The Mechanism of Low-Temperature Oxidation of Carbon Monoxide by Oxygen over the PdCl2–CuCl2/γ-Al2O3 Nanocatalyst

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
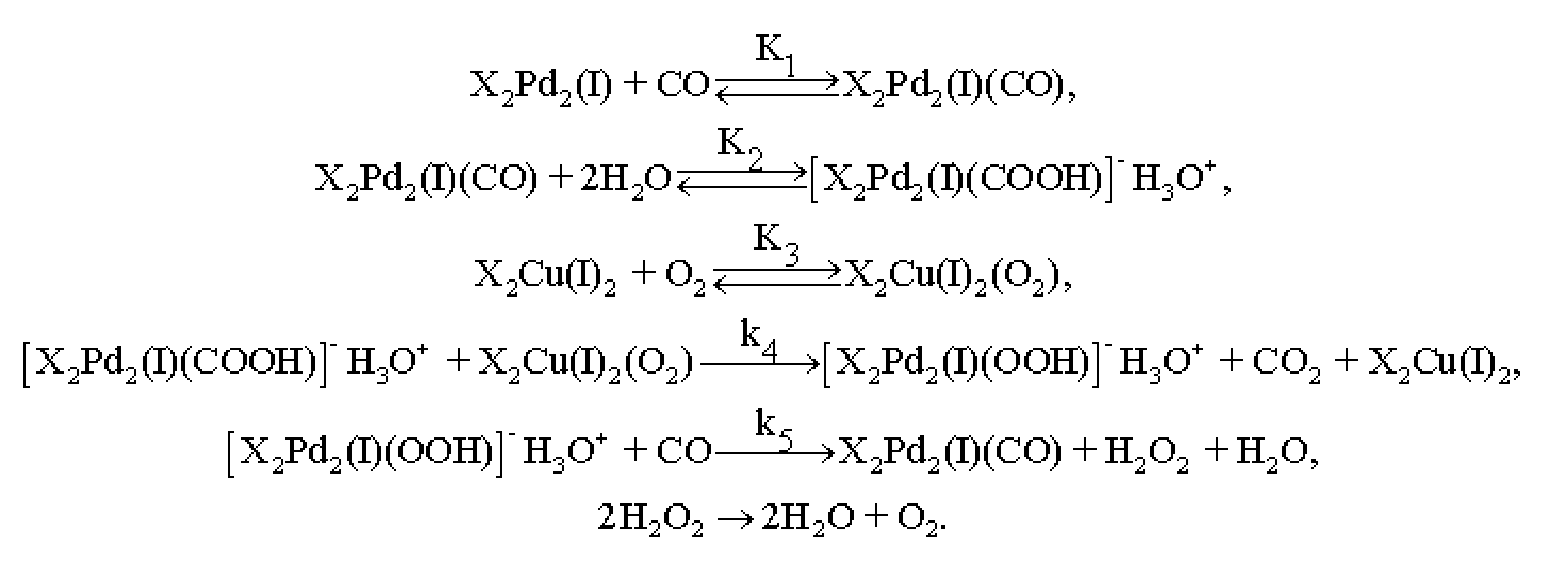{kind=link}
1. Introduction
2. Materials and Methods
3. Results and Discussion
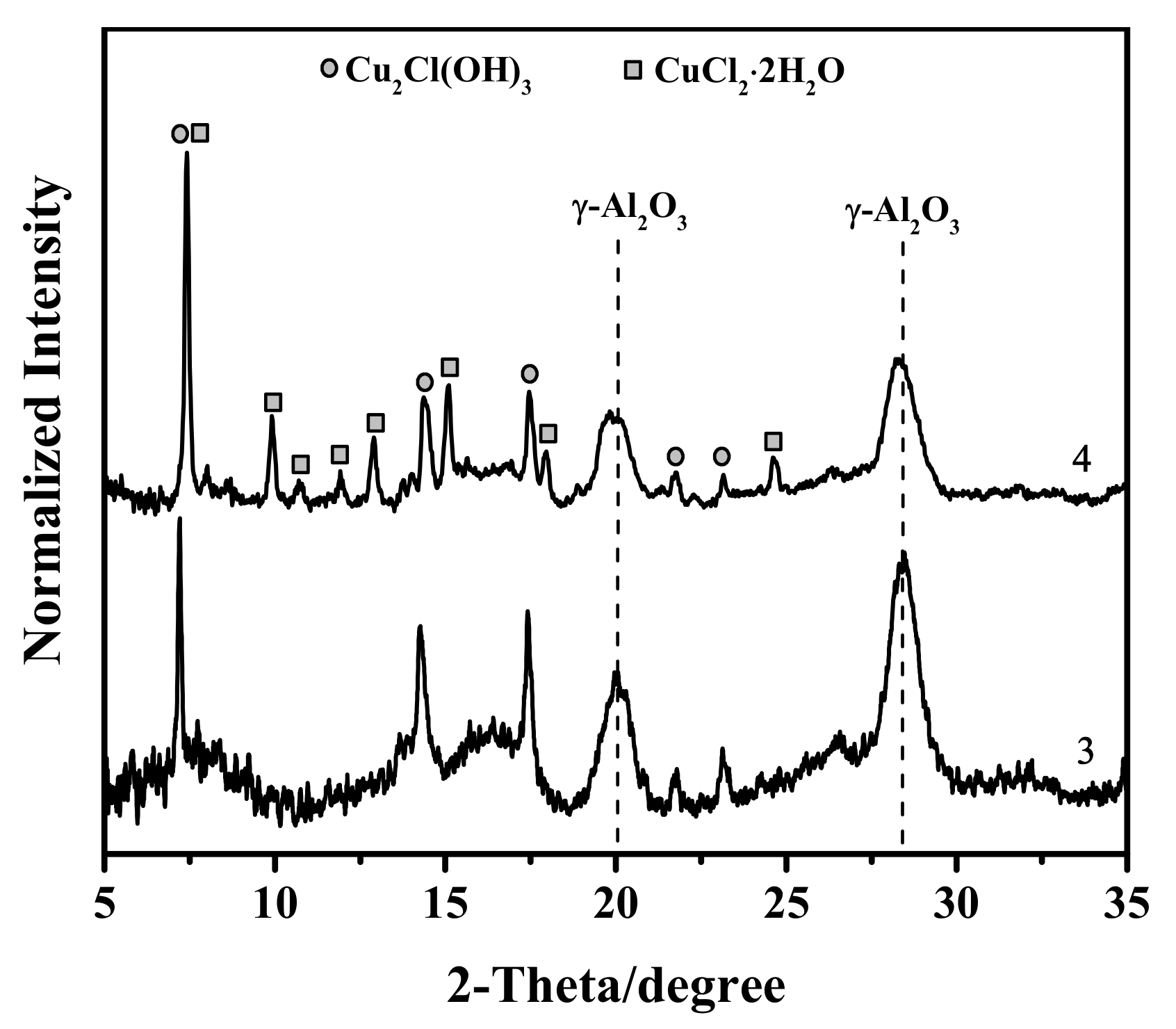
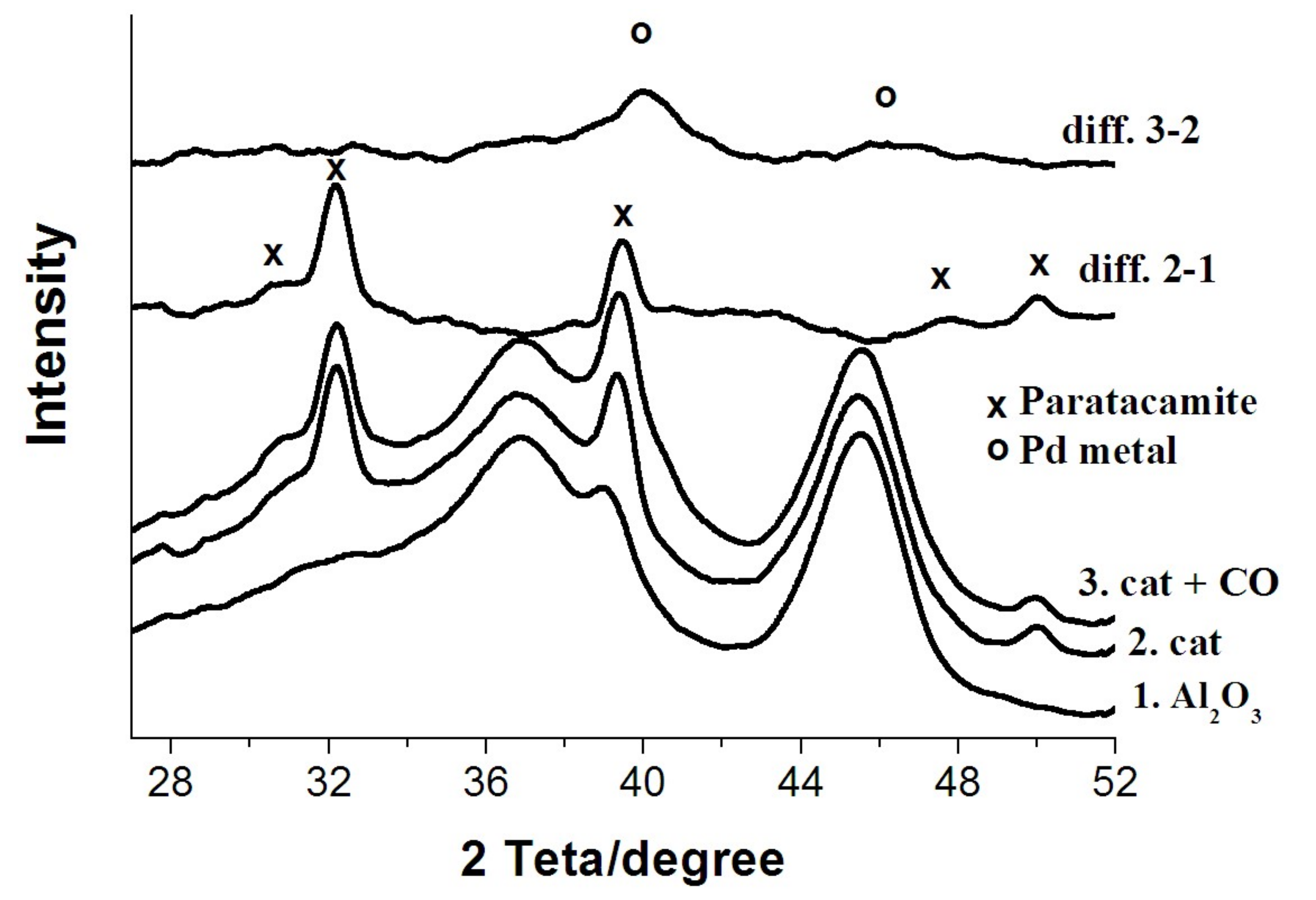
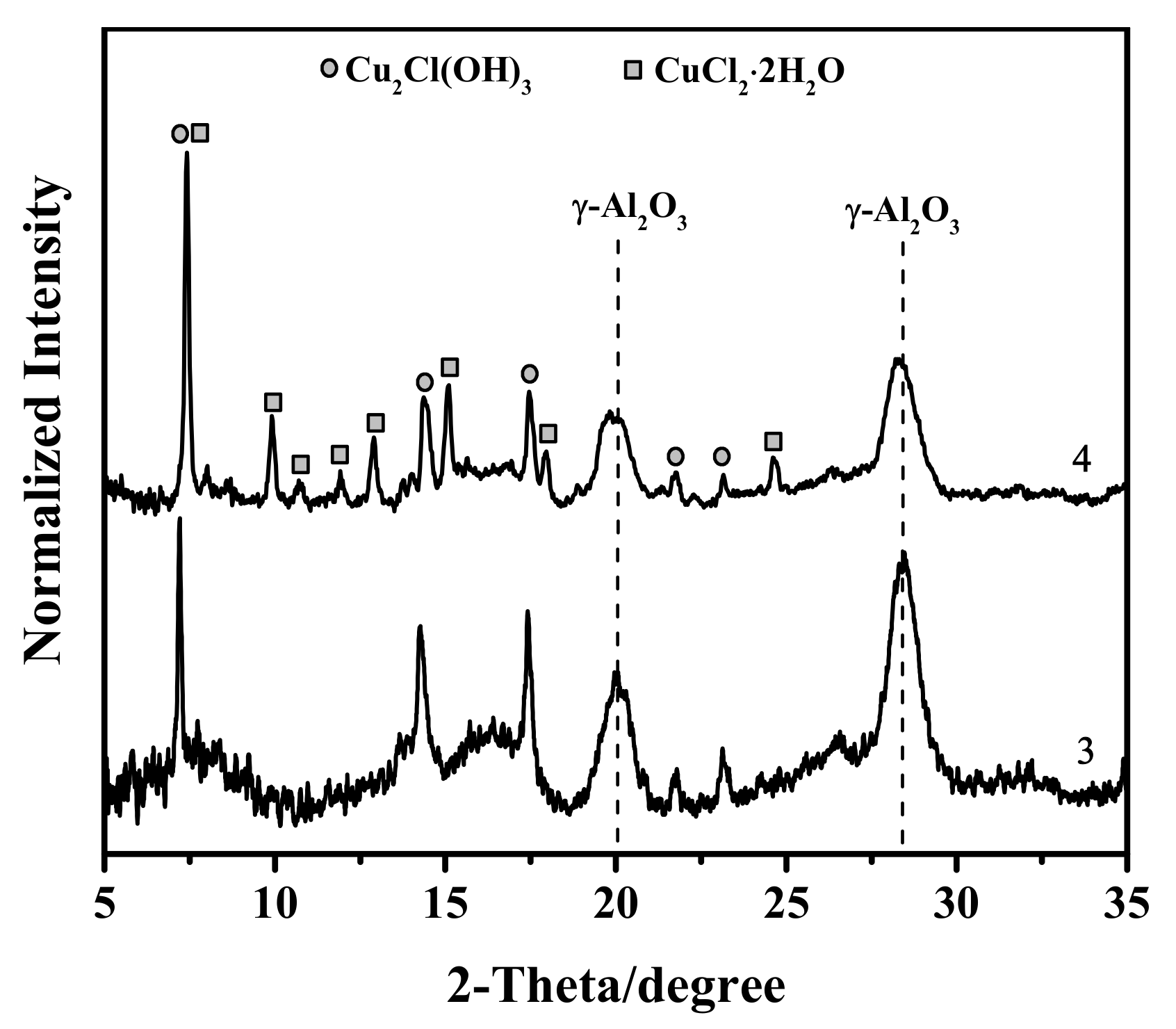
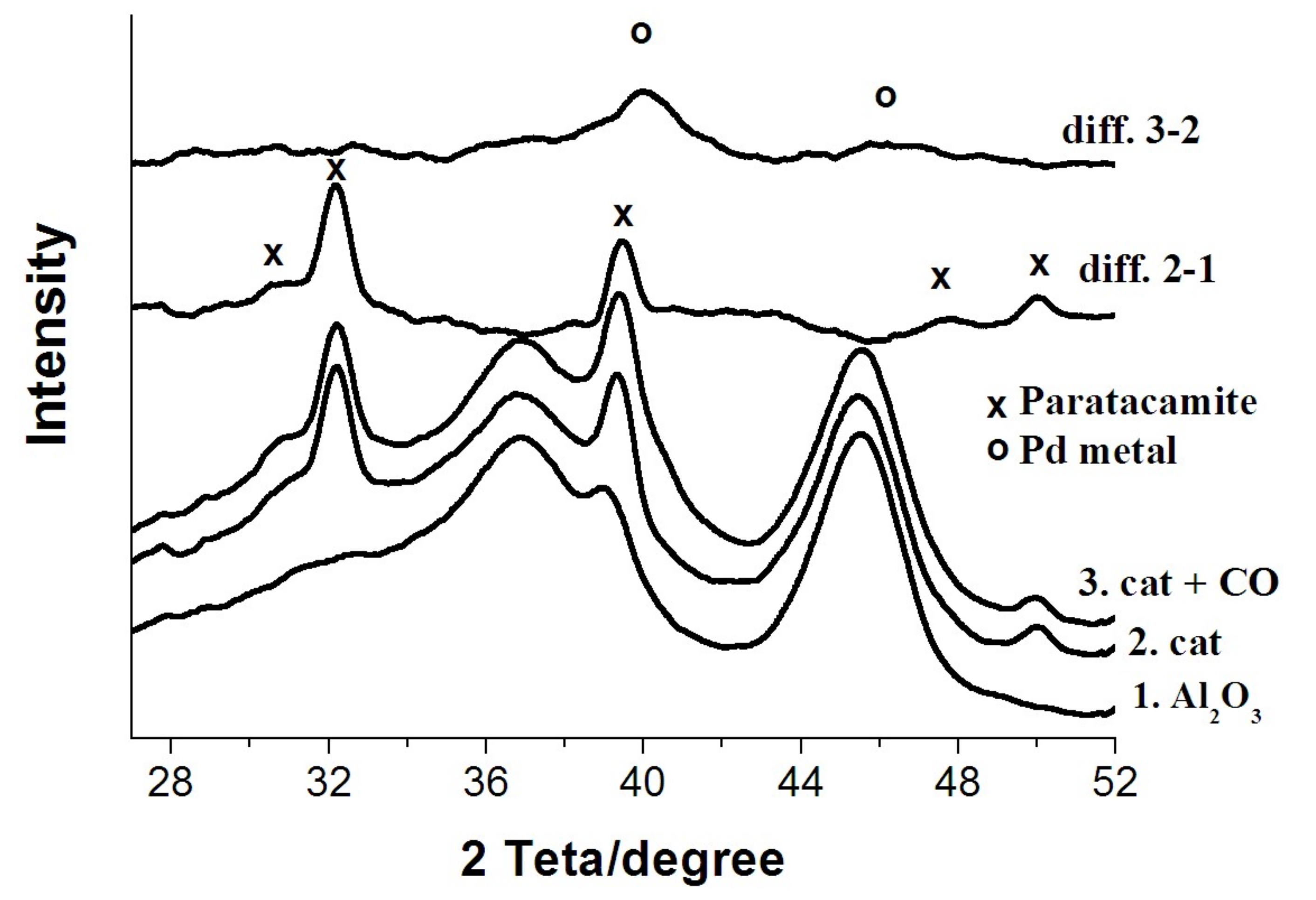
3.1. X-ray Diffraction Study
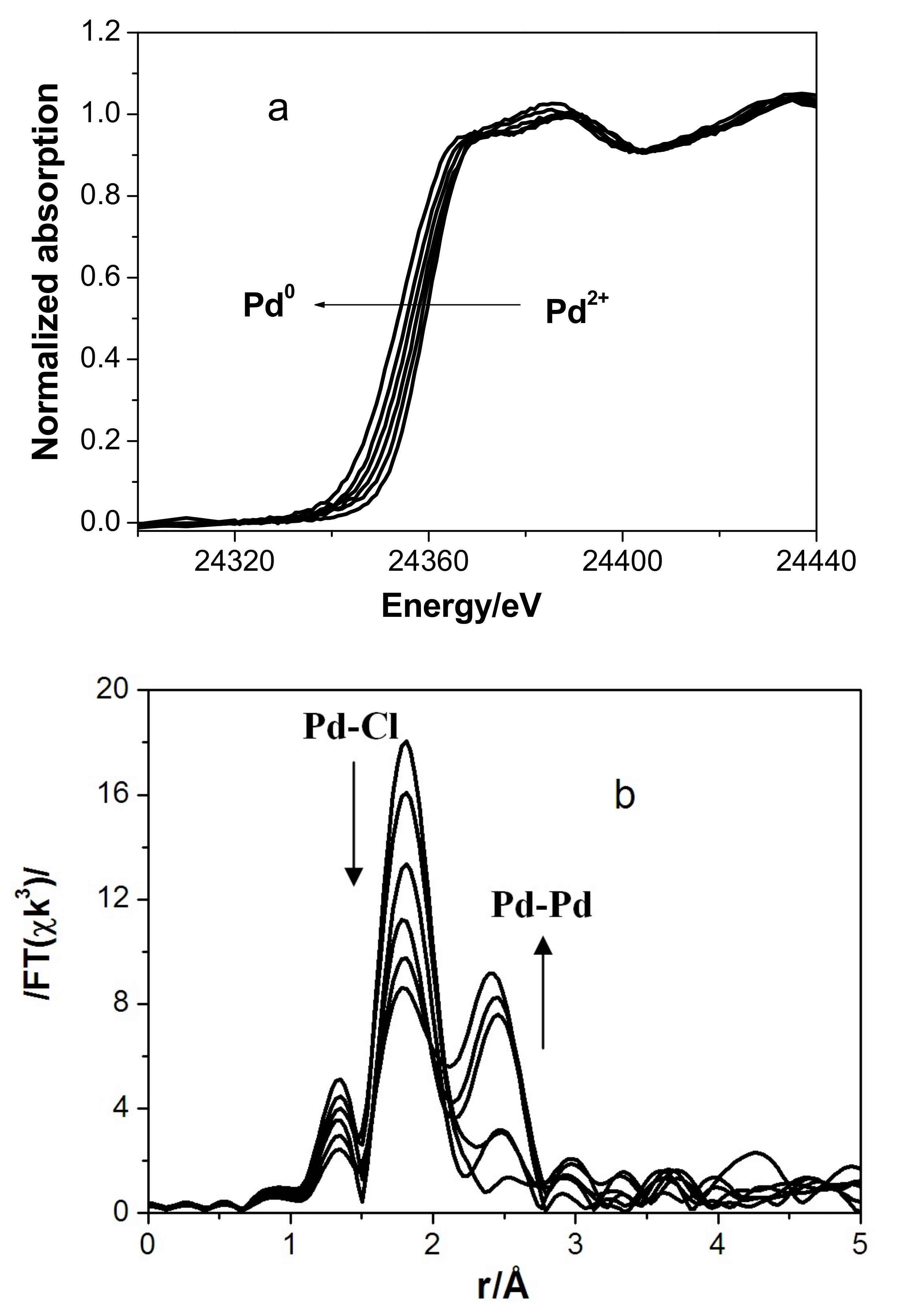
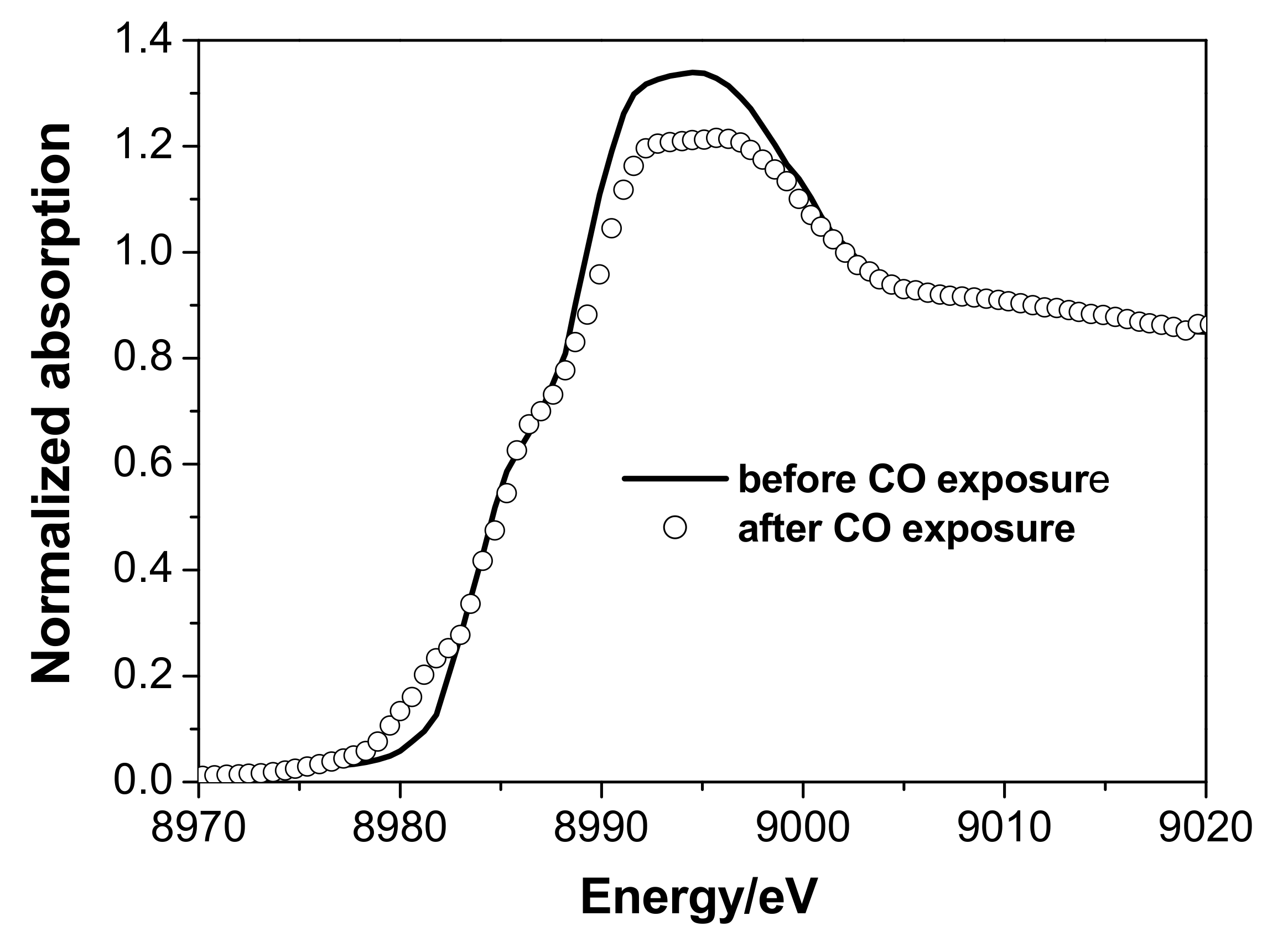
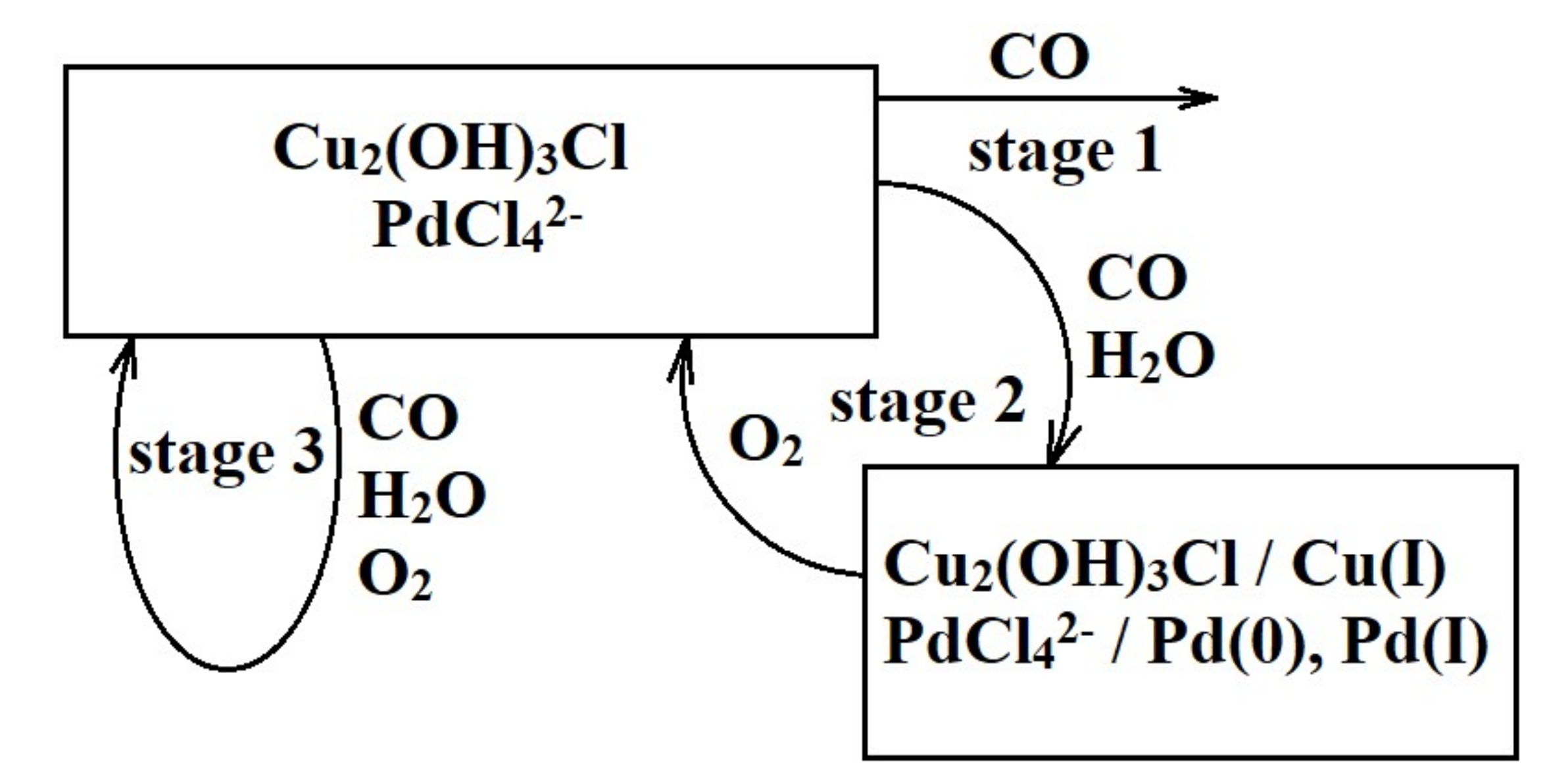
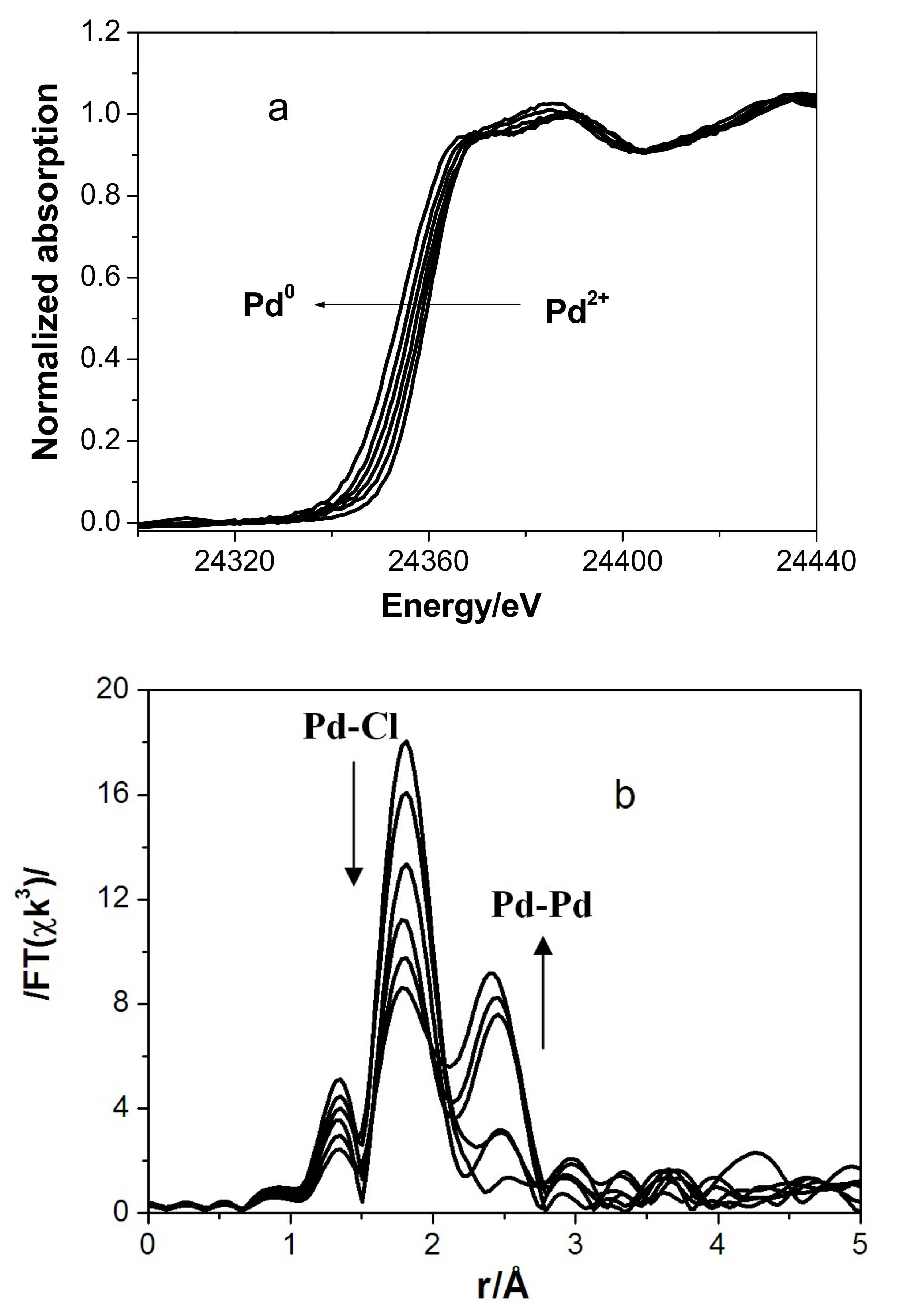
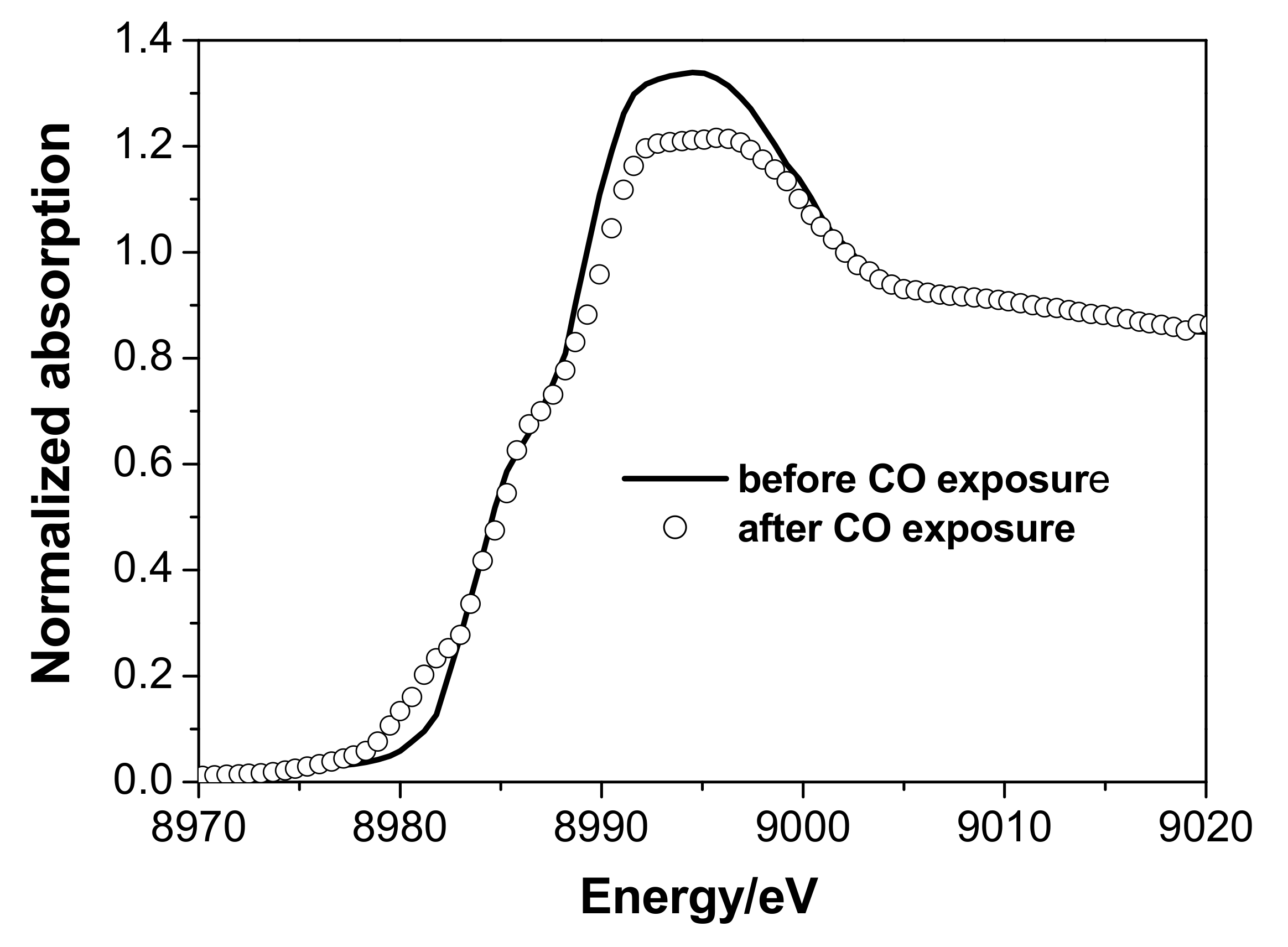
3.2. In Situ XAS Examination of the Catalyst
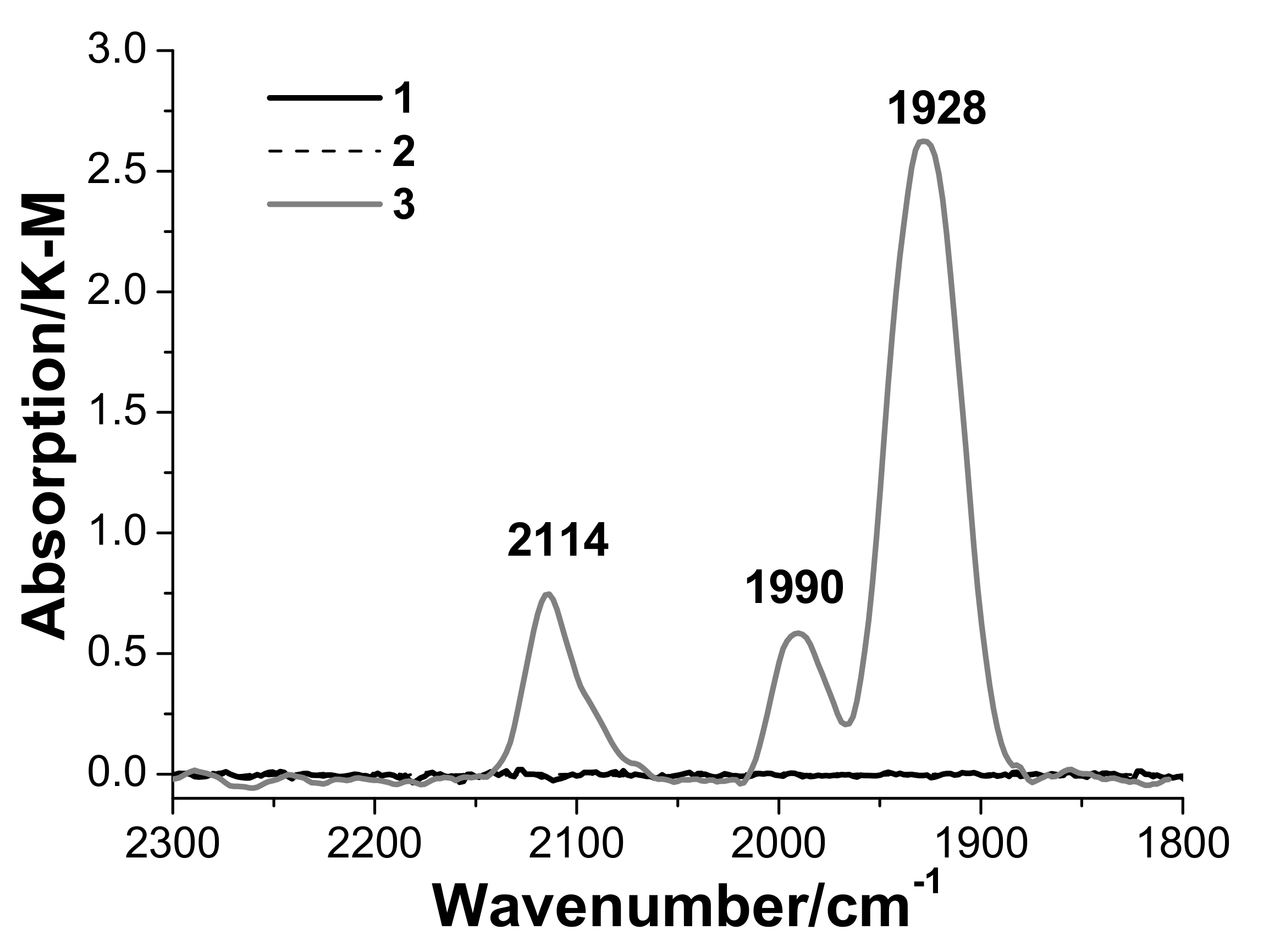
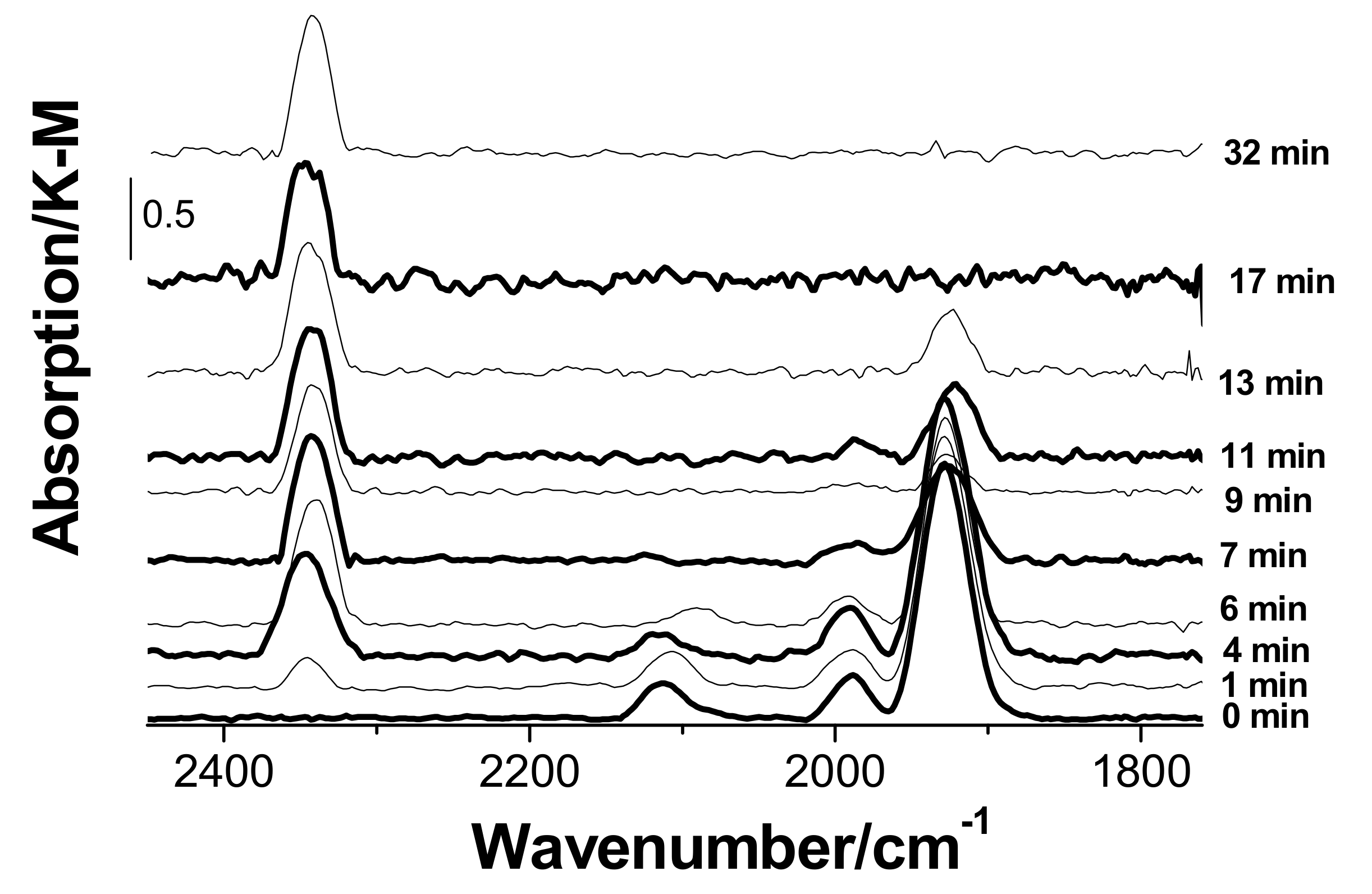
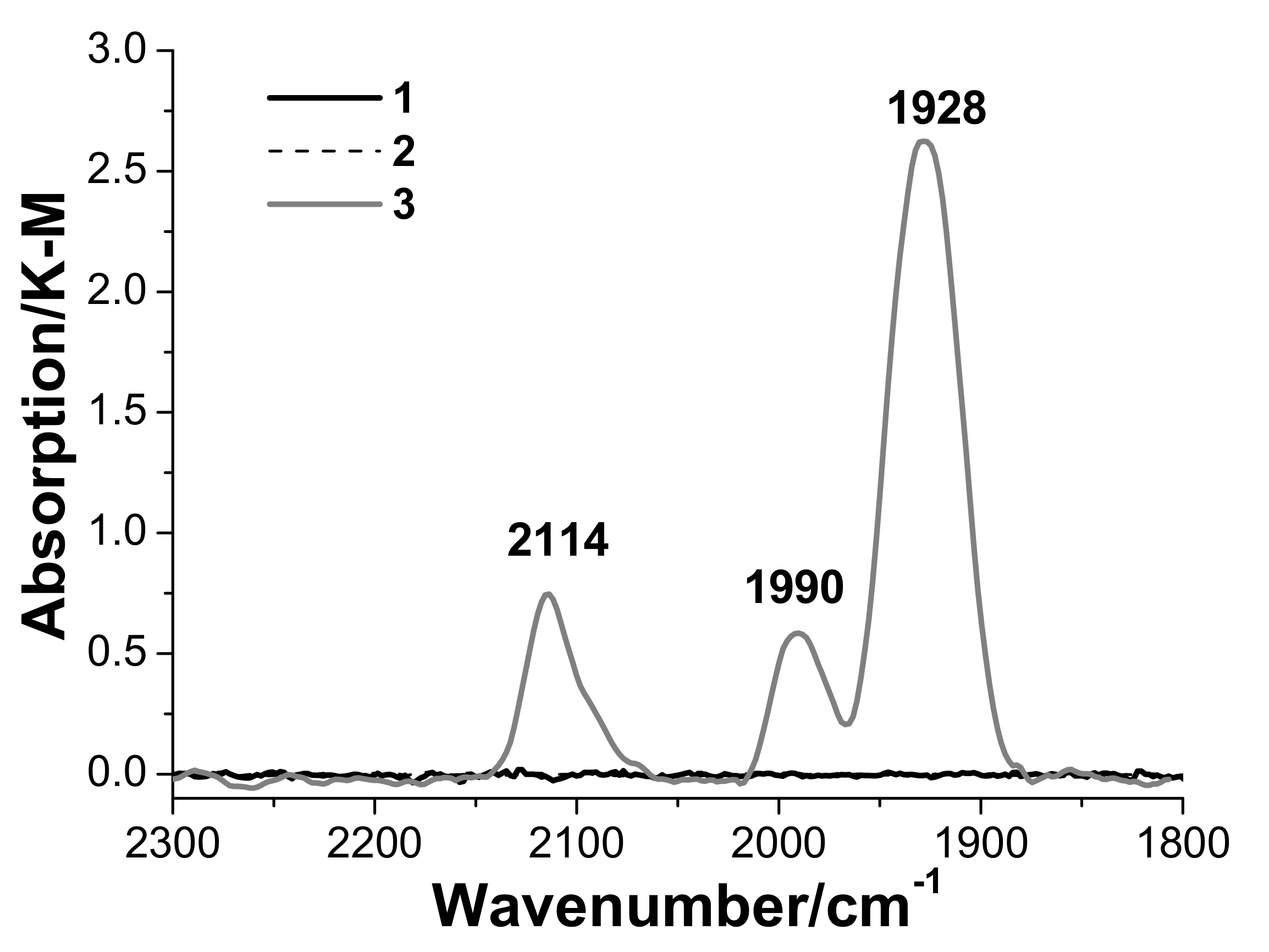
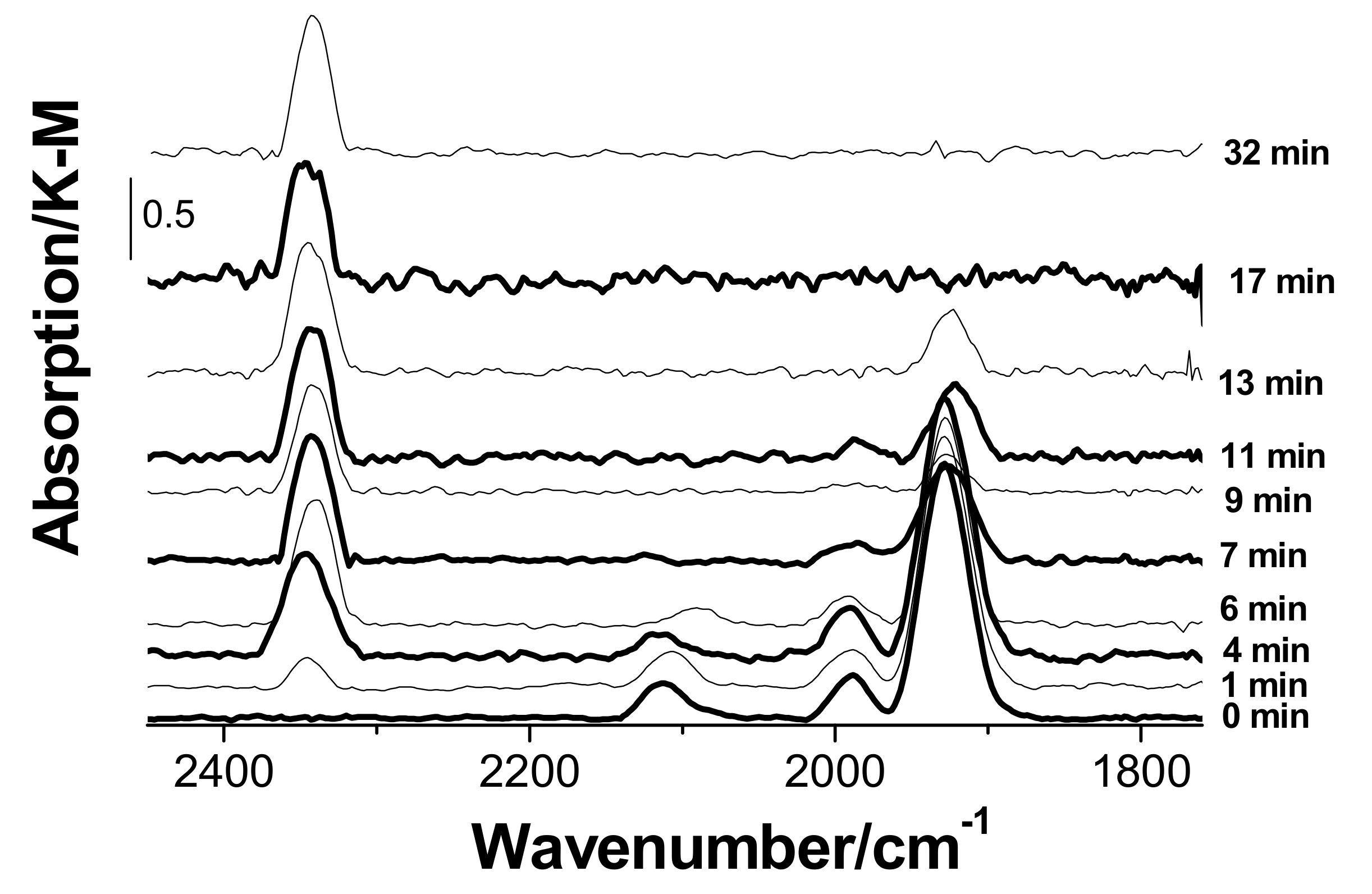
3.3. In Situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy DRIFTS Examination of the Catalyst
- (1)
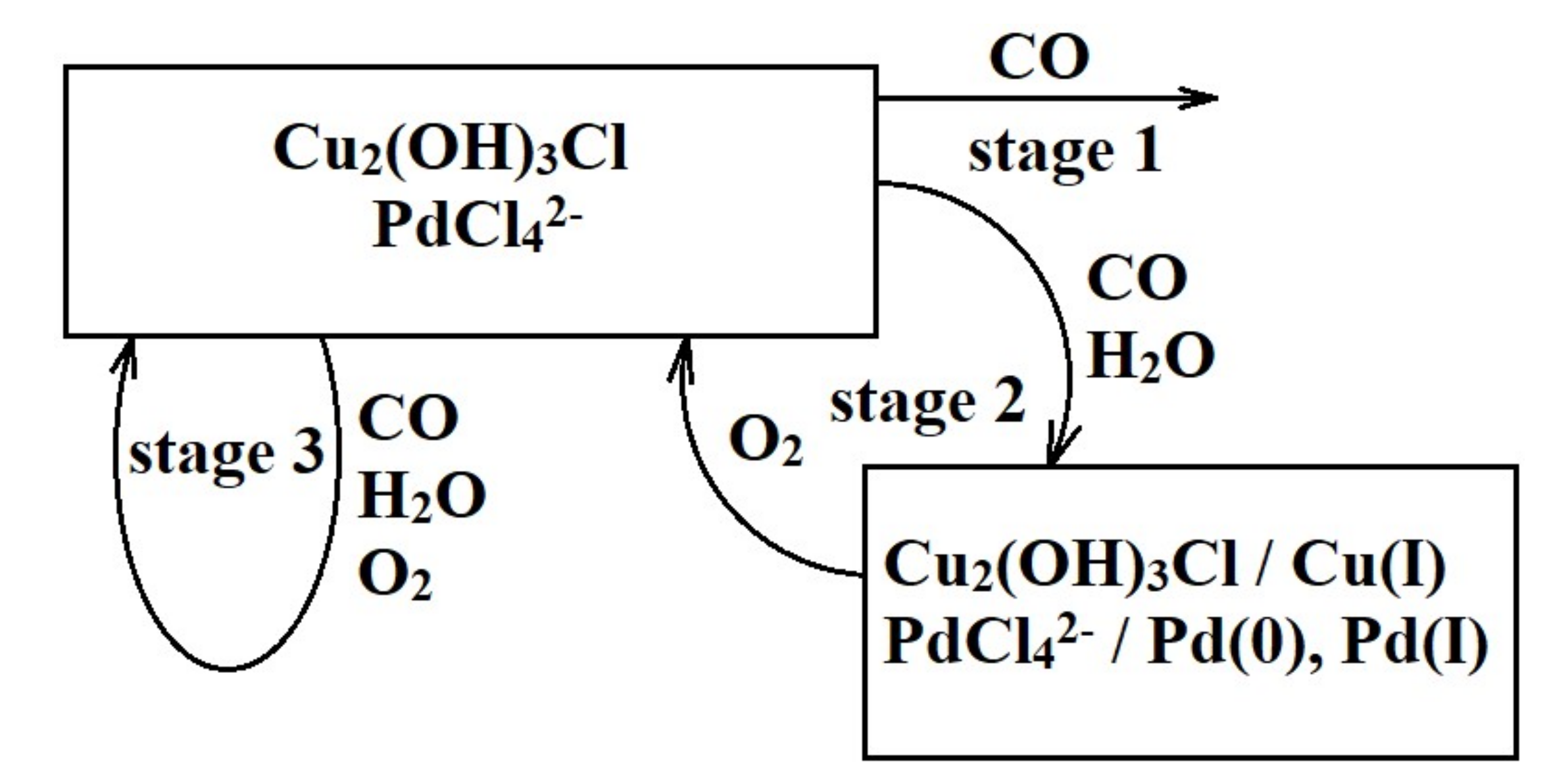
- There is no evidence of a direct copper–palladium interaction (such as the formation of mixed complexes) on the surface of a freshly prepared nanocatalyst;
- (2)
- When the catalyst is exposed to CO and water vapor (in the absence of oxygen), palladium(II) and copper(II) undergo slow reduction to yield Pd(I), Pd(0), and Cu(I). In the absence of palladium, copper in the form of hydrated copper(II) chloride and paratacamite nanophases on the support surface does not interact noticeably with CO and H2O at room temperature;
- (3)
- Palladium and copper carbonyl complexes with bridging and terminal carbonyl groups are formed as products of the interaction of the catalyst with CO and H2O. No CO2 was observed among the products;
- (4)
- Bringing the catalyst containing adsorbed CO into contact with oxygen causes rapid decomposition of the carbonyl complexes and the formation of carbon dioxide. Apparently, oxygen is directly involved in carbon dioxide formation steps;
- (5)
- The methods used in this study indicated no palladium(II) or copper(II) reduction under the conditions of catalytic carbon monoxide oxidation;
- (6)
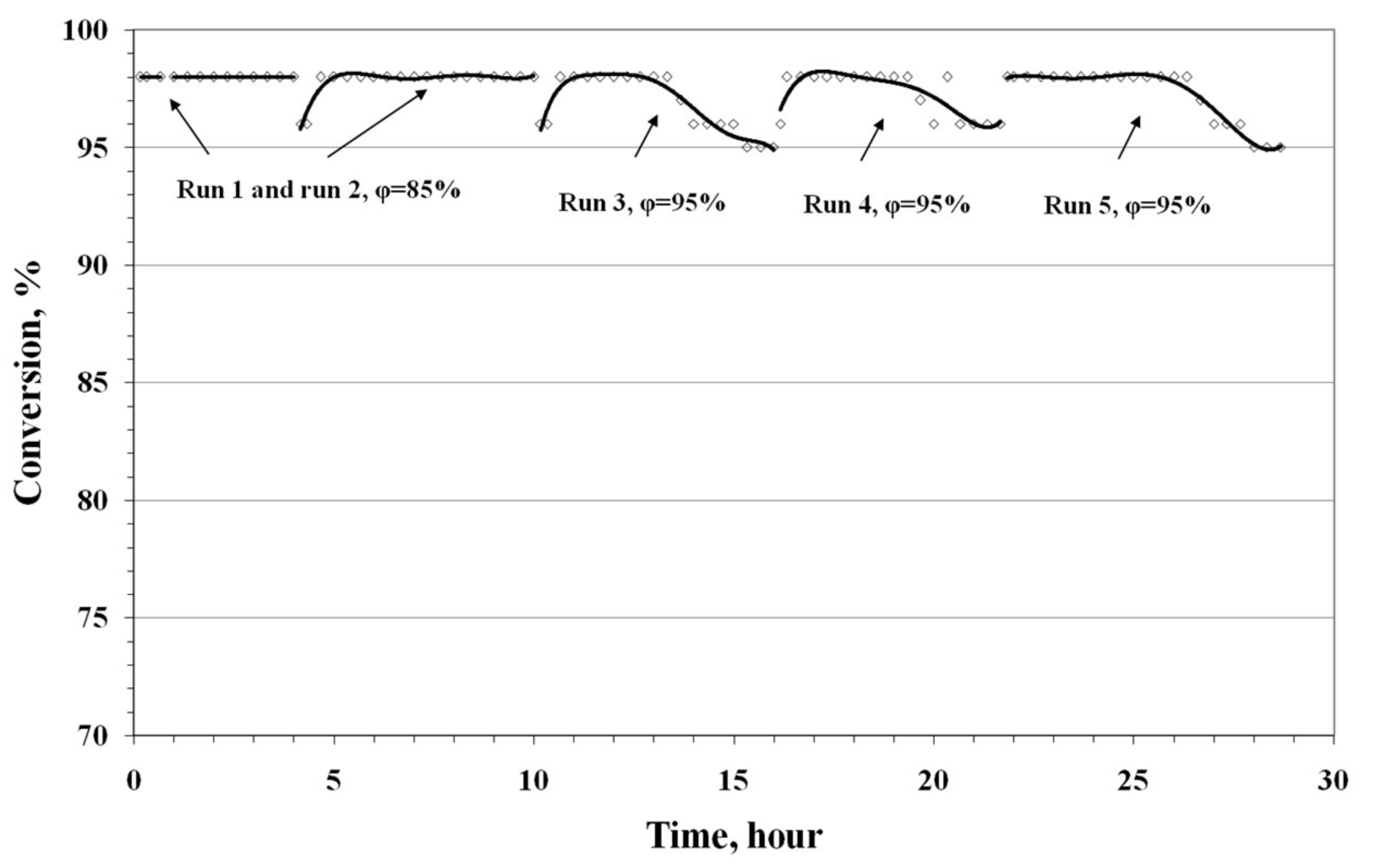
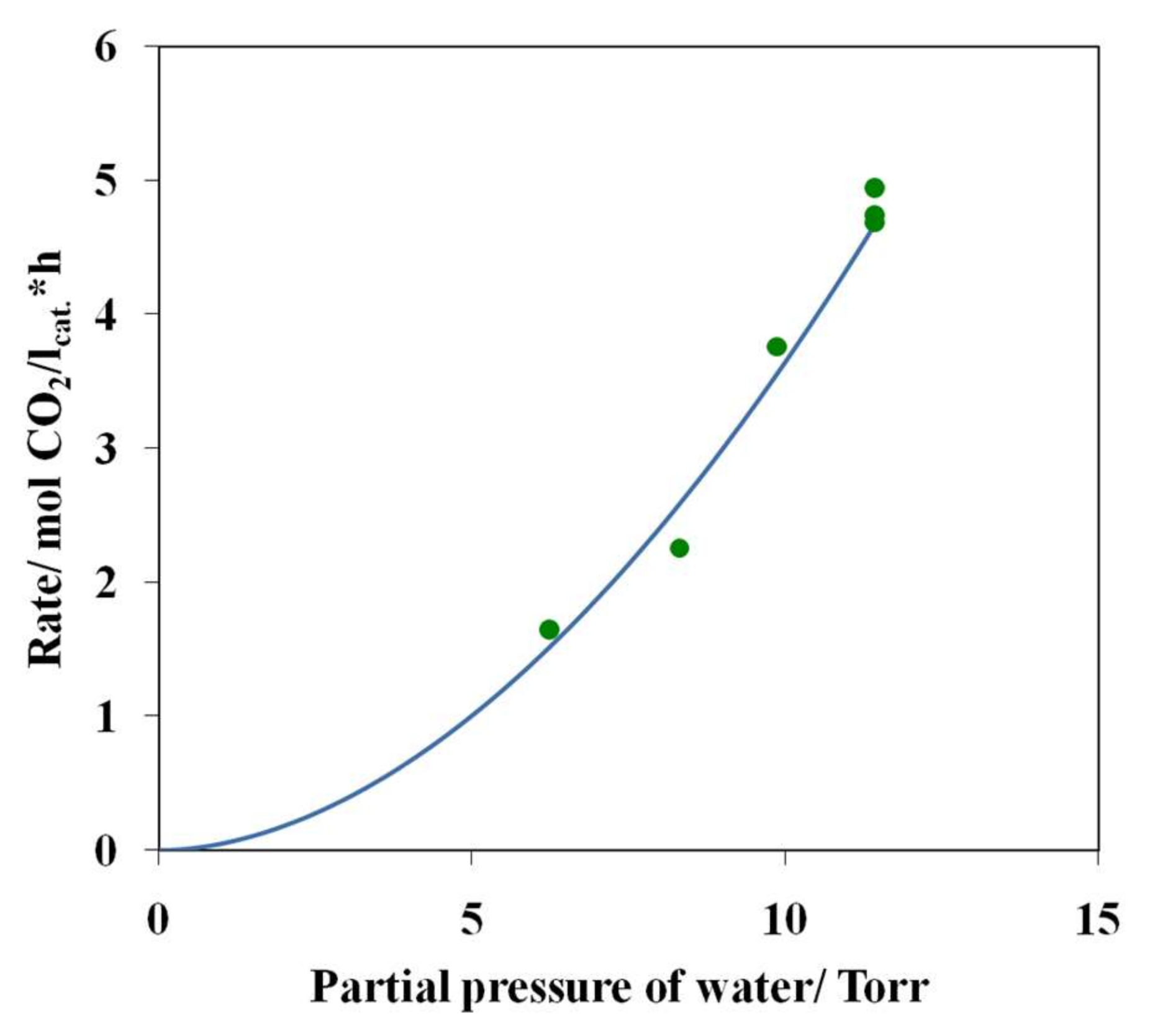
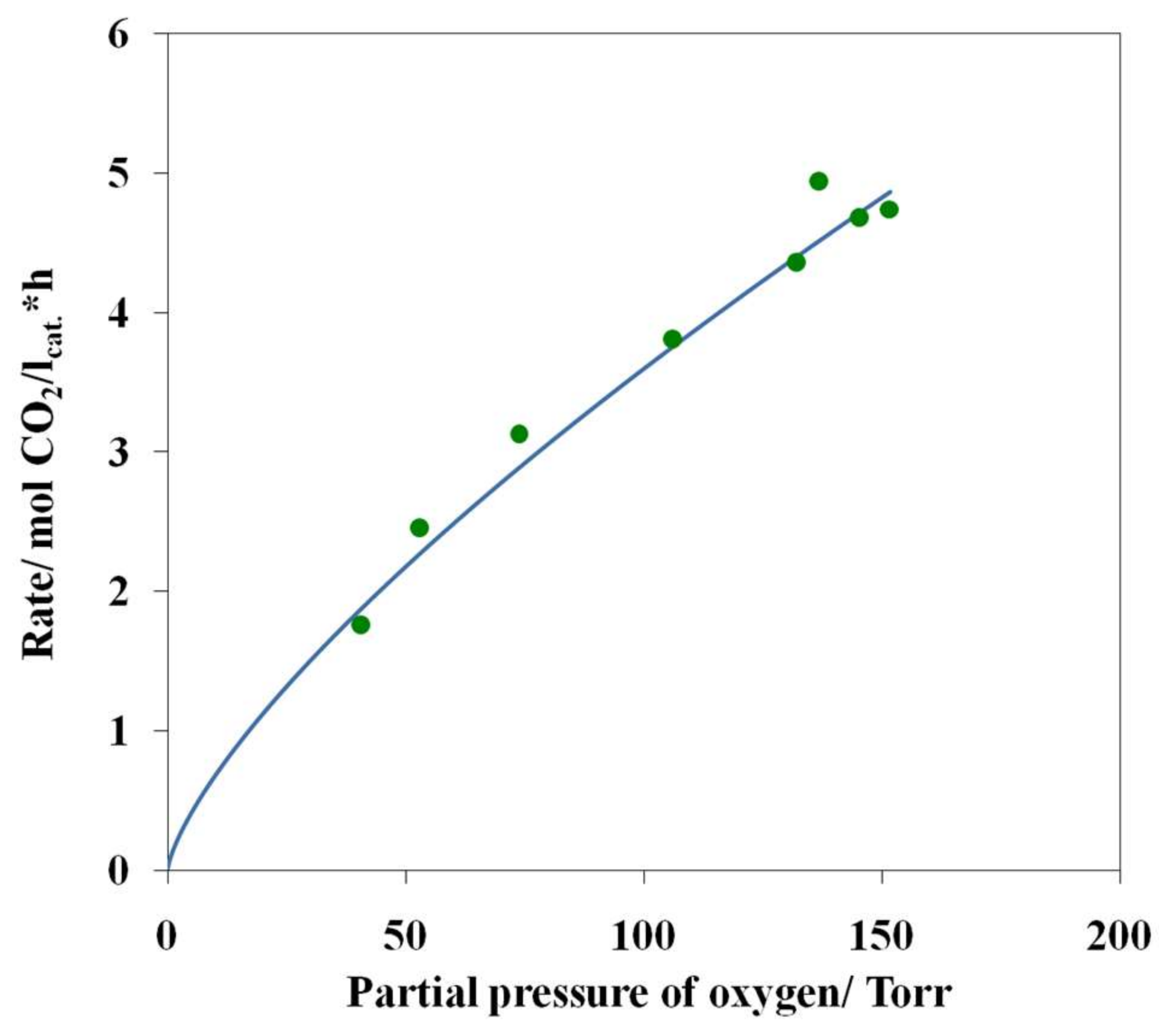
- The simultaneous presence of optimum amounts of oxygen and water vapor in the reaction mixture is a necessary condition for the active and steady operation of the low-temperature CO oxidation over the nanocatalyst PdCl2–CuCl2/γ-Al2O3.
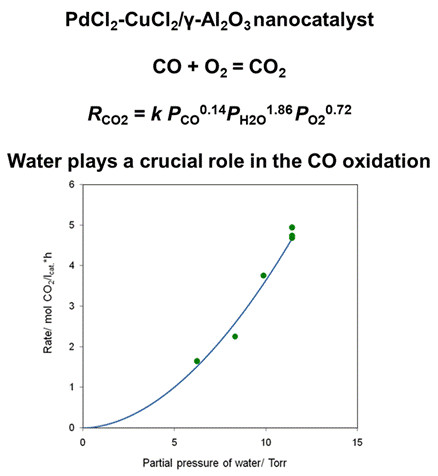
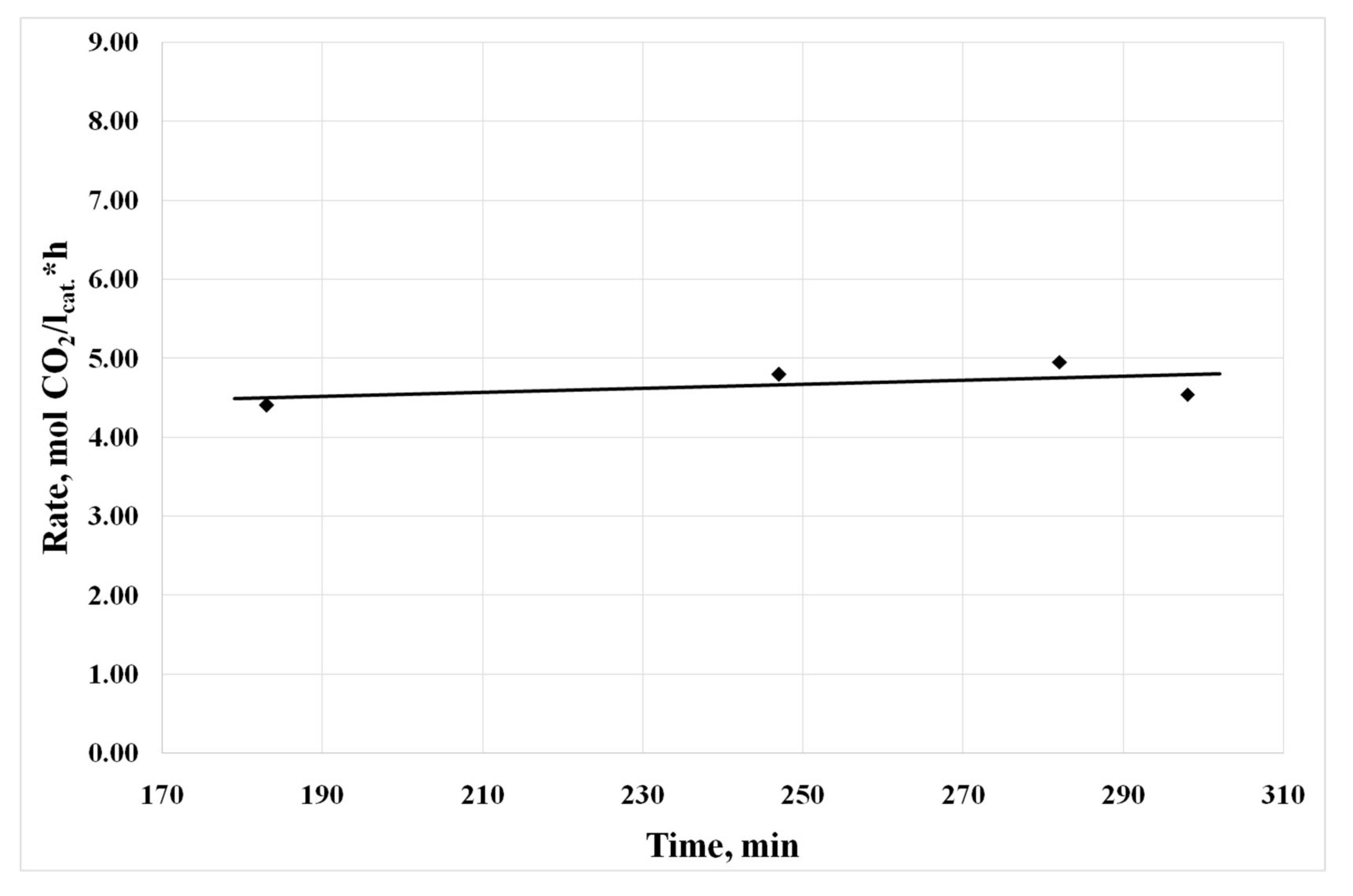
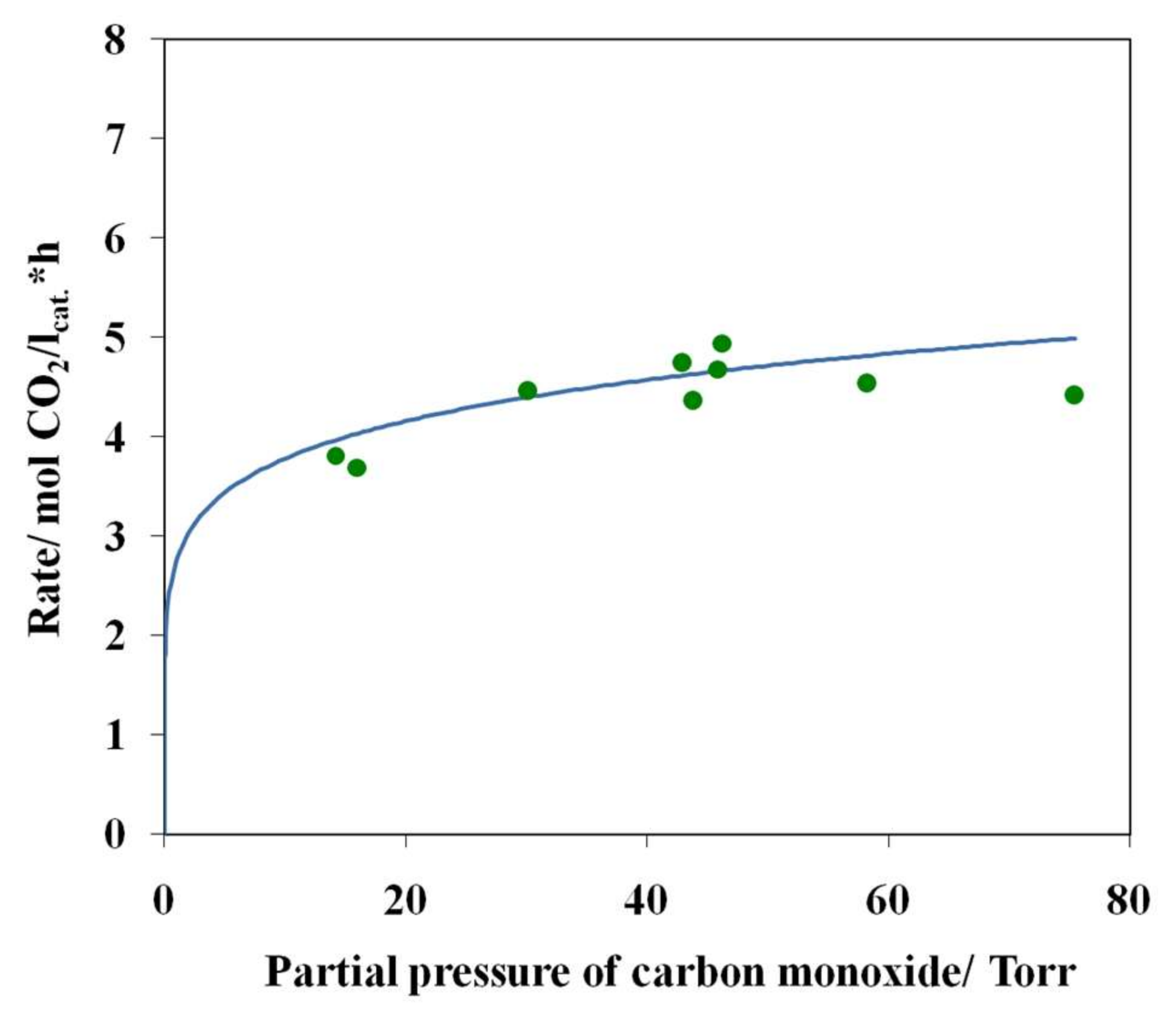
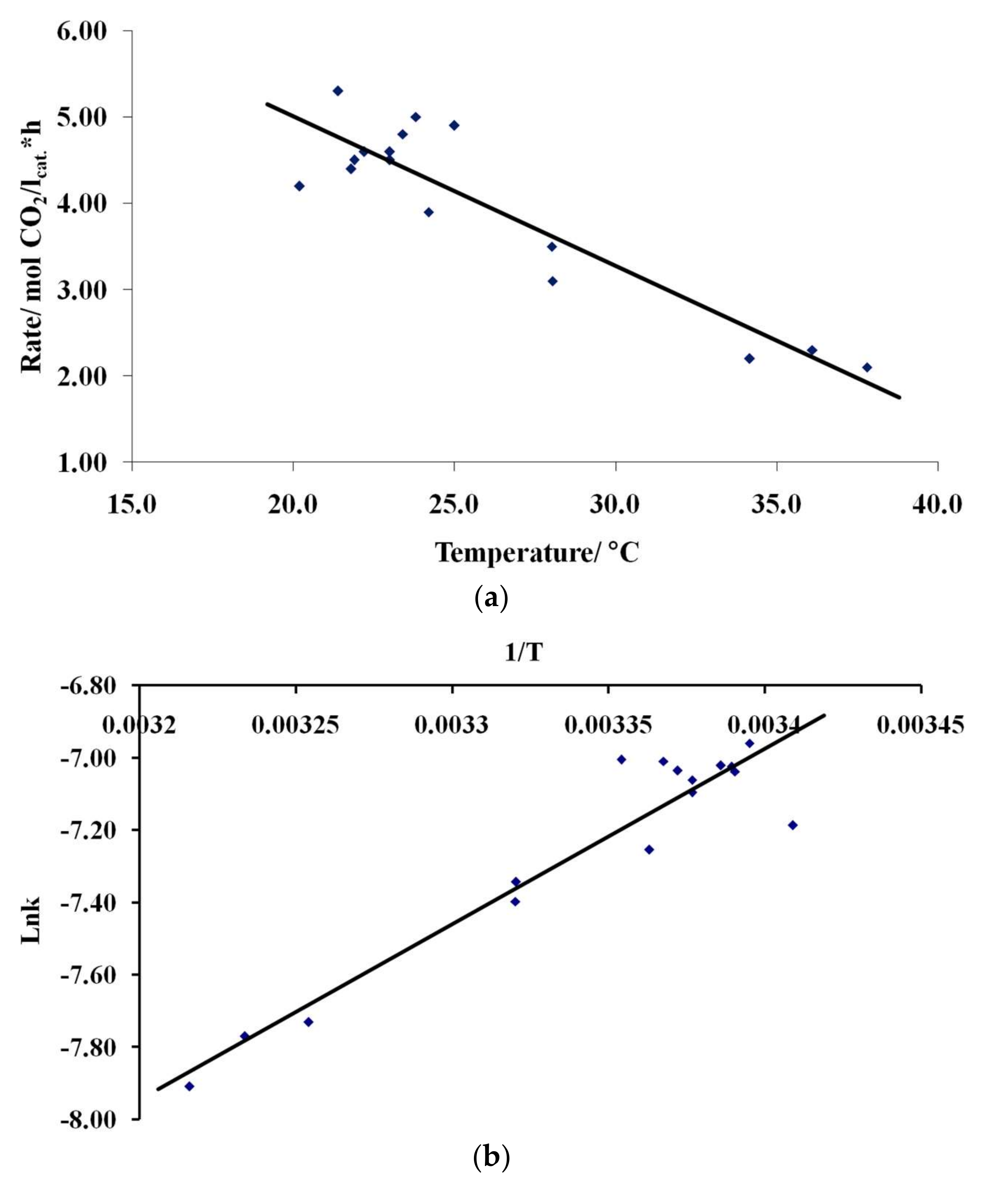
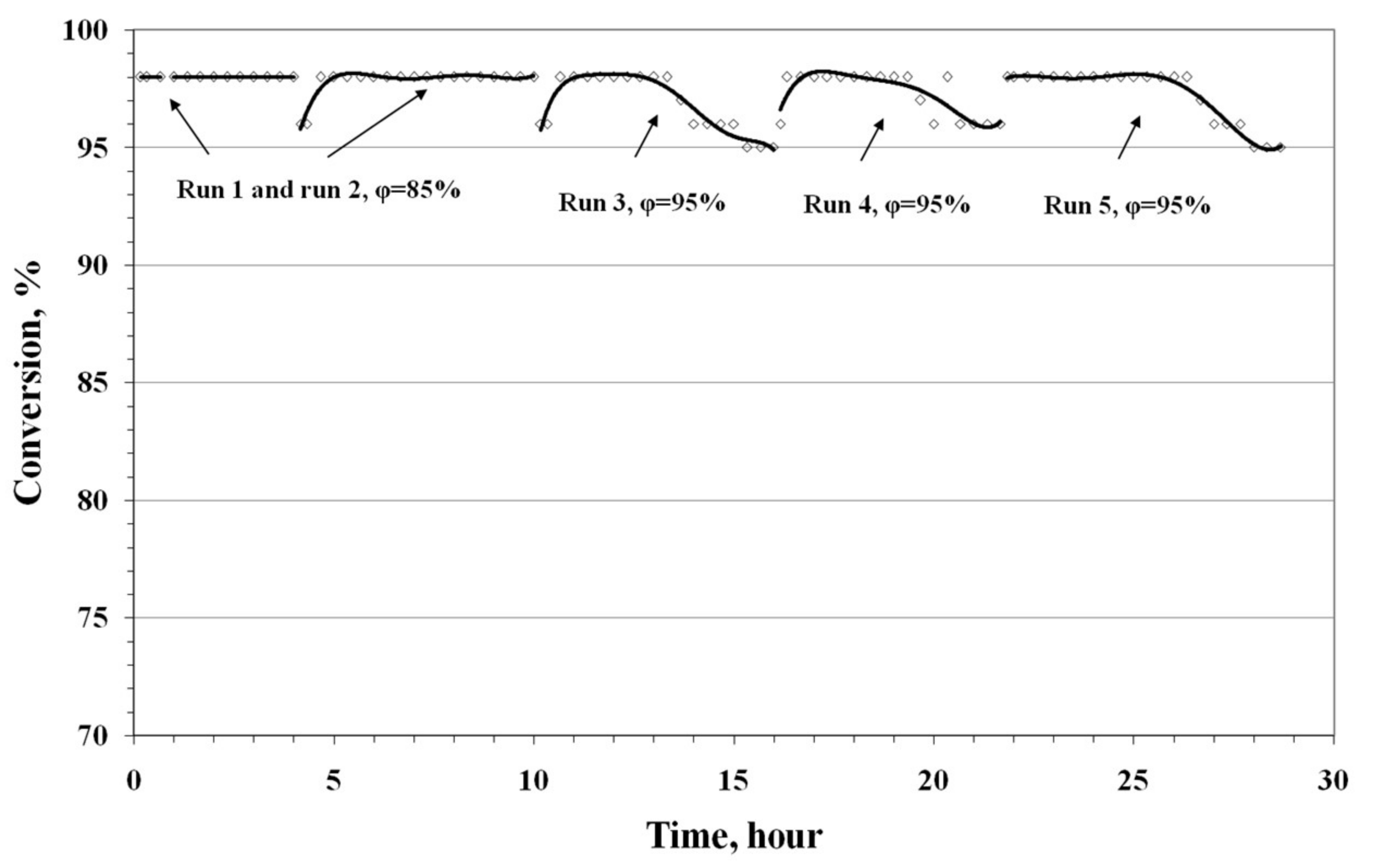
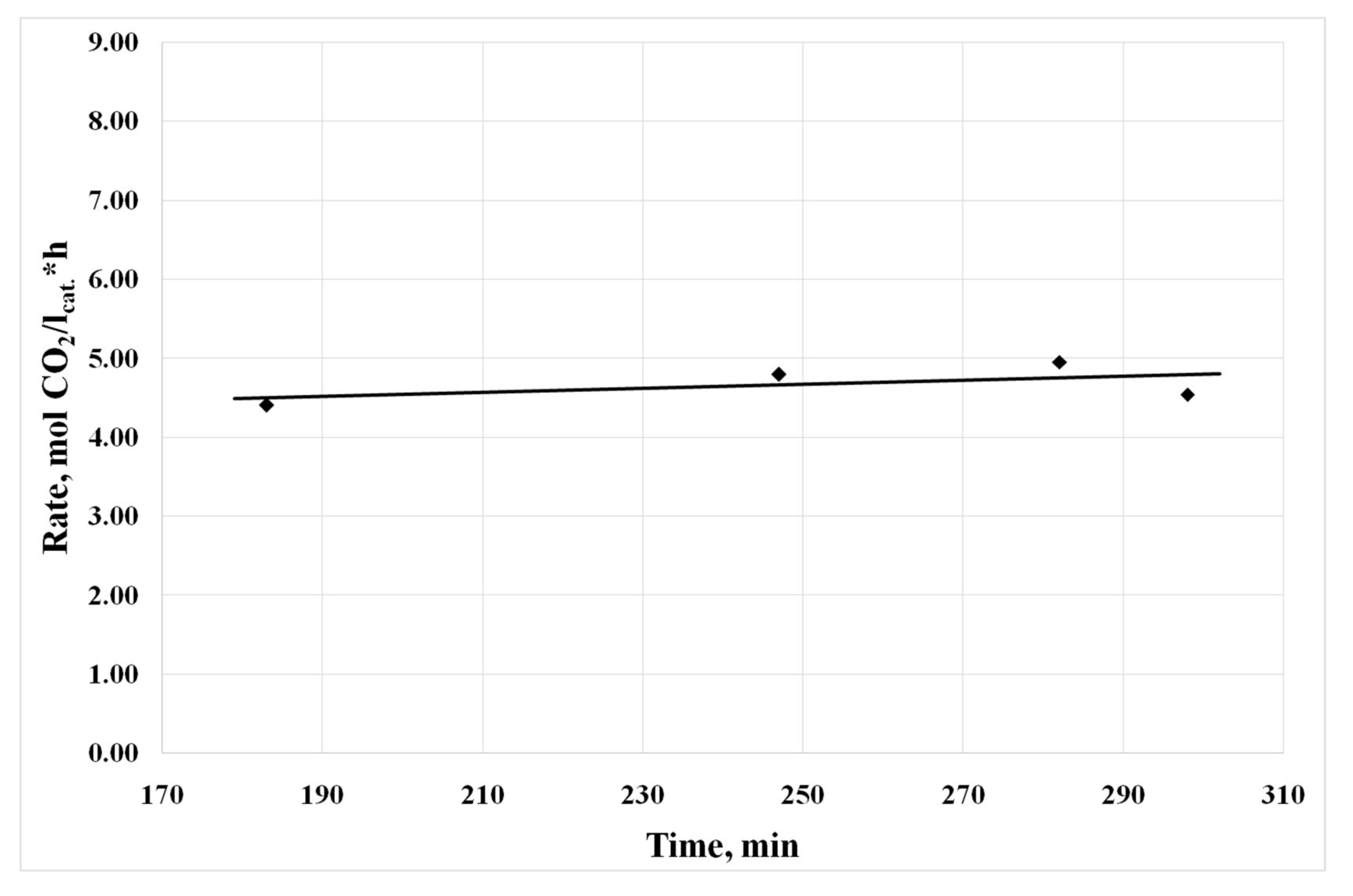
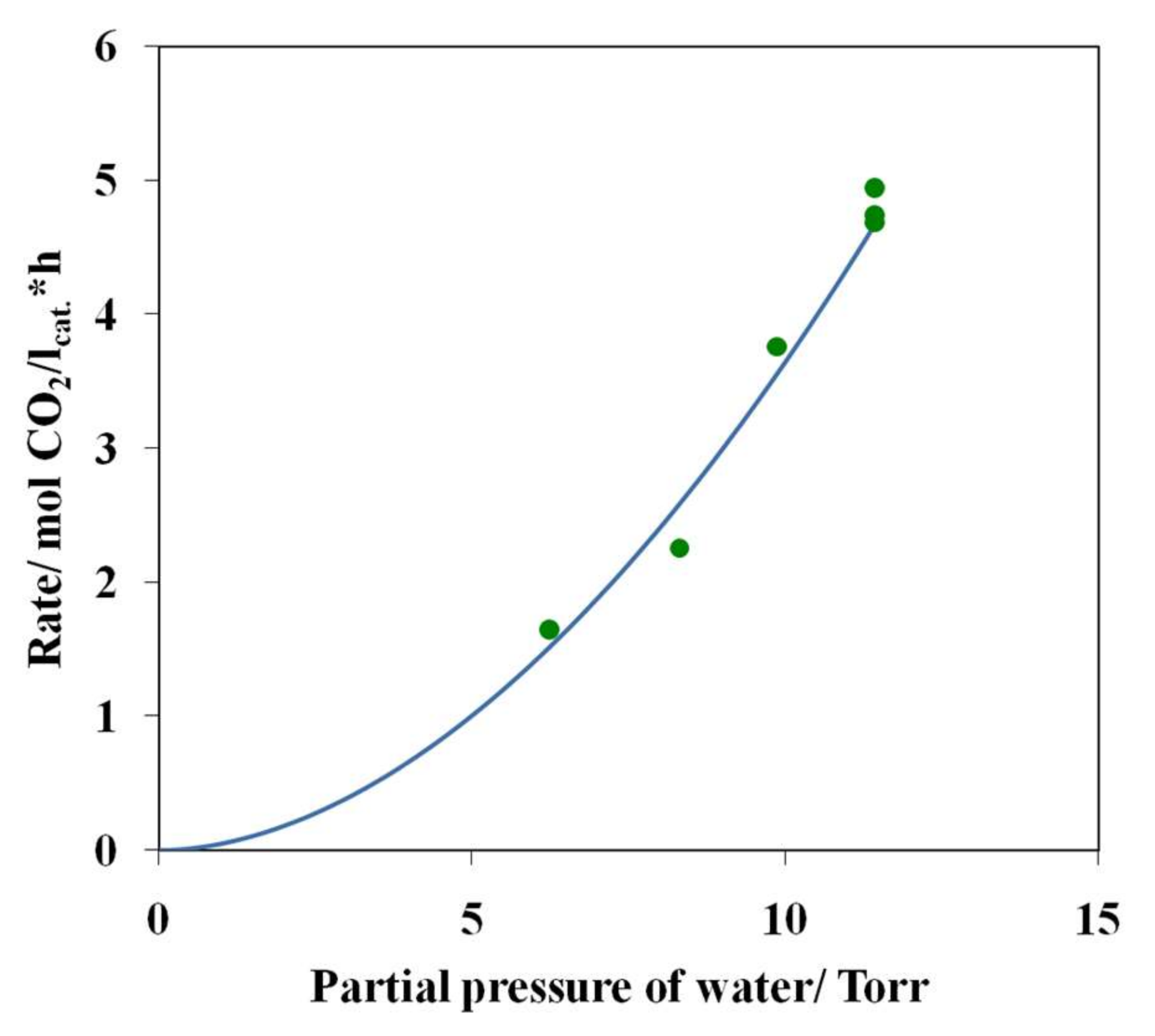
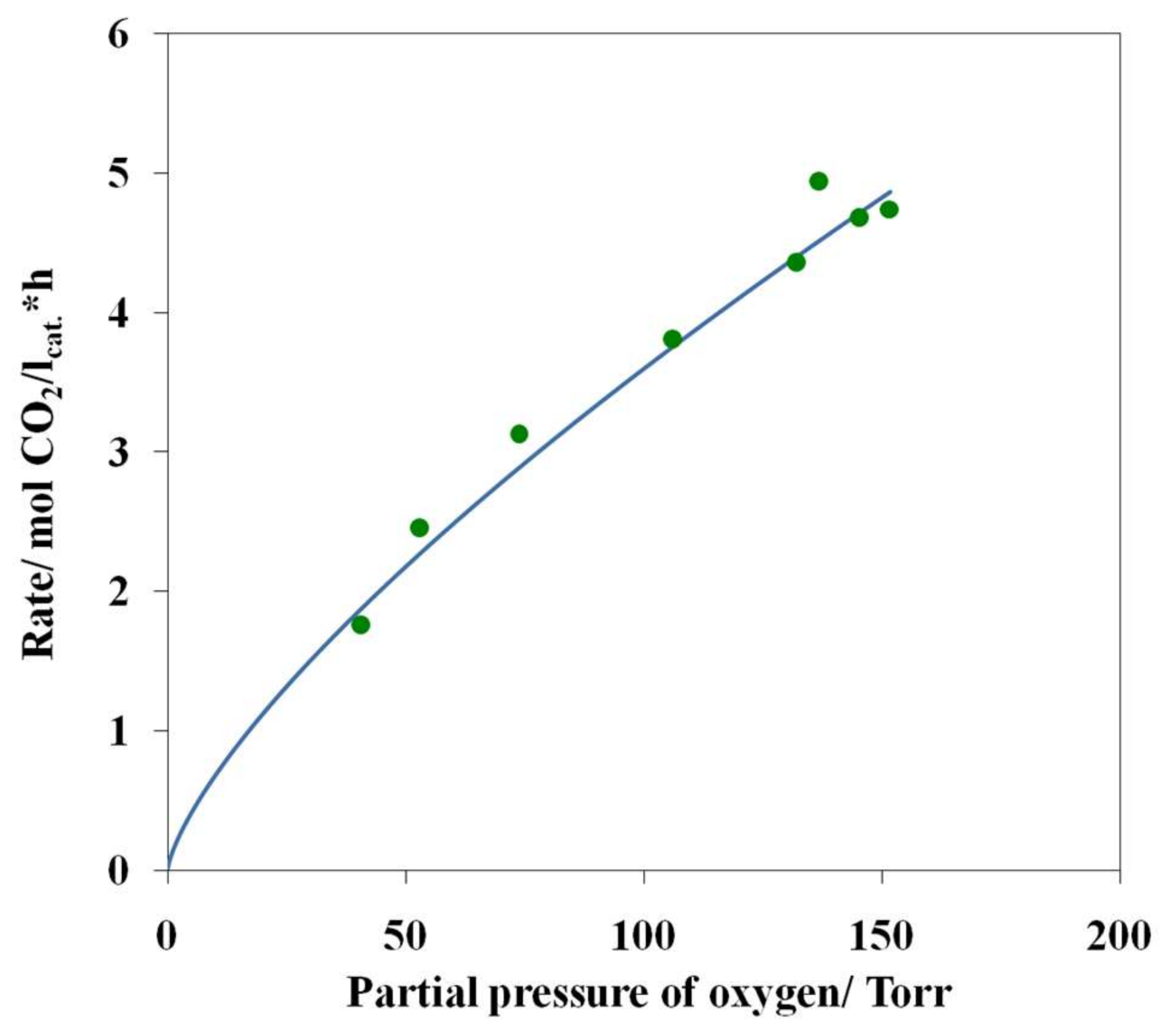
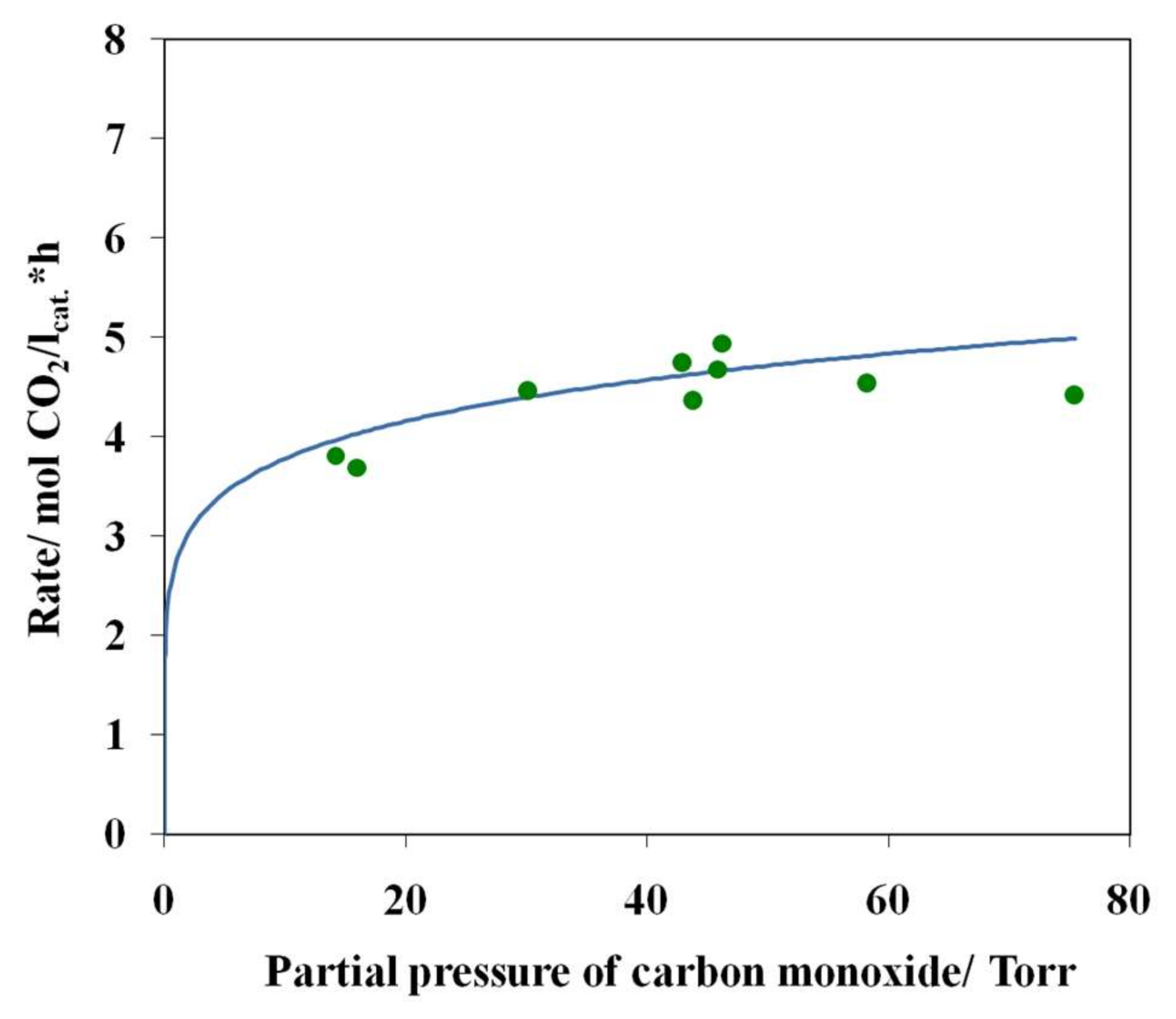
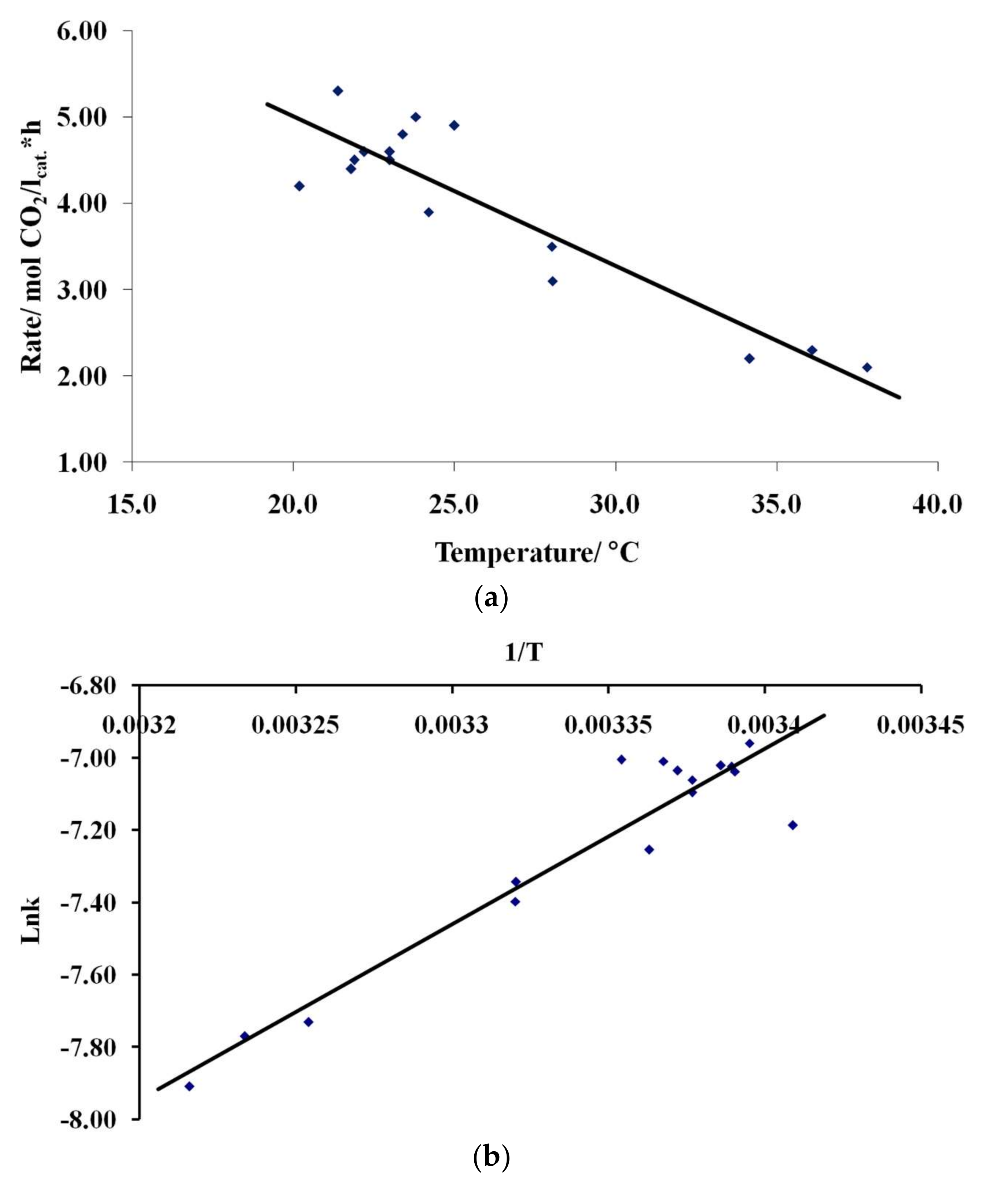
3.4. Results of Preliminary Kinetic Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Desai, M.N.; Butt, J.B.; Dranoff, J.S. Low-temperature oxidation of CO by a heterogenized Wacker catalyst. J. Catal. 1983, 79, 95–103. [Google Scholar] [CrossRef]
- Kuznetsova, L.I.; Matveev, K.I.; Zhizhina, E.G. Oxidation of carbon monoxide by dioxygen in the presence of palladium catalysts. Prospects for the development of new low-temperature reaction catalysts. Kinet. Catal. 1985, 25, 1029–1043. [Google Scholar]
- Rakitskaya, T.L.; Ennan, A.A.; Panina, V.Y. Catalysts for Low-Temperature Carbon Monoxide Oxidation; TSINTIKHIMNEFTEMASH: Moscow, Russian, 1991; p. 35. (In Russian) [Google Scholar]
- Golodov, V.A.; Sokolsky, D.V.; Noskova, N.F. Catalytic properties of complexes in solutions and on supports in the activation of simple molecules. J. Mol. Catal. 1977, 3, 51–60. [Google Scholar] [CrossRef]
- Golodov, V.A.; Sheludyakov, Y.L.; Di, R.I.; Fokanov, V.K. The role of intermediate carbonyl complexes in the reduction of Pd(II) and Cu(II) by carbon monoxide. Kinet. Catal. 1977, 18, 234–237. [Google Scholar]
- Golodov, V.A.; Kuksenko, E.L.; Taneeva, G.V.; Alekseev, A.M.; Geminova, M.V. Reduction of copper(II) salts by carbon monoxide in aqueous solutions of palladium(II) complexes. Kinet. Catal. 1984, 25, 268–272. [Google Scholar]
- Santos, V.P.; Carabineiro, S.A.C.; Bakker, J.J.W.; Soares, O.S.G.P.; Chen, X.; Pereira, M.F.R.; Órfão, J.J.M.; Figueiredo, J.L.; Gascon, J.; Kapteijn, F. Stabilized gold on cerium-modified cryptomelane: Highly active in low-temperature CO oxidation. J. Catal. 2014, 309, 58–65. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C.; Silva, A.M.T.; Dražić, G.; Tavares, P.B.; Figueiredo, J.L. Effect of chloride on the sinterization of Au/CeO2 catalysts. Catal. Today 2010, 154, 293–302. [Google Scholar] [CrossRef]
- Kakhniashvili, G.N.; Mishchenko, Y.A.; Dulin, D.A.; Isaeva, E.G.; Gelbshtein, A.I. On the mechanism of oxidation of carbon monoxide on supported catalysts. Kinet. Catal. 1985, 26, 134–140. [Google Scholar]
- Choi, K.I.; Vannice, M.A. CO oxidation over Pd and Cu catalysts I. Unreduced PdCl2 and CuCl2 dispersed on alumina or carbon. J. Catal. 1991, 127, 465–488. [Google Scholar] [CrossRef]
- Choi, K.I.; Vannice, M.A. CO oxidation over Pd and Cu catalysts II. Unreduced bimetallic PdCl2–CuCl2 dispersed on Al2O3 or carbon. J. Catal. 1991, 127, 489–511. [Google Scholar]
- Lee, J.S.; Park, E.D. In situ XAFS characterization of supported homogeneous catalysts. Top. Catal. 2002, 18, 67–72. [Google Scholar] [CrossRef]
- Kim, K.D.; Nam, I.S.; Chung, J.S.; Lee, J.S.; Ryu, S.G.; Yang, Y.S. Supported PdCl2–CuCl2 catalysts for carbon monoxide oxidation 1. Effects of catalyst composition and reaction conditions. Appl. Catal. B 1994, 5, 103–115. [Google Scholar] [CrossRef]
- Park, E.D.; Lee, J.S. Effect of surface treatment of the support on CO oxidation over carbone-supported wacker-type catalysts. J. Catal. 2000, 193, 5–15. [Google Scholar]
- Lee, J.S.; Choi, S.H.; Kim, K.D.; Nomura, M. Supported PdCl2–CuCl2 catalysts for carbon monoxide oxidation II. XAFS characterization. Appl. Catal. B 1996, 7, 199–212. [Google Scholar] [CrossRef]
- Choi, S.H.; Lee, J.S. XAFS characterization of supported PdCl2–CuCl2 catalysts for CO oxidation. React. Kinet. Catal. Lett. 1996, 57, 227–236. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Matsuzaki, T.; Ohdan, K.; Okamoto, Y. Structure and electronic state of PdCl2–CuCl2 catalysts supported on activated carbon. J. Catal. 1996, 161, 577–586. [Google Scholar]
- Koh, D.J.; Song, J.H.; Ham, S.W.; Nam, I.S.; Chang, R.W.; Park, E.D.; Lee, J.S.; Kim, Y.G. Low temperature oxidation of CO over supported PdCl2–CuCl2 catalysts. Korean J. Chem. Eng. 1997, 14, 486–490. [Google Scholar] [CrossRef]
- Park, E.D.; Lee, J.S. Effects of copper phase on CO oxidation over supported wacker-type catalysts. J. Catal. 1998, 180, 123–131. [Google Scholar] [CrossRef]
- Park, E.D.; Lee, J.S. Stabilization of molecular Pd species in a heterogenizedwacker-type catalyst for low temperature CO oxidation. Stud. Surf. Sci. Catal. 2000, 130, 2309–2314. [Google Scholar]
- Park, E.D.; Choi, S.H.; Lee, J.S. Active States of Pd and Cu in Carbon-Supported Wacker-Type Catalysts for Low-Temperature CO Oxidation. J. Phys. Chem. B 2000, 104, 5586–5594. [Google Scholar] [CrossRef]
- Moiseev, I.I. π-Complexes in Liquid-Phase Olefin Oxidations; Nauka: Moscow, Russian, 1970. (In Russian) [Google Scholar]
- Sheludyakov, Y.L.; Golodov, V.A. Reduction of Pd(II) by carbon monoxide in presence of additives in acidic and alkaline media. React. Kinet. And Catal. Lett. 1976, 4, 373–379. [Google Scholar]
- Titov, D.N.; Ustyugov, A.V.; Tkachenko, O.P.; Kustov, L.M.; Zubavichus, Y.V.; Veligzhanin, A.A.; Sadovskaya, N.V.; Oshanina, I.V.; Bruk, L.G.; Temkin, O.N. State of active components on the surface of the PdCl2–CuCl2/γ-Al2O3 catalyst for the low_temperature oxidation of carbon monoxide. Kinet. Catal. 2012, 53, 262–273. [Google Scholar] [CrossRef]
- Hammersley, A.P. FIT2D v9.129: Reference Manual; Version 3.1; ESRF Internal Report ESRF98HA01T; European Synchrotron Radiation Facility (ESRF): Grenoble, France, 1998; Available online: http://xray.tamu.edu/pdf/manuals/fit2d_ref_10.3.pdf (accessed on 30 March 2018).
- Chernyshov, A.A.; Veligzhanin, A.A.; Zubavichus, Y.V. Structural materials science end-station at the Kurchatov Synchrotron Radiation Source: Recent instrumentation upgrades and experimental results. Nucl. Instrum. Methods Phys. Res. Sect. A 2009, 603, 95–98. [Google Scholar] [CrossRef]
- Veligzhanin, A.A.; Zubavichus, Y.V.; Chernyshov, A.A.; Trigub, A.L.; Khlebnikov, A.S.; Nizovskii, A.I.; Khudorozhkov, A.K.; Beck, I.E.; Bukhtiyarov, V.I. An in-situ cell for investigation of the catalyst structure using synchrotron radiation. J. Struct. Chem. 2010, 51, S20–S27. [Google Scholar] [CrossRef]
- Kustov, L.M. New trends in IR-spectroscopic characterization of acid and basic sites in zeolites and oxide catalysts. Top. Catal. 1997, 4, 131–144. [Google Scholar] [CrossRef]
- Vargaftik, M.N.; Stromnova, T.A.; Moiseev, I.I. Carbonyl-complexes of palladium. J. Inorg. Chem. 1980, 25, 236–244. [Google Scholar]
- Temkin, O.N.; Bruk, L.G. Palladium(I) Complexes in Coordination Chemistry and Catalysis. Rus. Chem. Rev. 1983, 52, 206–243. [Google Scholar] [CrossRef]
- Davydov, A.A. Molecular Spectroscopy of Oxide Catalyst Surfaces; Wiley Interscience Publisher: Hoboken, NJ, USA, 2003; 466p. [Google Scholar]
- Goggin, P.L.; Goodfellow, R.J.; Herbert, I.R.; Orpen, A.J. Bridging by carbonyl vs. halide ligands: X-ray crystal structure of [NBu4n]2[Pd2Cl4(µ-CO)2]. J. Chem. Soc. Chem. Commun. 1981, 20, 1077–1079. [Google Scholar] [CrossRef]
- Shen, Y.; Lu, G.; Guo, Y.; Wang, Y.; Guo, Y.; Gong, X. Study on the catalytic reaction mechanism of low temperature oxidation of CO over Pd–Cu–Clx/Al2O3 catalyst. Catal. Today 2011, 175, 558–567. [Google Scholar] [CrossRef]
- Hosokawa, T.; Takano, M.; Murahashi, S.-L. The first isolation and characterization of a palladium−copper heterometallic complex Bearing μ4-oxo atom derived from molecular oxygen. J. Am. Chem. Soc. 1996, 118, 3990–3991. [Google Scholar] [CrossRef]
- Hosokawa, T.; Murahashi, S.-L. Palladium-copper-DMF complexes involved in the oxidation of alkenes. J. Organomet. Chem. 1998, 551, 387–389. [Google Scholar] [CrossRef]
- Eremin, D.B.; Ananikov, V.P. Understanding Active Species in Catalytic Transformations: From Molecular Catalysis to Nanoparticles, Leaching, “Cocktails” of Catalysts and Dynamic Systems. Coord. Chem. Rev. 2017, 346, 2–19. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Gordeev, E.G.; Egorov, M.P.; Sakharov, A.M.; Zlotin, S.G.; Redina, E.A.; Isaeva, V.I.; Kustov, L.M.; Gening, M.L.; Nifantiev, N.E. Challenges in the Development Organic and Hybrid Molecular Systems. Mend. Commun. 2016, 26, 365–374. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Eremin, D.B.; Yakukhnov, S.A.; Dilman, A.D.; Levin, V.V.; Egorov, M.P.; Karlov, S.S.; Kustov, L.M.; Tarasov, A.L.; Greish, A.A.; et al. Organic and hybrid systems: From science to practice. Mendeleev Commun. 2017, 27, 425–438. [Google Scholar] [CrossRef]
- Kuchurov, I.V.; Zharkov, M.N.; Fershtat, L.L.; Makhova, N.N.; Zlotin, S.G. Prospective Symbiosis of Green Chemistry and Energetic Materials. ChemSusChem 2017, 10, 3914. [Google Scholar] [CrossRef] [PubMed]
- Kustov, L.M. New organic–inorganic hybrid molecular systems and highly organized materials in catalysis. Rus. J. Phys. Chem. 2015, 89, 2006–2021. [Google Scholar] [CrossRef]
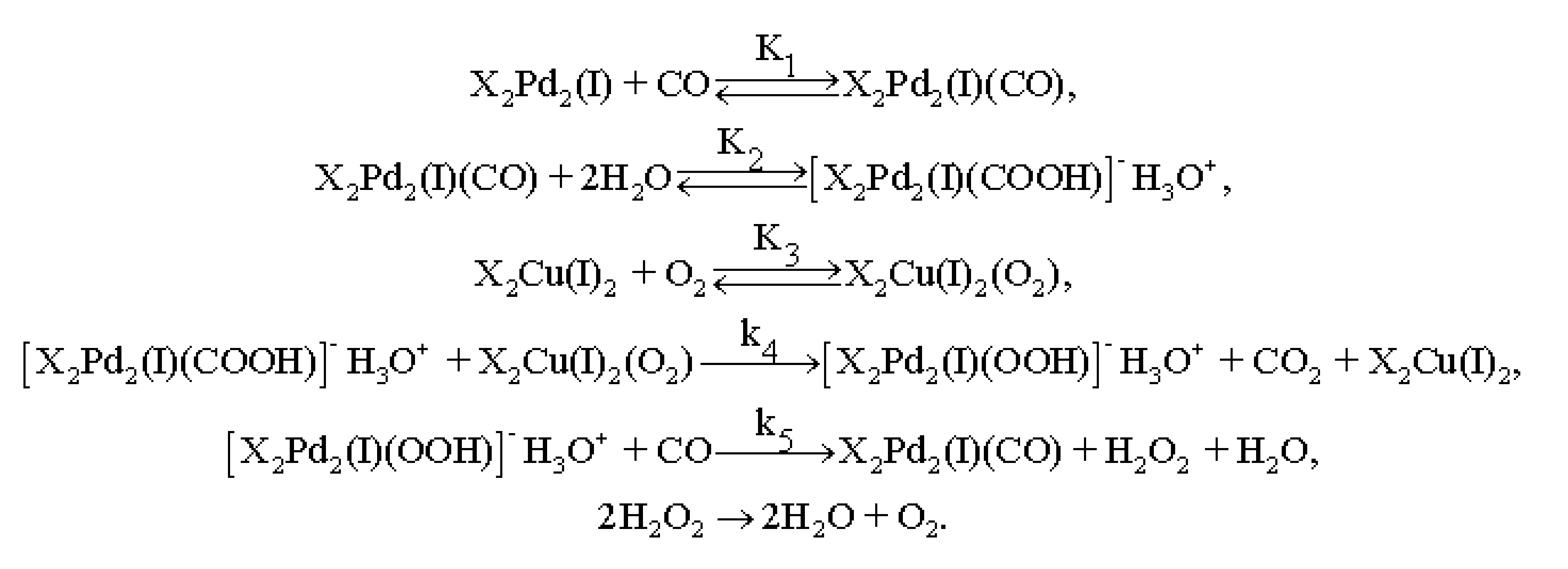
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruk, L.; Titov, D.; Ustyugov, A.; Zubavichus, Y.; Chernikova, V.; Tkachenko, O.; Kustov, L.; Murzin, V.; Oshanina, I.; Temkin, O. The Mechanism of Low-Temperature Oxidation of Carbon Monoxide by Oxygen over the PdCl2–CuCl2/γ-Al2O3 Nanocatalyst. Nanomaterials 2018, 8, 217. https://doi.org/10.3390/nano8040217
Bruk L, Titov D, Ustyugov A, Zubavichus Y, Chernikova V, Tkachenko O, Kustov L, Murzin V, Oshanina I, Temkin O. The Mechanism of Low-Temperature Oxidation of Carbon Monoxide by Oxygen over the PdCl2–CuCl2/γ-Al2O3 Nanocatalyst. Nanomaterials. 2018; 8(4):217. https://doi.org/10.3390/nano8040217
Chicago/Turabian StyleBruk, Lev, Denis Titov, Alexander Ustyugov, Yan Zubavichus, Valeriya Chernikova, Olga Tkachenko, Leonid Kustov, Vadim Murzin, Irina Oshanina, and Oleg Temkin. 2018. "The Mechanism of Low-Temperature Oxidation of Carbon Monoxide by Oxygen over the PdCl2–CuCl2/γ-Al2O3 Nanocatalyst" Nanomaterials 8, no. 4: 217. https://doi.org/10.3390/nano8040217
APA StyleBruk, L., Titov, D., Ustyugov, A., Zubavichus, Y., Chernikova, V., Tkachenko, O., Kustov, L., Murzin, V., Oshanina, I., & Temkin, O. (2018). The Mechanism of Low-Temperature Oxidation of Carbon Monoxide by Oxygen over the PdCl2–CuCl2/γ-Al2O3 Nanocatalyst. Nanomaterials, 8(4), 217. https://doi.org/10.3390/nano8040217