Uranyl Sulfate Nanotubules Templated by N-phenylglycine



Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Single Crystal X-ray Diffraction Studies
2.3. High-Temperature Powder X-ray Diffraction Studies
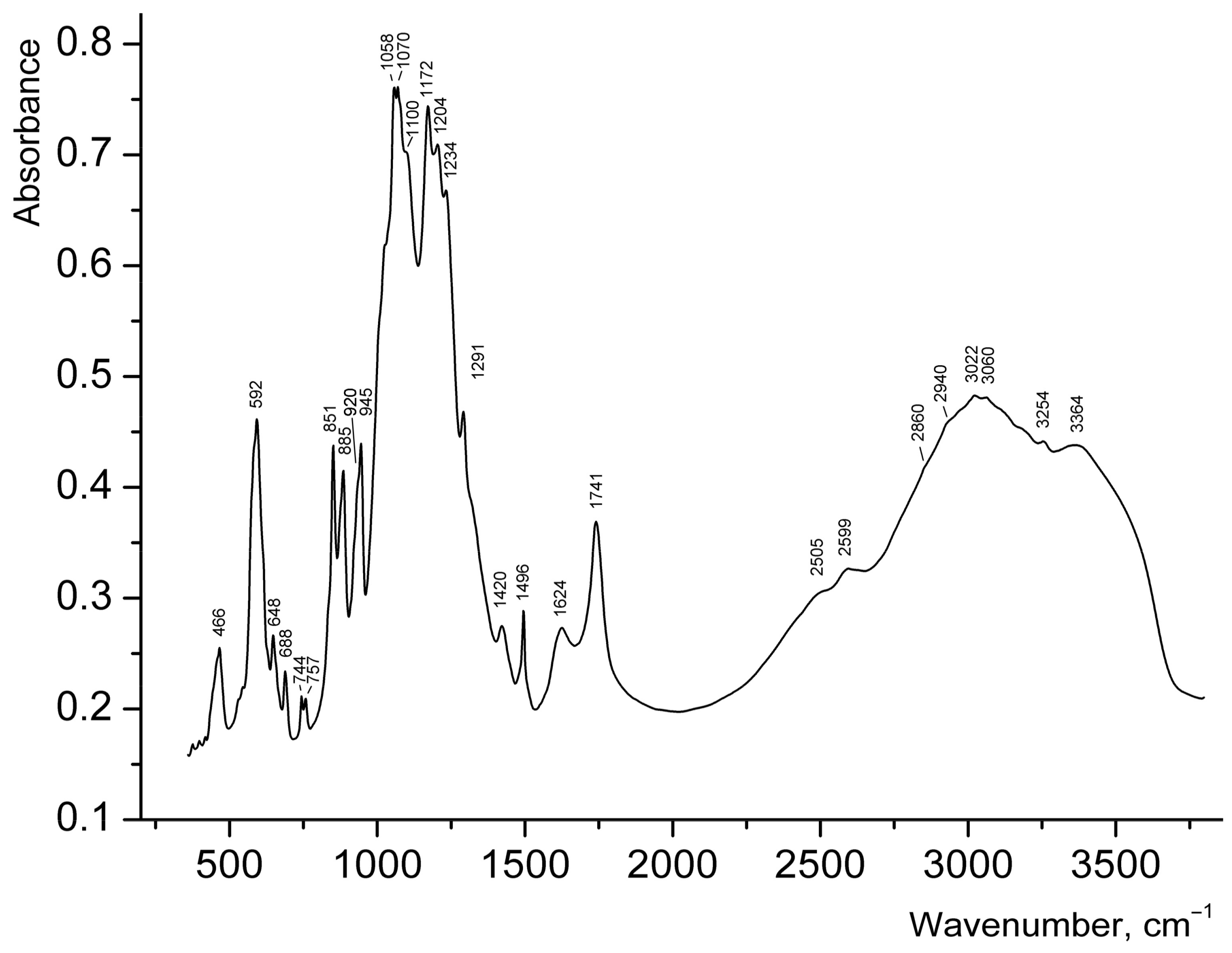
2.4. Infrared Spectroscopy
2.5. Crystal Surface Microtopography
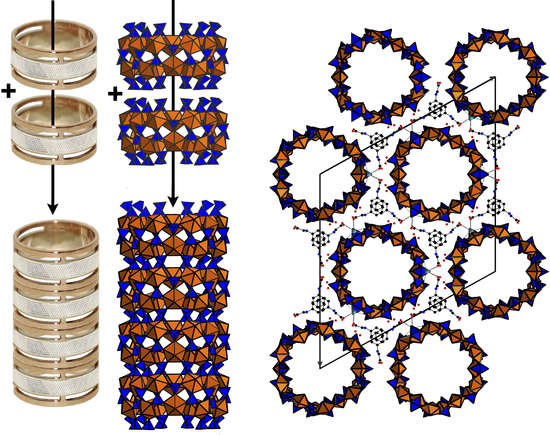
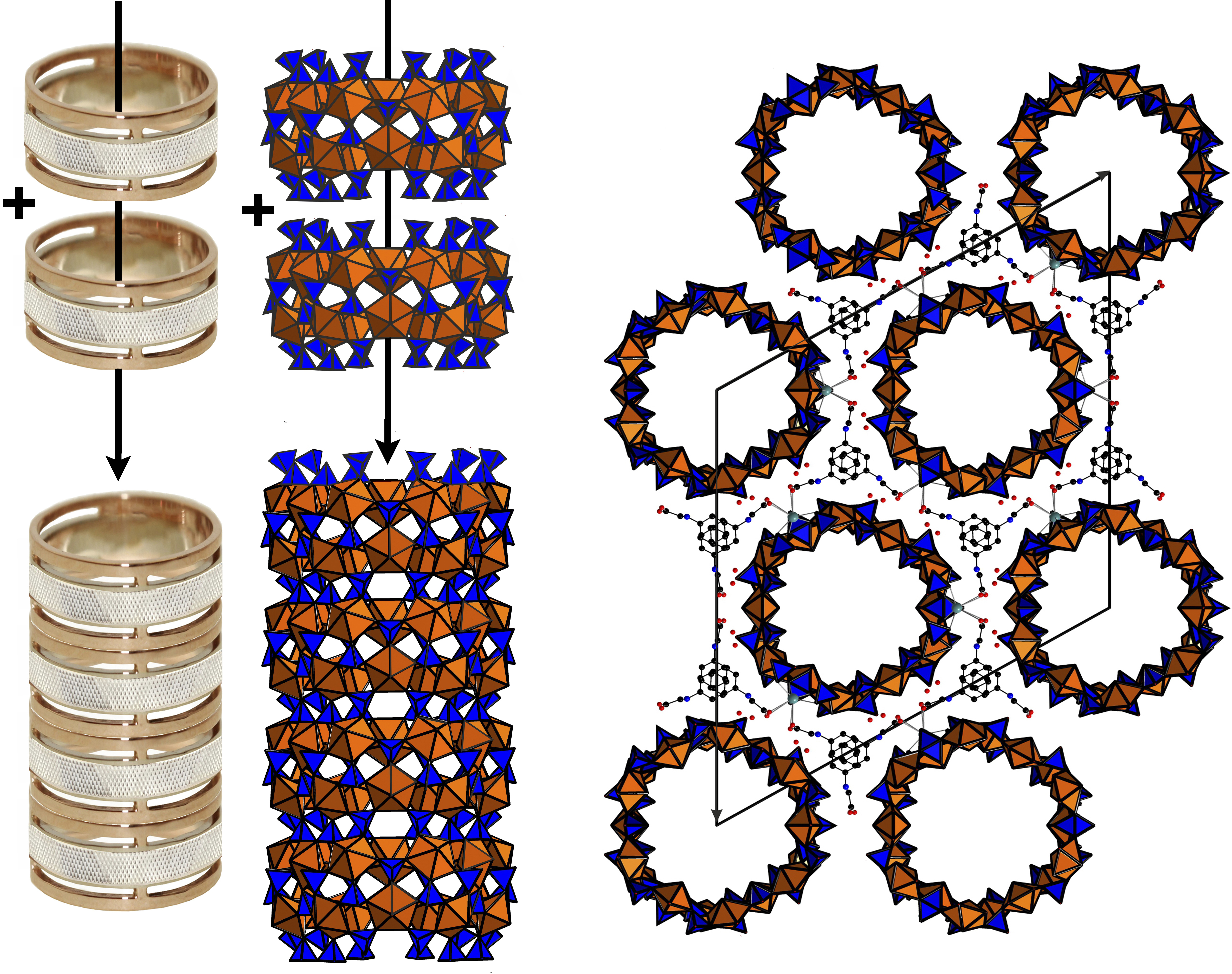
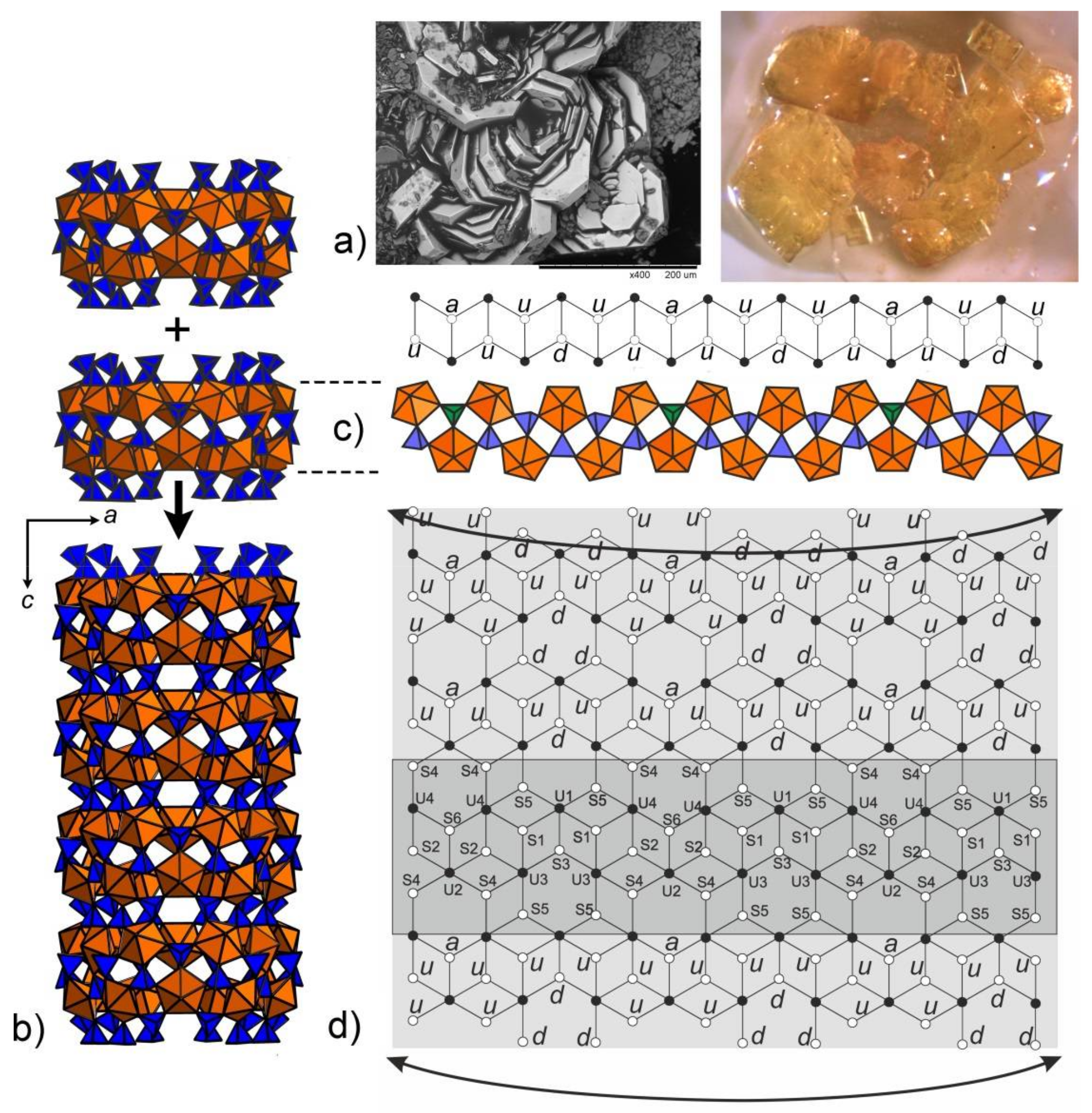
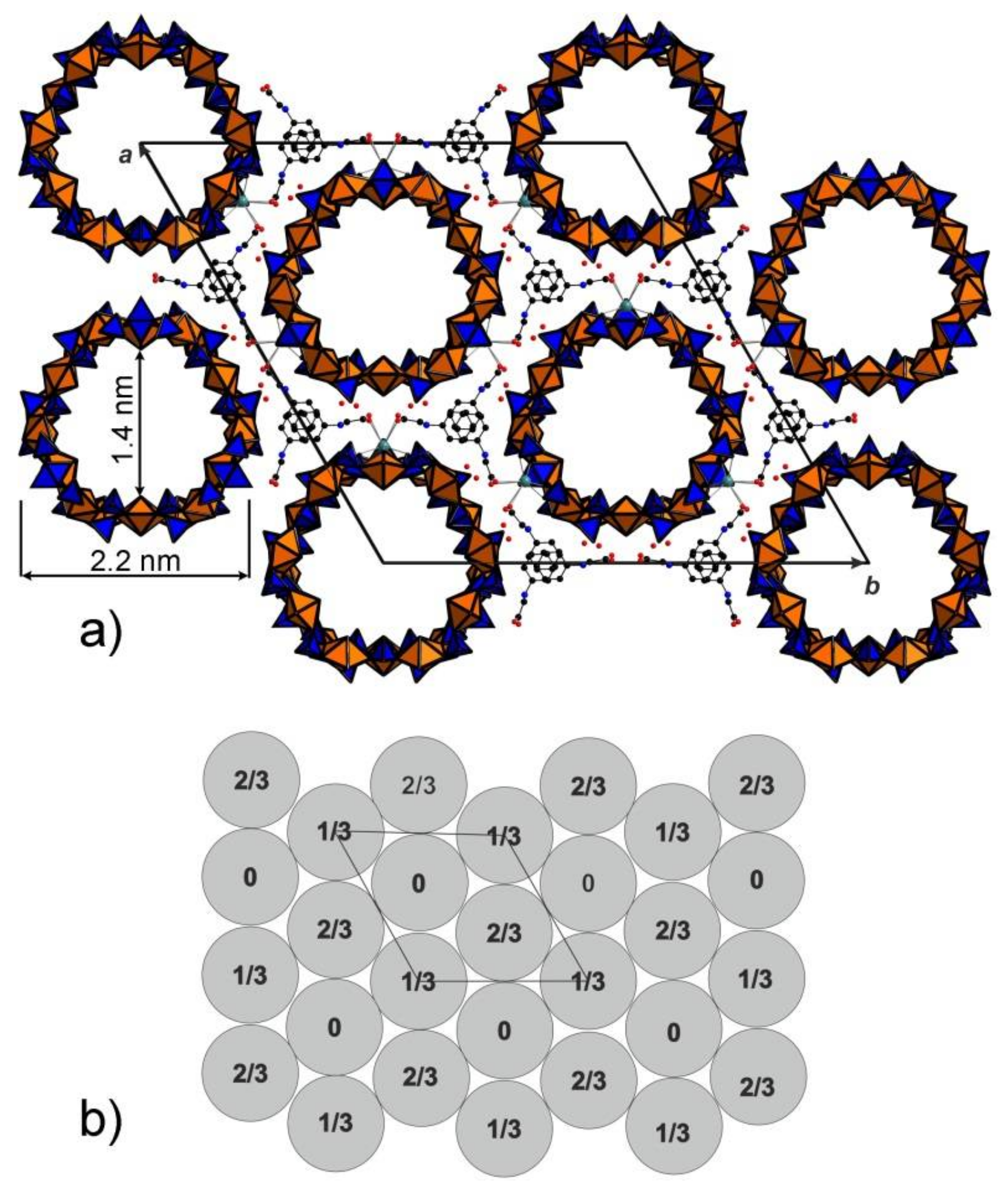
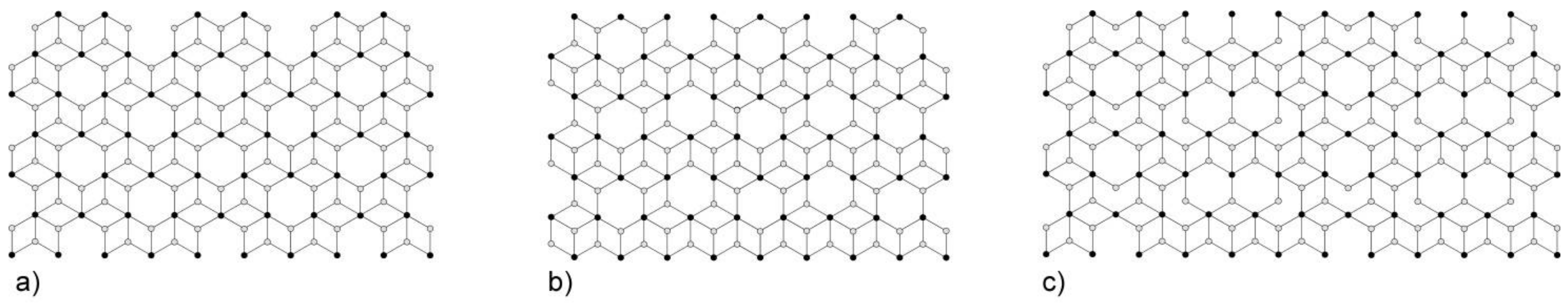
3. Results and Discussion
4. Final Remarks
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Finch, R.; Murakami, T. Systematics and paragenesis of uranium minerals. Rev. Mineral. Geochem. 1999, 38, 91–179. [Google Scholar]
- Jensen, K.A.; Ewing, R.C. The Okelobondo natural fission reactor, southeast Gabon: Geology, mineralogy, and retardation of nuclear-reaction products. Geol. Soc. Am. Bull. 2011, 113, 32–62. [Google Scholar] [CrossRef]
- Kampf, A.R.; Plášil, J.; Kasatkin, A.V.; Marty, J.; Čejka, J. Fermiite, Na4(UO2)(SO4)3·3H2O and oppenheimerite, Na2(UO2)(SO4)2·3H2O, two new uranyl sulfate minerals from the Blue Lizard mine, San Juan County, Utah, USA. Mineral. Mag. 2015, 79, 1123–1142. [Google Scholar] [CrossRef]
- Albrecht-Schmitt, T.E. Actinide materials adopt curvature: Nanotubules and nanospheres. Angew. Chem. Int. Ed. 2005, 44, 4836–4838. [Google Scholar] [CrossRef] [PubMed]
- Krivovichev, S.V.; Burns, P.C.; Tananaev, I.G. (Eds.) Structural Chemistry of Inorganic Actinide Compounds; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Siidra, O.I.; Nazarchuk, E.V.; Bocharov, S.N.; Depmeier, W.; Kayukov, R.A. Microporous uranyl chromates successively formed by evaporation from acidic solution. Z. Krist.-Cryst. Mater. 2018, 233, 1–8. [Google Scholar] [CrossRef]
- Krivovichev, S.V.; Kahlenberg, V.; Kaindl, R.; Mersdorf, E.; Tananaev, I.G.; Myasoedov, B.F. Nanoscale tubules in uranyl selenates. Angew. Chem. Int. Ed. 2005, 44, 1134–1136. [Google Scholar] [CrossRef] [PubMed]
- Alekseev, E.V.; Krivovichev, S.V.; Depmeier, W. A crown ether as template for microporous and nanostructured uranium compounds. Angew. Chem. Int. Ed. 2008, 47, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Krivovichev, S.V.; Kahlenberg, V.; Tananaev, I.G.; Kaindl, R.; Mersdorf, E.; Myasoedov, B.F. Highly porous uranyl selenate nanotubules. J. Am. Chem. Soc. 2005, 127, 1072–1073. [Google Scholar] [CrossRef] [PubMed]
- Adelani, P.O.; Albrecht-Schmitt, T.E. Differential ion exchange in elliptical uranyl diphosphonate nanotubules. Angew. Chem. Int. Ed. 2010, 49, 8909–8911. [Google Scholar] [CrossRef] [PubMed]
- Unruh, D.K.; Gojdas, K.; Libo, A.; Forbes, T.Z. Development of metal–organic nanotubes exhibiting low-temperature, reversible exchange of confined “ice channels”. J. Am. Chem. Soc. 2013, 135, 7398–7401. [Google Scholar] [CrossRef] [PubMed]
- Thuéry, P.; Harrowfield, J. Variations on the honeycomb topology: From triangular- and square-grooved networks to tubular assemblies in uranyl tricarballylate complexes. Cryst. Growth Des. 2017, 17, 963–966. [Google Scholar] [CrossRef]
- Gattermann, L. Laboratory Methods of Organic Chemistry; The Macmillan Company: New York, NY, USA, 1937. [Google Scholar]
- Hawthorne, F.C.; Krivovichev, S.V.; Burns, P.C. The crystal chemistry of sulfate minerals. Rev. Mineral. Geochem. 2000, 40, 1–112. [Google Scholar] [CrossRef]
- Nazarchuk, E.V.; Siidra, O.I.; Kayukov, R.A. Synthesis and crystal-chemical features of two new uranyl chromates with the structures derived from [(UO2)(T6+O4)(H2O)n]0 (T = Cr6+, S6+, Se6+, n = 0–2). Radiochemistry 2016, 58, 571–577. [Google Scholar] [CrossRef]
- Halasyamani, P.S.; Francis, R.J.; Walker, S.M.; O’Hare, D. New layered uranium(VI) molybdates: Syntheses and structures of (NH3(CH2)3NH3)(H3O)2(UO2)3(MoO4)5, C(NH2)3(UO2)(OH)(MoO4), (C4H12N2)(UO2)(MoO4)2, and (C5H14N2)(UO2)(MoO4)2·H2O. Inorg. Chem. 1999, 38, 271–279. [Google Scholar] [CrossRef]
- Grigor’ev, M.S.; Fedoseev, A.M.; Budantseva, N.A. Crystal structure of the mixed-valence neptunium compound Na6[(NpVO2)2(NpVIO2)(MoO4)5]·13H2O. Russ. J. Coord. Chem. 2003, 29, 877–879. [Google Scholar] [CrossRef]
- Krivovichev, S.V.; Kahlenberg, V. Synthesis and crystal structures of α- and β-Mg2[(UO2)3(SeO4)5](H2O)16. Z. Anorg. Allg. Chem. 2004, 630, 2736–2742. [Google Scholar] [CrossRef]
- Krivovichev, S.V.; Kahlenberg, V. Synthesis and crystal structures of M2[(UO2)3(SeO4)5](H2O)16 (M = Co, Zn). J. Alloys Compd. 2005, 395, 41–47. [Google Scholar] [CrossRef]
- Heacock, R.A.; Marion, L. The infrared spectra of secondary amines and their salts. Can. J. Chem. 1956, 34, 1782–1795. [Google Scholar] [CrossRef]
- Colthup, N.B.; Daly, L.H.; Wiberley, S.E. Introduction to Infrared and Raman Spectroscopy; Academic Press: New York, NY, USA, 1990; 547 p. [Google Scholar]
- Wilkins, R.W.T.; Mateen, A.; West, G.W. The spectroscopic study of oxonium ions in minerals. Am. Mineral. 1974, 59, 811–819. [Google Scholar]
- Čejka, J. Infrared spectroscopy and thermal analysis of the uranyl minerals. Rev. Mineral. 1999, 38, 521–622. [Google Scholar]
- Chukanov, N.V.; Chervonnyi, A.D. Infrared Spectroscopy of Minerals and Related Compounds; Springer: Cham, Switzerland, 2016; 1109 p. [Google Scholar]
- Hoekstra, H.R. Vibrational spectra. In Gmelin Handbook of Inorganic Chemistry Uranium Supplementum; Springer: Berlin, Germany, 1982; Volume 15, pp. 211–240. [Google Scholar]
- Zhang, X.; Brauer, N.B.; Berden, G.; Rijs, A.M. Mid-Infrared spectroscopy of molecular ions in helium nanodroplets. J. Chem. Phys. 2012, 136, 044305. [Google Scholar] [CrossRef] [PubMed]
- Tremel, W. Inorganic nanotubes. Angew. Chem. Int. Ed. 1999, 38, 2175–2179. [Google Scholar] [CrossRef]
- Tenne, R. Inorganic nanotubes and fullerene-like materials. Chem. Eur. J. 2002, 8, 5297–5304. [Google Scholar] [CrossRef]
- Krivovichev, S.V. Actinyl compounds with hexavalent elements (S, Cr, Se, Mo)—Structural diversity, nanoscale chemistry, and cellular automata modeling. Eur. J. Inorg. Chem. 2010, 2010, 2594–2603. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | |
| Crystal system | trigonal |
| Space group | R3m (No. 160) |
| a (Å) | 44.001(10) |
| c (Å) | 10.367(2) |
| V (Å3) | 17382(9) |
| Z | 1 |
| ρcalc (g cm−3) | 2.543 |
| Crystal size (mm3) | 0.15 × 0.15 × 0.10 |
| Data Collection | |
| Diffractometer | Bruker X8 APEX II (CCD) |
| Radiation, λ (Å) | MoKα, 0.71073 |
| μ (mm−1) | 12.91 |
| θ range (°) | 1.85–28.00 |
| No. of measured reflections | 37916 |
| Total number of reflections (Rint) | 8480 (0.04) |
| Unique reflections with |Fo| ≥ 4σF | 8212 |
| Refinement | |
| Refinement method | Full-matrix least-squares on F2 |
| Weighting coefficients a, b | 0.03680, 271.9710 |
| Data/restraints/parameters | 8480/1/455 |
| R1, wR2 (|Fo| ≥ 4σF) | 0.028, 0.076 |
| R1, wR2 (all data) | 0.029, 0.077 |
| GoF | 1.058 |
| largest diff. peak and hole (e Å−3) | 2.951, −0.987 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siidra, O.I.; Nazarchuk, E.V.; Charkin, D.O.; Chukanov, N.V.; Depmeier, W.; Bocharov, S.N.; Sharikov, M.I. Uranyl Sulfate Nanotubules Templated by N-phenylglycine. Nanomaterials 2018, 8, 216. https://doi.org/10.3390/nano8040216
Siidra OI, Nazarchuk EV, Charkin DO, Chukanov NV, Depmeier W, Bocharov SN, Sharikov MI. Uranyl Sulfate Nanotubules Templated by N-phenylglycine. Nanomaterials. 2018; 8(4):216. https://doi.org/10.3390/nano8040216
Chicago/Turabian StyleSiidra, Oleg I., Evgeny V. Nazarchuk, Dmitry O. Charkin, Nikita V. Chukanov, Wulf Depmeier, Sergey N. Bocharov, and Mikhail I. Sharikov. 2018. "Uranyl Sulfate Nanotubules Templated by N-phenylglycine" Nanomaterials 8, no. 4: 216. https://doi.org/10.3390/nano8040216
APA StyleSiidra, O. I., Nazarchuk, E. V., Charkin, D. O., Chukanov, N. V., Depmeier, W., Bocharov, S. N., & Sharikov, M. I. (2018). Uranyl Sulfate Nanotubules Templated by N-phenylglycine. Nanomaterials, 8(4), 216. https://doi.org/10.3390/nano8040216