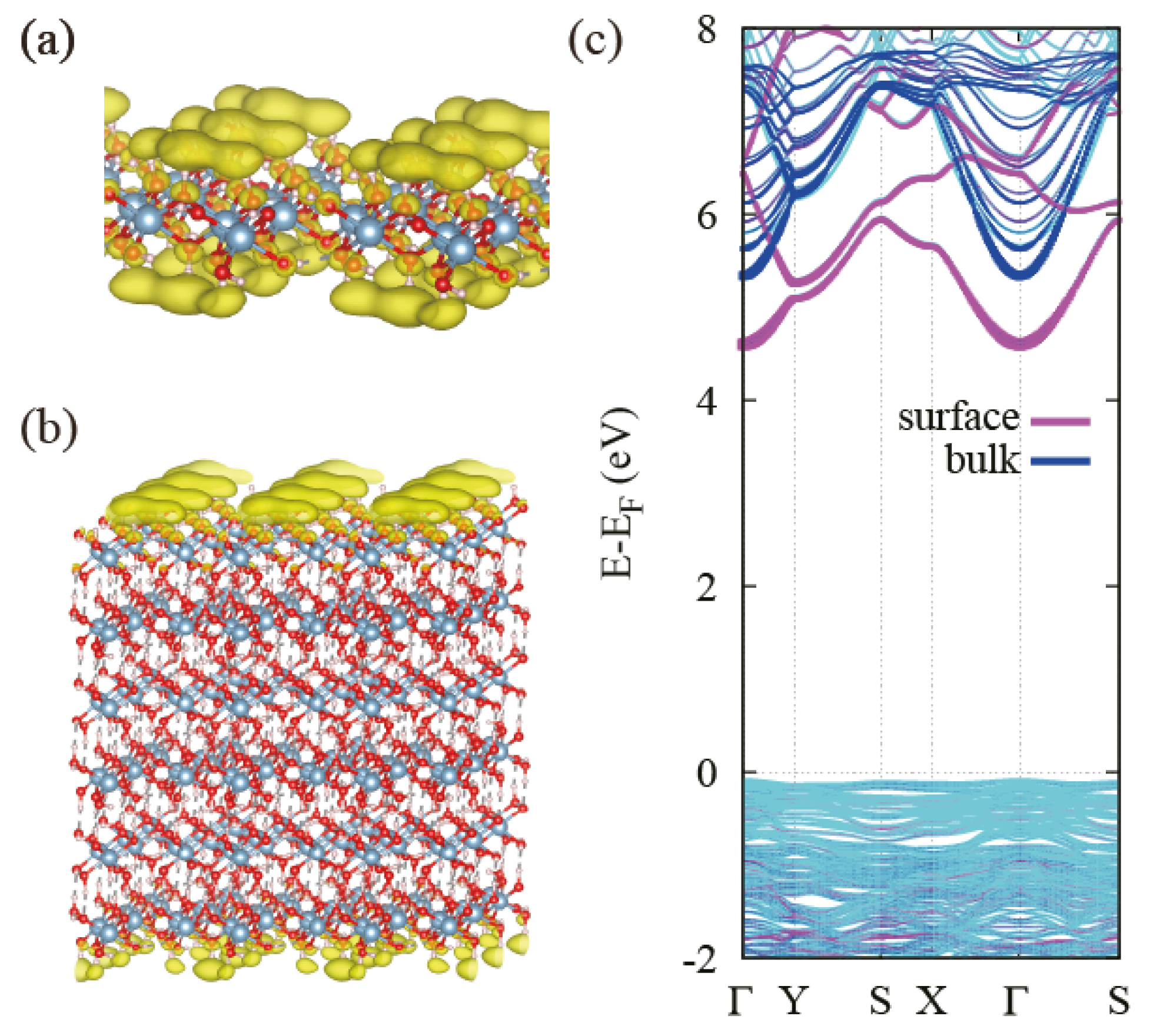
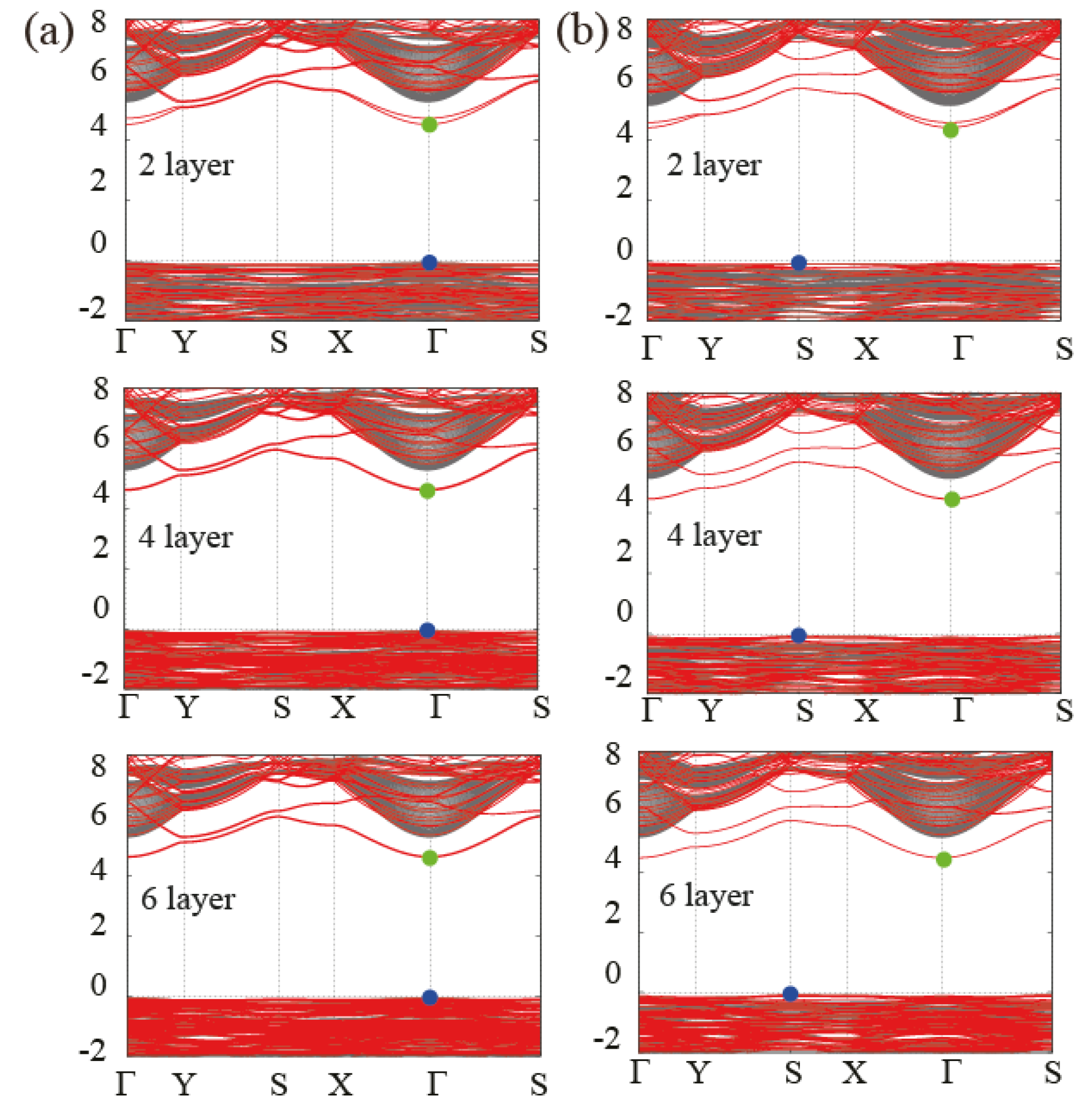
3.3. Behavior of the Band Gap in Bulk and Few-Layer Al(OH)
3.4. Alkali-Halide Intercalated Al(OH)
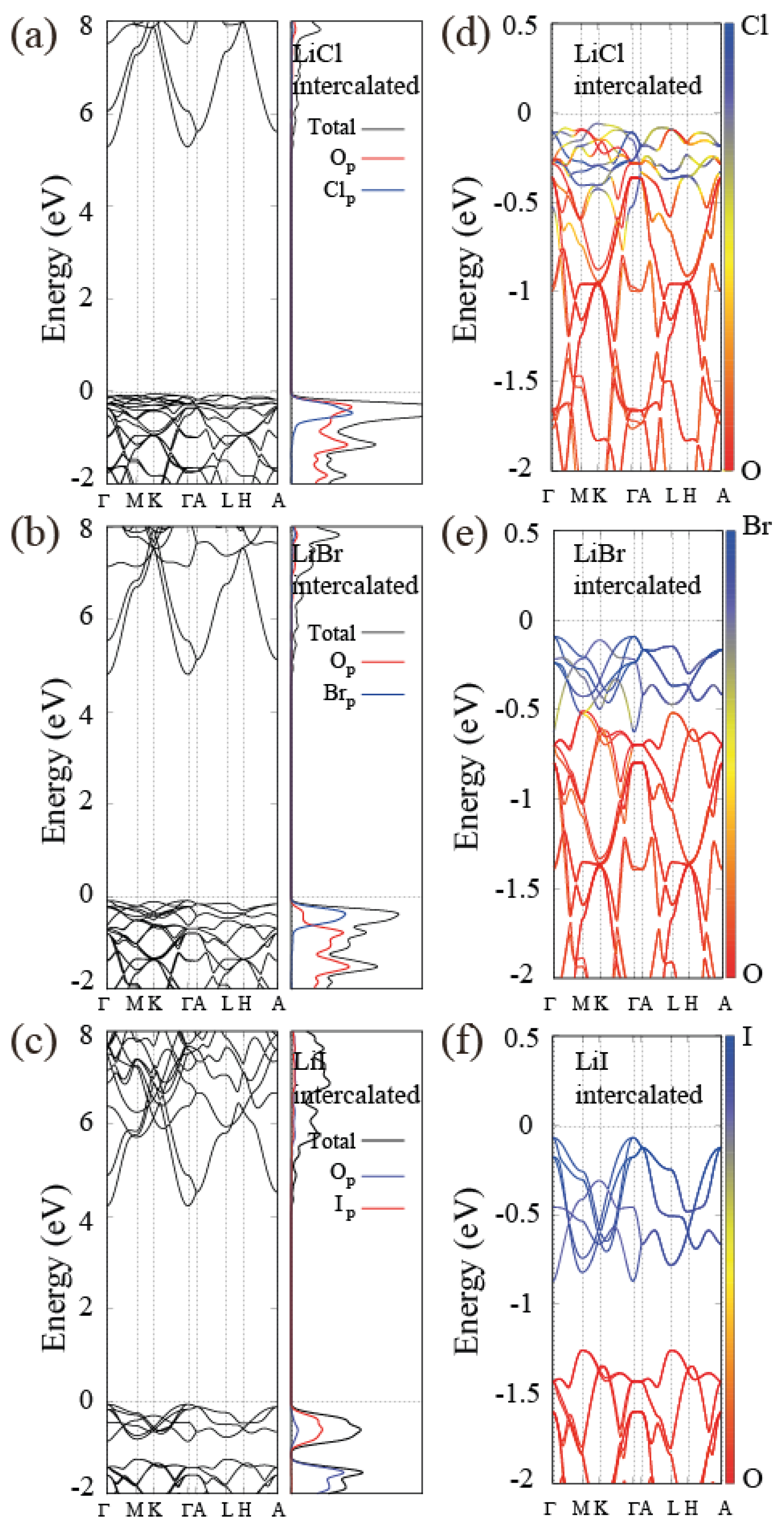
3.5. Electronic Band Structure of Alkali-Halide Intercalated Al(OH)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Song, Z.; Li, T.; Li, X.; Ogbazghi, A.Y.; Feng, R.; Dai, Z.; Marchenkov, A.N.; Conrad, E.H.; First, P.N.; et al. Ultrathin Epitaxial Graphite: 2D Electron Gas Properties and a Route toward Graphene-based Nanoelectronics. J. Phys. Chem. B 2004, 108, 19912–19916. [Google Scholar] [CrossRef]
- Berger, C.; Song, Z.; Li, X.; Wu, X.; Brown, N.; Naud, C.; Mayou, D.; Li, T.; Hass, J.; Marchenkov, A.N.; et al. Electronic Confinement and Coherence in Patterned Epitaxial Graphene. Science 2006, 312, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Reina, A.; Shi, Y.; Park, H.; Li, L.J.; Lee, Y.H.; Kong, J. Enhancing the conductivity of transparent graphene films via doping. Nanotechnology 2010, 21, 285205. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cai, W.; An, J.; Kim, S.; Nah, J.; Yang, D.; Piner, R.; Velamakanni, A.; Jung, I.; Tutuc, E.; et al. Large-Area Synthesis of High-Quality and Uniform Graphene Films on Copper Foils. Science 2009, 324, 1312–1314. [Google Scholar] [CrossRef] [PubMed]
- Blase, X.; Rubio, A.; Louie, S.G.; Cohen, M.L. Quasiparticle band structure of bulk hexagonal boron nitride and related systems. Phys. Rev. B 1995, 51, 6868–6875. [Google Scholar] [CrossRef]
- Henck, H.; Pierucci, D.; Fugallo, G.; Avila, J.; Cassabois, G.; Dappe, Y.J.; Silly, M.G.; Chen, C.; Gil, B.; Gatti, M.; et al. Direct observation of the band structure in bulk hexagonal boron nitride. Phys. Rev. B 2017, 95, 085410. [Google Scholar] [CrossRef]
- Tegeler, E.; Kosuch, N.; Wiech, G.; Faessler, A. On the electronic structure of hexagonal boron nitride. Phys. Status Solidi B 1979, 91, 223–231. [Google Scholar] [CrossRef]
- Singh, R.S.; Tay, R.Y.; Chow, W.L.; Tsang, S.H.; Mallick, G.; Teo, E.H.T. Band gap effects of hexagonal boron nitride using oxygen plasma. Appl. Phys. Lett. 2014, 104, 163101. [Google Scholar] [CrossRef]
- Ooi, N.; Rairkar, A.; Lindsley, L.; Adams, J.B. Electronic structure and bonding in hexagonal boron nitride. J. Phys. Condens. Matter 2006, 18, 97. [Google Scholar] [CrossRef]
- Liu, G.B.; Xiao, D.; Yao, Y.; Xu, X.; Yao, W. Electronic structures and theoretical modelling of two-dimensional group-VIB transition metal dichalcogenides. Chem. Soc. Rev. 2015, 44, 2643–2663. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Tongay, S.; Li, J.; Wu, J. Monolayer semiconducting transition metal dichalcogenide alloys: Stability and band bowing. J. Appl. Phys. 2013, 113, 143703. [Google Scholar] [CrossRef]
- Rasmussen, F.A.; Thygesen, K.S. Computational 2D Materials Database: Electronic Structure of Transition-Metal Dichalcogenides and Oxides. J. Phys. Chem. C 2015, 119, 13169–13183. [Google Scholar] [CrossRef]
- Yeh, P.C.; Jin, W.; Zaki, N.; Zhang, D.; Liou, J.T.; Sadowski, J.T.; Al-Mahboob, A.; Dadap, J.I.; Herman, I.P.; Sutter, P.; et al. Layer-dependent electronic structure of an atomically heavy two-dimensional dichalcogenide. Phys. Rev. B 2015, 91, 041407. [Google Scholar] [CrossRef]
- Pomerantseva, E.; Gogotsi, Y. Two-dimensional heterostructures for energy storage. Nat. Energy 2017, 2, 17089. [Google Scholar] [CrossRef]
- Kumar, P.; Abuhimd, H.; Wahyudi, W.; Li, M.; Ming, J.; Li, L.J. Review—Two-Dimensional Layered Materials for Energy Storage Applications. ECS J. Solid State Sci. Technol. 2016, 5, Q3021–Q3025. [Google Scholar] [CrossRef]
- Kumar, A.; Xu, Q. Two-Dimensional Layered Materials as Catalyst Supports. ChemNanoMat 2018, 4, 28–40. [Google Scholar] [CrossRef]
- Cesteros, Y.; Salagre, P.; Medina, F.; Sueiras, J.E. Several Factors Affecting Faster Rates of Gibbsite Formation. Chem. Mater. 1999, 11, 123–129. [Google Scholar] [CrossRef]
- Cesteros, Y.; Salagre, P.; Medina, F.; Sueiras, J.E. A New Route to the Synthesis of Fine-Grain Gibbsite. Chem. Mater. 2001, 13, 2595–2600. [Google Scholar] [CrossRef]
- Louaer, S.; Wang, Y.; Guo, L. Fast Synthesis and Size Control of Gibbsite Nanoplatelets, Their Pseudomorphic Dehydroxylation, and Efficient Dye Adsorption. ACS Appl. Mater. Interfaces 2013, 5, 9648–9655. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Chow, P.S.; Chen, F.; Feng, S.; Tan, R.B. Synthesis of submicron gibbsite platelets by organic-free hydrothermal crystallization process. J. Cryst. Growth 2006, 292, 136–142. [Google Scholar] [CrossRef]
- Kumara, C.; Ng, W.; Bandara, A.; Weerasooriya, R. Nanogibbsite: Synthesis and characterization. J. Colloid Interface Sci. 2010, 352, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmceller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Rothbauer, R.; Zigan, F.; O’danieL, H. Verfeinerung der struktur des bayerits, Al(OH)3. Z. Kristallogr. 1967, 125, 317–331. [Google Scholar] [CrossRef]
- First-principles study of the OH-stretching modes of gibbsite. Am. Mineral. 2006, 91, 115–119.
- Jung, J.H.; Park, C.H.; Ihm, J. A Rigorous Method of Calculating Exfoliation Energies from First Principles. Nano Lett. 2018, 18, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Ben Aziza, Z.; Pierucci, D.; Henck, H.; Silly, M.G.; David, C.; Yoon, M.; Sirotti, F.; Xiao, K.; Eddrief, M.; Girard, J.C.; et al. Tunable quasiparticle band gap in few-layer GaSe/graphene van der Waals heterostructures. Phys. Rev. B 2017, 96, 035407. [Google Scholar] [CrossRef]
- Zhang, Y.; Chang, T.R.; Zhou, B.; Cui, Y.T.; Yan, H.; Liu, Z.; Schmitt, F.; Lee, J.; Moore, R.; Chen, Y.; et al. Direct observation of the transition from indirect to direct bandgap in atomically thin epitaxial MoSe2. Nat. Nanotechnol. 2013, 9, 111. [Google Scholar] [CrossRef] [PubMed]
- Coy Diaz, H.; Avila, J.; Chen, C.; Addou, R.; Asensio, M.C.; Batzill, M. Direct Observation of Interlayer Hybridization and Dirac Relativistic Carriers in Graphene/MoS2 van der Waals Heterostructures. Nano Lett. 2015, 15, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Poeppelmeier, K.R.; Hwu, S.J. Synthesis of lithium dialuminate by salt imbibition. Inorg. Chem. 1987, 26, 3297–3302. [Google Scholar] [CrossRef]
- Besserguenev, A.V.; Fogg, A.M.; Francis, R.J.; Price, S.J.; O’Hare, D.; Isupov, V.P.; Tolochko, B.P. Synthesis and Structure of the Gibbsite Intercalation Compounds [LiAl2(OH)6]X {X = Cl, Br, NO3} and [LiAl2(OH)6]CljjaessH2O Using Synchrotron X-ray and Neutron Powder Diffraction. Chem. Mater. 1997, 9, 241–247. [Google Scholar] [CrossRef]
- Fogg, A.M.; O’Hare, D. Study of the Intercalation of Lithium Salt in Gibbsite Using Time-Resolved in Situ X-ray Diffraction. Chem. Mater. 1999, 11, 1771–1775. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cal./Exp. | Bayerite | Gibbsite | Single Layer |
|---|---|---|---|
| a(Å) | 5.03/5.06 | 5.03/5.08 | 5.08 |
| b(Å) | 8.66/8.67 | 8.65/8.68 | 8.68 |
| c(Å) | 9.12/9.42 | 9.53/9.74 | - |
| () | 90.45/90.27 | 92.34/94.54 | - |
| V(Å) | 397.27/413.25 | 414.50/427.98 | - |
| Alkali-Halide Intercalated Al(OH) | |||
|---|---|---|---|
| Bayerite/Gibbsite | Li[Al(OH)]Cl | Li[Al(OH)]Br | Li[Al(OH)]I |
| a(Å) | 5.12/5.12 | 5.12/5.13 | 5.12/5.12 |
| c(Å) | 13.90/14.21 | 14.58/14.62 | 15.02/15.52 |
| V(Å) | 315.72/322.03 | 331.05/332.75 | 341.44/351.93 |
| BE (eV) | −8.80/−8.64 | −8.39/−8.23 | −7.81/−7.80 |
| Band gap (eV) | 5.37/5.35 | 4.90/4.90 | 4.51/4.30 |
| Bayerite/Gibbsite | Na[Al(OH)]Cl | Na[Al(OH)]Br | Na[Al(OH)]I |
| a(Å) | 5.28/5.28 | 5.28/5.28 | 5.28/5.28 |
| c(Å) | 13.46/13.64 | 13.94/14.34 | 14.69/15.17 |
| V(Å) | 324.71/329.84 | 336.86/346.67 | 355.22/366.03 |
| BE (eV) | −7.13/−6.95 | −6.62/−6.50 | −6.06/−6.00 |
| Band gap (eV) | 5.12/5.15 | 4.80/4.84 | 4.45/4.32 |
| Bayerite/Gibbsite | K[Al(OH)]Cl | K[Al(OH)]Br | K[Al(OH)]I |
| a(Å) | 5.48/5.48 | 5.47/5.48 | 5.47/5.48 |
| c(Å) | 12.90/13.04 | 13.58/13.93 | 14.35/14.75 |
| V(Å) | 335.00/339.15 | 351.89/362.48 | 372.15/384.08 |
| BE (eV) | −4.22/−4.01 | −3.74/−3.59 | −3.21/−3.14 |
| Band gap (eV) | 4.98/4.90 | 4.63/4.62 | 4.25/4.20 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Jung, J.H.; Ihm, J. Theoretical Study of Aluminum Hydroxide as a Hydrogen-Bonded Layered Material. Nanomaterials 2018, 8, 375. https://doi.org/10.3390/nano8060375
Kim D, Jung JH, Ihm J. Theoretical Study of Aluminum Hydroxide as a Hydrogen-Bonded Layered Material. Nanomaterials. 2018; 8(6):375. https://doi.org/10.3390/nano8060375
Chicago/Turabian StyleKim, Dongwook, Jong Hyun Jung, and Jisoon Ihm. 2018. "Theoretical Study of Aluminum Hydroxide as a Hydrogen-Bonded Layered Material" Nanomaterials 8, no. 6: 375. https://doi.org/10.3390/nano8060375
APA StyleKim, D., Jung, J. H., & Ihm, J. (2018). Theoretical Study of Aluminum Hydroxide as a Hydrogen-Bonded Layered Material. Nanomaterials, 8(6), 375. https://doi.org/10.3390/nano8060375