Electrospun Composites of Polycaprolactone and Porous Silicon Nanoparticles for the Tunable Delivery of Small Therapeutic Molecules

 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Fabrication of pSi NPs
2.3. CPT Loading of pSi NPs and PCL Composite Materials
2.4. Water Contact Angle (WCA) Measurements
2.5. Electrospinning of PCL and NaOH Treatments
2.6. Scanning Electron Microscopy (SEM)
2.7. X-ray Photoelectron Spectroscopy (XPS)
2.8. Energy-Dispersive X-ray Spectroscopy (EDX)
2.9. Fluorescence Microscopy
2.10. Drug Release
2.11. Cytotoxicity Assay
3. Results and Discussion
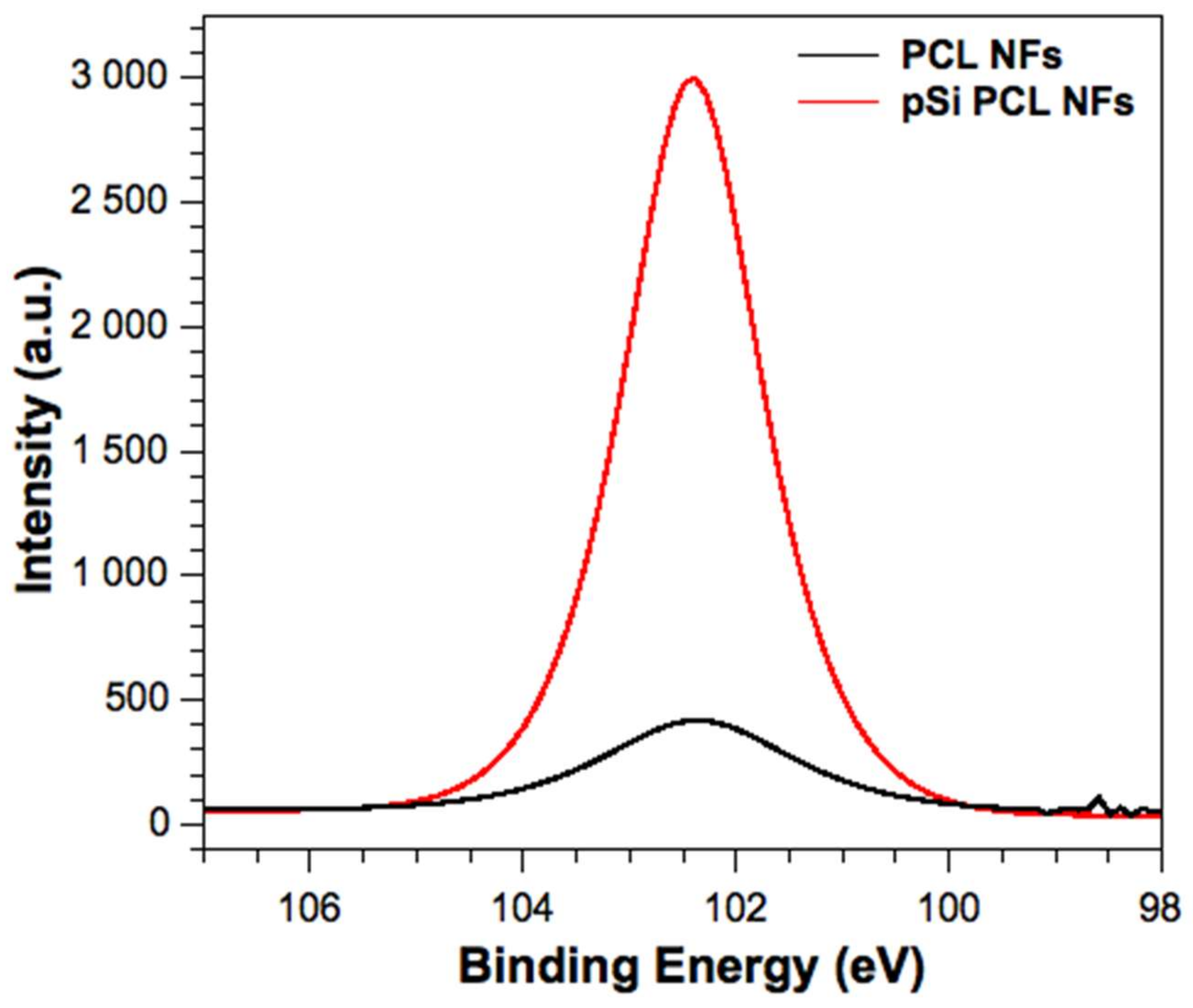
3.1. Material Characterization
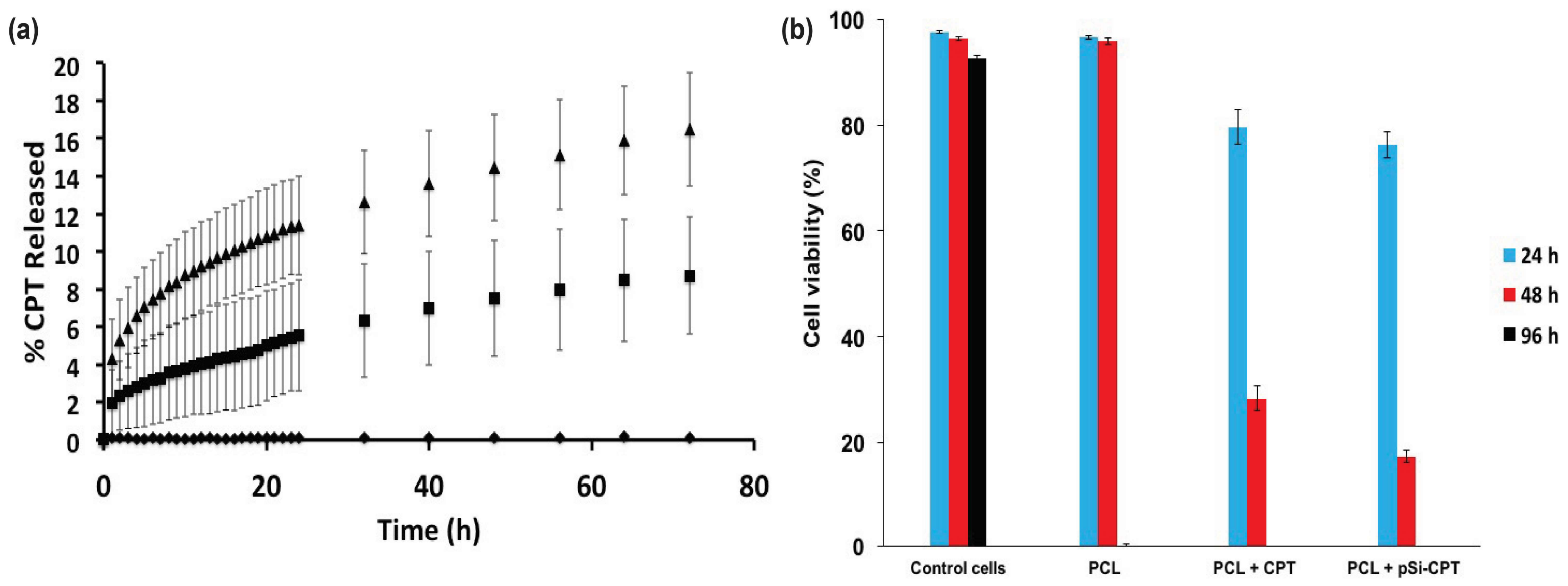
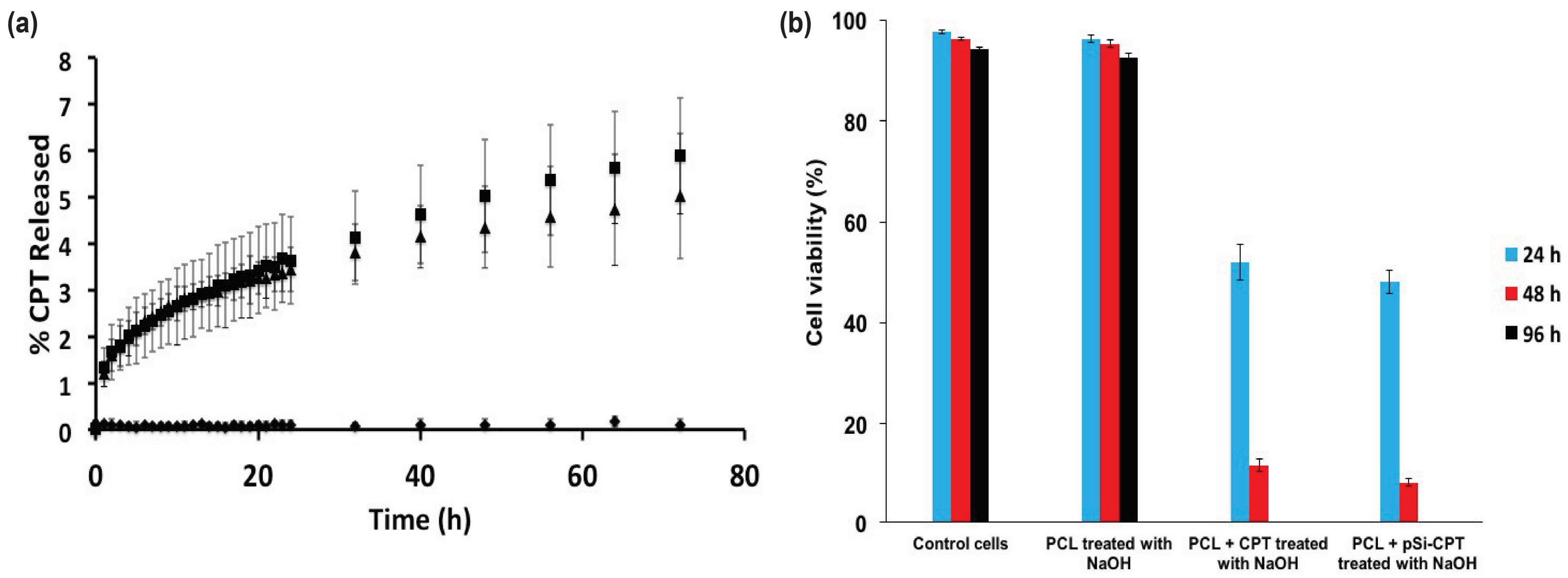
3.2. Drug Loading and Release
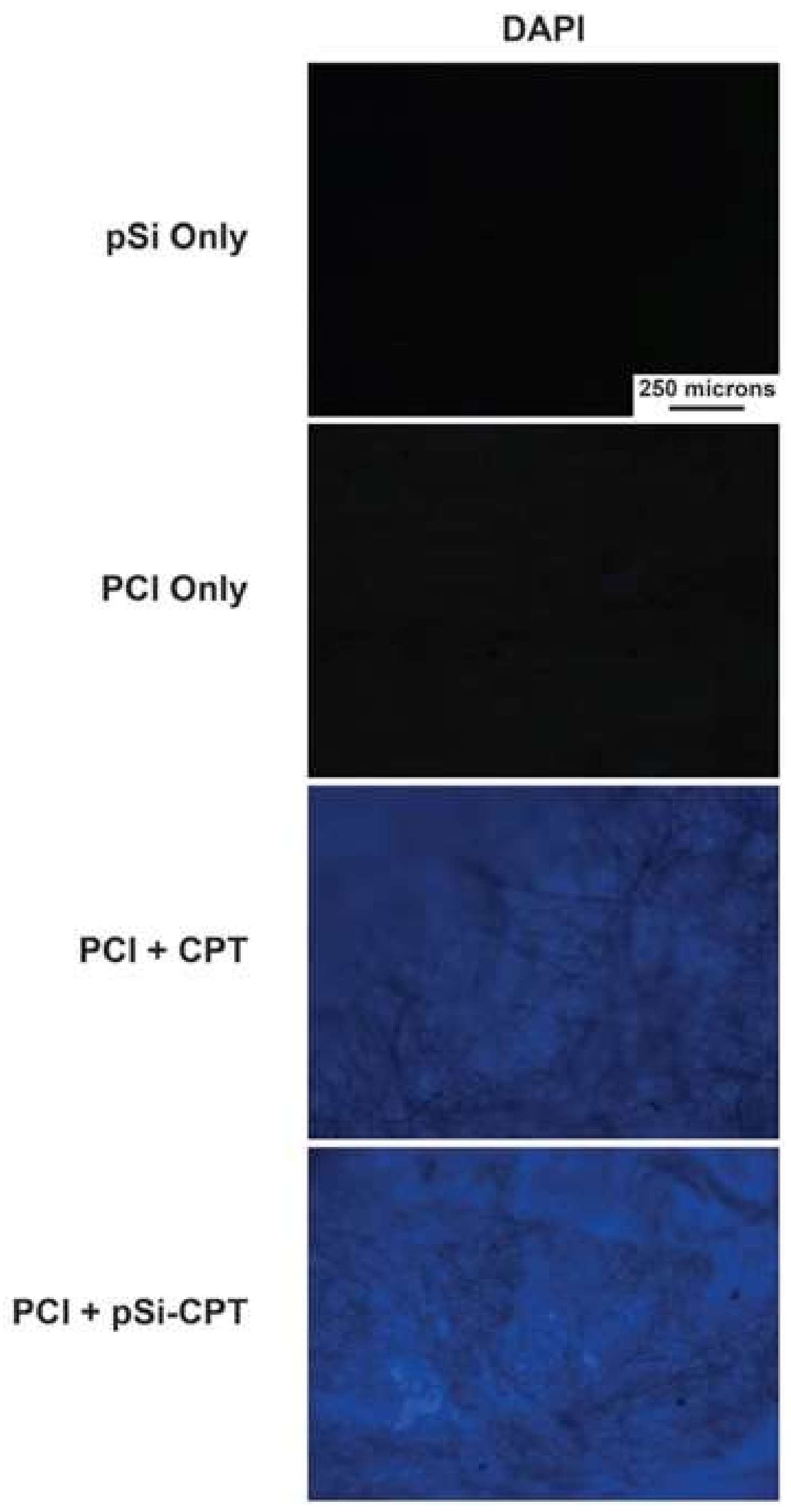
3.3. Cell Viability Assay
3.4. General Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Endres, M.; Hutmacher, D.; Salgado, A.; Kaps, C.; Ringe, J.; Reis, R.; Sittinger, M.; Brandwood, A.; Scantz, J.T. Osteogenic Induction of Human Bone Marrow-Derived Mesenchymal Progenitor Cells in Novel Synthetic Polymer—Hydrogel Matrices. Tissue Eng. 2003, 9, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical Applications of Biodegradable Polymers. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S.R.; Rodrigues, G.; Martins, G.G.; Roberto, M.A.; Mafra, M.; Henriques, C.M.R.; Silva, J.C. In vitro and in vivo evaluation of electrospun nanofibers of PCL, chitosan and gelatin: A comparative study. Mater. Sci. Eng. C 2015, 46, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Chakrapani, V.Y.G.A.; Giridev, V.R.; Madhusoothanan, M.; Sekaran, G. Electrospinning of type I collagen and PCL nanofibers using acetic acid. J. Appl. Polym. Sci. 2012, 125, 3221–3227. [Google Scholar] [CrossRef]
- Cho, S.J.; Jung, S.M.; Kang, M.; Shin, H.S.; Youk, J.H. Preparation of hydrophilic PCL nanofiber scaffolds via electrospinning of PCL/PVP-b-PCL block copolymers for enhanced cell biocompatibility. Polymer 2015, 69, 95–102. [Google Scholar] [CrossRef]
- Prosecká, E.; Rampichová, M.; Litvinec, A.; Tonar, Z.; Králíková, M.; Vojtová, L.; Kochová, P.; Plencner, M.; Buzgo, M.; Míková, A.; et al. Collagen/hydroxyapatite scaffold enriched with polycaprolactone nanofibers, thrombocyte-rich solution and mesenchymal stem cells promotes regeneration in large bone defect in vivo. J. Biomed. Mater. Res. A 2015, 103, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, R.; Madhaiyan, K.; Sundarrajan, S.; Gora, A.; Venugopal, J.R.; Ramakrishna, S. Cross-linking of protein scaffolds for therapeutic applications: PCL nanofibers delivering riboflavin for protein cross-linking. J. Mater. Chem. B 2014, 2, 1626–1633. [Google Scholar] [CrossRef]
- Khil, M.-S.; Bhattarai, S.R.; Kim, H.-Y.; Kim, S.-Z.; Lee, K.-H. Novel fabricated matrix via electrospinning for tissue engineering. J. Biomed. Mater. Res. B Appl. Biomater. 2005, 72, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, H.; Shin, Y.M.; Terai, H.; Vacanti, J.P. A biodegradable nanofiber scaffold by electrospinning and its potential for bone tissue engineering. Biomaterials 2003, 24, 2077–2082. [Google Scholar] [CrossRef]
- Woodhead Publishing Series in Biomaterials. In Porous Silicon for Biomedical Applications; Woodhead Publishing: Cambridge, UK, 2014.
- McInnes, S.J.P.; Szili, E.J.; Al-Bataineh, S.A.; Xu, J.; Alf, M.E.; Gleason, K.K.; Short, R.D.; Voelcker, N.H. Combination of iCVD and Porous Silicon for the Development of a Controlled Drug Delivery System. ACS Appl. Mater. Interfaces 2012, 4, 3566–3574. [Google Scholar] [CrossRef] [PubMed]
- Canham, L. Handbook of Porous Silicon; Springer International Publishing: Basel, Switzerland, 2014. [Google Scholar]
- Low, S.P.; Voelcker, N.H.; Canham, L.T.; Williams, K.A. The biocompatibility of porous silicon in tissues of the eye. Biomaterials 2009, 30, 2873–2880. [Google Scholar] [CrossRef] [PubMed]
- Bimbo, L.M.; Sarparanta, M.; Santos, H.A.; Airaksinen, A.J.; Mäkilä, E.; Laaksonen, T.; Peltonen, L.; Lehto, V.-P.; Hirvonen, J.; Salonen, J. Biocompatibility of Thermally Hydrocarbonized Porous Silicon Nanoparticles and their Biodistribution in Rats. ACS Nano 2010, 4, 3023–3032. [Google Scholar] [CrossRef] [PubMed]
- Acquaroli, L.N.; Kuchel, T.; Voelcker, N.H. Towards implantable porous silicon biosensors. RSC Adv. 2014, 4, 34768–34773. [Google Scholar] [CrossRef]
- Sailor, M.J.; Park, J.-H. Hybrid Nanoparticles for Detection and Treatment of Cancer. Adv. Mater. 2012, 24, 3779–3802. [Google Scholar] [CrossRef] [PubMed]
- Pace, S.; Vasani, R.B.; Cunin, F.; Voelcker, N.H. Study of the optical properties of a thermoresponsive polymer grafted onto porous silicon scaffolds. New J. Chem. 2013, 37, 228–235. [Google Scholar] [CrossRef]
- Santos, H.A.; Mäkilä, E.; Airaksinen, A.J.; Bimbo, L.M.; Hirvonen, J. Porous silicon nanoparticles for nanomedicine: Preparation and biomedical applications. Nanomedicine 2014, 9, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Salonen, J.; Laitinen, L.; Kaukonen, A.M.; Tuura, J.; Björkqvist, M.; Heikkilä, T.; Vähä-Heikkilä, K.; Hirvonen, J.; Lehto, V.-P. Mesoporous silicon microparticles for oral drug delivery: Loading and release of five model drugs. J. Control. Release 2005, 108, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Kashanian, S.; Harding, F.; Irani, Y.; Klebe, S.; Marshall, K.; Loni, A.; Canham, L.; Fan, D.; Williams, K.A.; Voelcker, N.H.; et al. Evaluation of mesoporous silicon/polycaprolactone composites as ophthalmic implants. Acta Biomater. 2010, 6, 3566–3572. [Google Scholar] [CrossRef] [PubMed]
- Irani, Y.D.K.S.; McInnes, S.J.P.; Jasieniak, M.; Voelcker, N.H.; Williams, K.A. Oral Mucosal Epithelial Cells Grown on Porous Silicon Membrane for Transfer to the Rat Eye. Sci. Rep. 2017, 7, 10042. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.; McInnes, S.J.P.; Macdonald, T.J.; Nann, T.; Voelcker, N.H. Porous silicon nanoparticles as a nanophotocathode for photoelectrochemical water splitting. RSC Adv. 2015, 5, 85978. [Google Scholar] [CrossRef]
- Macdonald, T.J.; Xu, J.; Elmas, S.; Mange, Y.J.; Skinner, W.M.; Xu, H.; Nann, T. NiO Nanofibers as a Candidate for a Nanophotocathode. Nanomaterials 2014, 4, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Batmunkh, M.; Macdonald, T.J.; Shearer, C.J.; Baterdene, M.; Wang, Y.; Biggs, M.J.; Parkin, I.P.; Nann, T.; Shapter, J.G. Carbon Nanotubes in TiO2 Nanofiber Photoelectrodes for High Performance Perovskite Solar Cells. Adv. Sci. 2017, 4, 1600504. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-M.; Zhang, Y.-Z.; Kotaki, M.; Ramakrishna, S. A review on polymer nanofibers by electrospinning and their applications in nanocomposites. Compos. Sci. Technol. 2003, 63, 2223–2253. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Ebara, M.; Aoyagi, T. A Smart Hyperthermia Nanofiber with Switchable Drug Release for Inducing Cancer Apoptosis. Adv. Funct. Mater. 2013, 23, 5753–5761. [Google Scholar] [CrossRef]
- Squires, A.H.; Hersey, J.S.; Grinstaff, M.W.; Meller, A. A Nanopore-Nanofiber Mesh Biosensor to Control DNA Translocation. J. Am. Chem. Soc. 2013, 135, 16304–16307. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, T.J.; Tune, D.D.; Dewi, M.R.; Gibson, C.T.; Shapter, J.G.; Nann, T. A TiO2 Nanofiber–Carbon Nanotube-Composite Photoanode for Improved Efficiency in Dye-Sensitized Solar Cells. ChemSusChem 2015. [Google Scholar] [CrossRef]
- Ambroz, F.; Macdonald, T.J.; Nann, T. Trends in Aluminium Based Intercalation Batteries. Adv. Energy Mater. 2017, 7, 7. [Google Scholar] [CrossRef]
- Delalat, B.; Harding, F.; Gundsambuu, B.; De-Juan-Pardo, E.M.; Wunner, F.M.; Wille, M.-L.; Jasieniak, M.; Malatesta, K.A.L.; Griesser, H.J.; Simula, A.; et al. 3D printed lattices as an activation and expansion platform for T cell therapy. Biomaterials 2017, 140, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, L.; Zhu, K. Coaxial electrospinning for encapsulation and controlled release of fragile water-soluble bioactive agents. Drug Deliv. Res. Asia Pac. Reg. 2014, 193, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, X.; Ma, P.; Wang, X.; Jing, X. The release behavior of doxorubicin hydrochloride from medicated fibers prepared by emulsion-electrospinning. Eur. J. Pharm. Biopharm. 2008, 70, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Hrib, J.; Sirc, J.; Hobzova, R.; Hampejsova, Z.; Bosakova, Z.; Munzarova, M.; Michalek, J. Nanofibers for drug delivery—Incorporation and release of model molecules, influence of molecular weight and polymer structure. Beilstein J. Nanotechnol. 2015, 6, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Saraf, A.; Baggett, L.S.; Raphael, R.M.; Kasper, F.K.; Mikos, A.G. Regulated non-viral gene delivery from coaxial electrospun fiber mesh scaffolds. J. Control. Release 2010, 143, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.C.; Ribeiro, C.; Sencadas, V.; Botelho, G.; Ribelles, J.L.G.; Lanceros-Mendez, S. Influence of fiber diameter and crystallinity on the stability of electrospun poly(l-lactic acid) membranes to hydrolytic degradation. Polym. Test. 2012, 31, 770–776. [Google Scholar] [CrossRef]
- Araujo, J.V.; Martins, A.; Leonor, I.B.; Pinho, E.D.; Reis, R.L.; Neves, N.M. Surface controlled biomimetic coating of polycaprolactone nanofiber meshes to be used as bone extracellular matrix analogues. J. Biomater. Sci. Polym. Ed. 2008, 19, 1261–1278. [Google Scholar] [CrossRef] [PubMed]
- Sætern, A.M.; Nguyen, N.B.; Bauer-Brandl, A.; Brandl, M. Effect of hydroxypropyl-a-cyclodextrin-complexation and pH on solubility of camptothecin. Int. J. Pharm. 2004, 284, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Stead, S.O.; McInnes, S.J.P.; Kireta, S.; Rose, P.D.; Jesudason, S.; Rojas-Canales, D.; Warther, D.; Cunin, F.; Durand, J.-O.; Drogemuller, C.J.; et al. Manipulating human dendritic cell phenotype and function with targeted porous silicon nanoparticles. Biomaterials 2018, 155, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.R.; Banerjee, S.; Bonin, K.; Guthold, M. Determining the mechanical properties of electrospun poly-e-caprolactone (PCL) nanofibers using AFM and a novel fiber anchoring technique. Mater. Sci. Eng. C 2016, 59, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.Y.; Hufnagel, T.C.; Lim, C.T.; Leong, K.W. Mechanical properties of single electrospun drug-encapsulated nanofibres. Nanotechnology 2006, 17, 3880–3891. [Google Scholar] [CrossRef] [PubMed]
- Oran, U.; Ünveren, E.; Wirth, T.; Unger, W.E. Poly-dimethyl-siloxane (PDMS) contamination of polystyrene (PS) oligomers samples: A comparison of time-of-flight static secondary ion mass spectrometry (TOF-SSIMS) and X-ray photoelectron spectroscopy (XPS) results. Appl. Surf. Sci. 2004, 227, 318–324. [Google Scholar] [CrossRef]
- Reneker, D.H.; Yarin, A.L. Electrospinning jets and polymer nanofibers. Polymer 2008, 49, 2387–2425. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Zero-Order | First Order | Higuchi | Hixson-Crowell | Ritger-Peppas | ‘n’ |
|---|---|---|---|---|---|---|
| PCL + CPT | 0.899 | 0.925 | 0.992 | 0.558 | 0.981 | 0.74 |
| PCL + CPT 1-h NaOH | 0.890 | 0.909 | 0.991 | 0.559 | 0.987 | 0.70 |
| PCL + pSi-CPT | 0.806 | 0.886 | 0.966 | 0.425 | 0.999 | 0.63 |
| PCL + pSi-CPT 1-h NaOH | 0.823 | 0.843 | 0.974 | 0.531 | 0.998 | 0.67 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McInnes, S.J.P.; Macdonald, T.J.; Parkin, I.P.; Nann, T.; Voelcker, N.H. Electrospun Composites of Polycaprolactone and Porous Silicon Nanoparticles for the Tunable Delivery of Small Therapeutic Molecules. Nanomaterials 2018, 8, 205. https://doi.org/10.3390/nano8040205
McInnes SJP, Macdonald TJ, Parkin IP, Nann T, Voelcker NH. Electrospun Composites of Polycaprolactone and Porous Silicon Nanoparticles for the Tunable Delivery of Small Therapeutic Molecules. Nanomaterials. 2018; 8(4):205. https://doi.org/10.3390/nano8040205
Chicago/Turabian StyleMcInnes, Steven J. P., Thomas J. Macdonald, Ivan P. Parkin, Thomas Nann, and Nicolas H. Voelcker. 2018. "Electrospun Composites of Polycaprolactone and Porous Silicon Nanoparticles for the Tunable Delivery of Small Therapeutic Molecules" Nanomaterials 8, no. 4: 205. https://doi.org/10.3390/nano8040205
APA StyleMcInnes, S. J. P., Macdonald, T. J., Parkin, I. P., Nann, T., & Voelcker, N. H. (2018). Electrospun Composites of Polycaprolactone and Porous Silicon Nanoparticles for the Tunable Delivery of Small Therapeutic Molecules. Nanomaterials, 8(4), 205. https://doi.org/10.3390/nano8040205