Preparation, Modification, Characterization, and Biosensing Application of Nanoporous Gold Using Electrochemical Techniques

 ,
,  and
and 
Abstract
1. Introduction
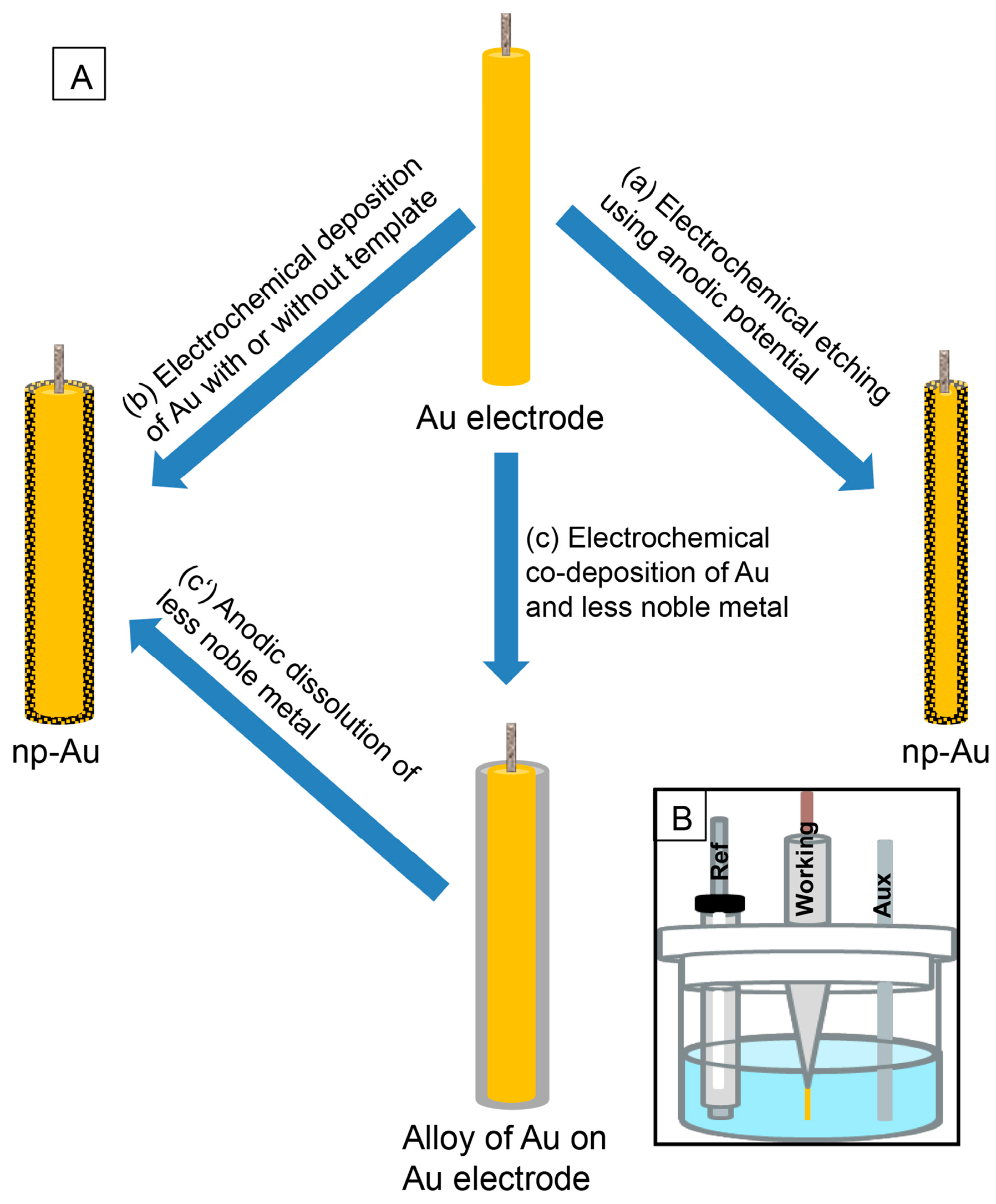
2. Preparation of np-Au Using Electrochemical Techniques
2.1. Etching of Au Electrode
2.2. Electrodeposition of Au
2.3. Electrochemical Dissolution of Less Noble Metals from Alloy
2.3.1. Alloy Preparation
2.3.2. Nano/Micro-Structured Alloy Preparation
2.3.3. Electrochemical Dealloying
3. Post-Annealing of np-Au
4. Self-Supported np-Au Electrode
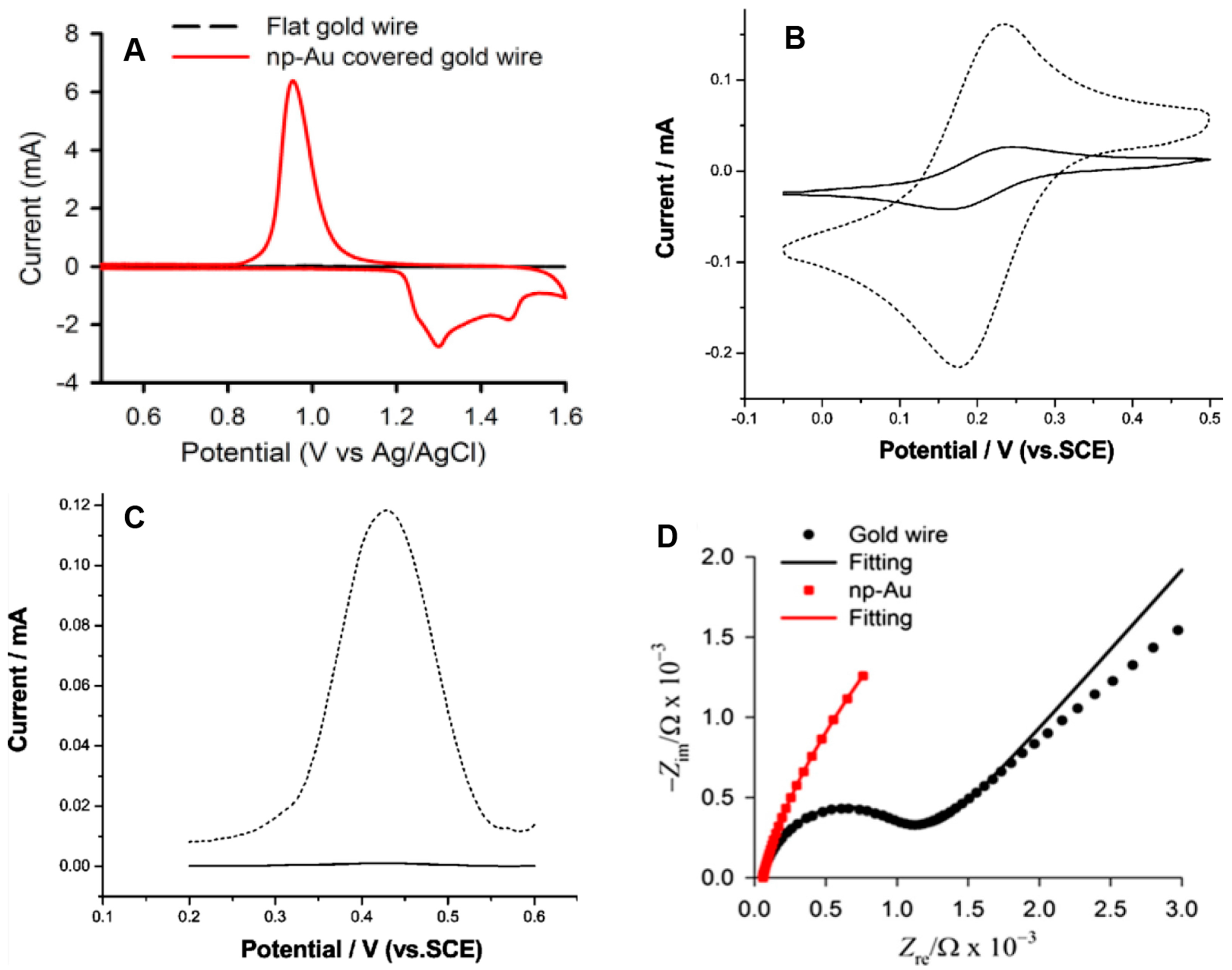
5. Electrochemical Characterization of np-Au
6. Electrochemical Biosensing
6.1. DNA Sensor
6.1.1. Aptamer-Based Electrochemical Sensors
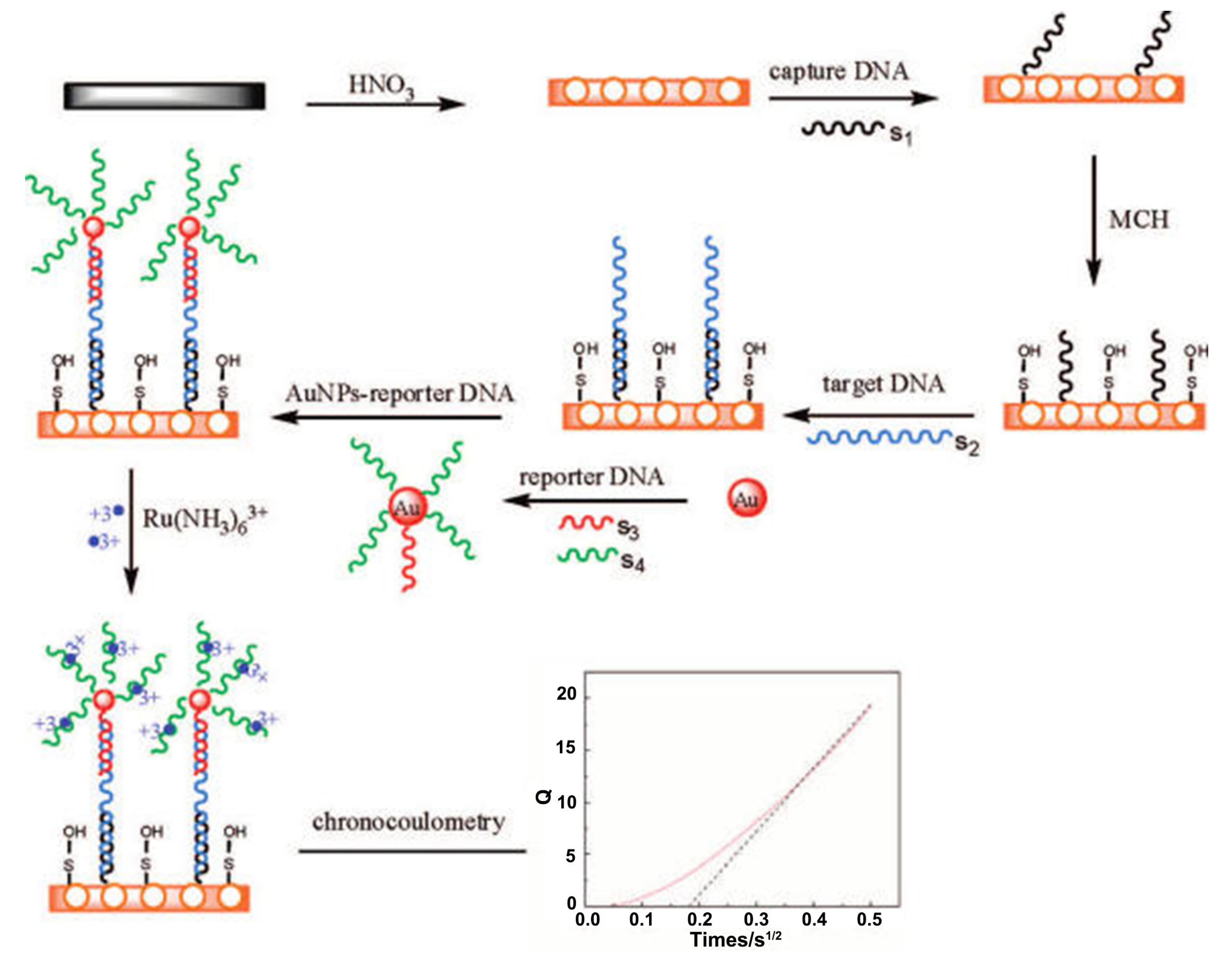
6.1.2. DNA Hybridization-Based Electrochemical Sensors
6.2. Enzymatic Sensor
6.2.1. Glucose as an Analyte
6.2.2. Other Small Molecules as Analyte
6.3. Immunosensor
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stine, K.J.; Jefferson, K.; Shulga, O.V. Nanoporous gold for enzyme immobilization. In Enzyme Stabilization and Immobilization: Methods and Protocols; Minteer, S.D., Ed.; Springer: New York, NY, USA, 2017; pp. 37–60. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Zdravkov, B.; Čermák, J.; Šefara, M.; Janků, J. Pore classification in the characterization of porous materials: A perspective. Open Chem. 2007, 5, 385–395. [Google Scholar] [CrossRef]
- Sharma, A.; Bhattarai, J.K.; Alla, A.J.; Demchenko, A.V.; Stine, K.J. Electrochemical annealing of nanoporous gold by application of cyclic potential sweeps. Nanotechnology 2015, 26, 085602. [Google Scholar] [CrossRef] [PubMed]
- Seker, E.; Reed, M.L.; Begley, M.R. Nanoporous gold: Fabrication, characterization, and applications. Materials 2009, 2, 2188–2215. [Google Scholar] [CrossRef]
- Collinson, M.M. Nanoporous gold electrodes and their applications in analytical chemistry. ISRN Anal. Chem. 2013, 2013, 692484. [Google Scholar] [CrossRef]
- Matharu, Z.; Daggumati, P.; Wang, L.; Dorofeeva, T.S.; Li, Z.; Seker, E. Nanoporous-gold-based electrode morphology libraries for investigating structure-property relationships in nucleic acid based electrochemical biosensors. ACS Appl. Mater. Interfaces 2017, 9, 12959–12966. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bhattarai, J.K.; Nigudkar, S.S.; Pistorio, S.G.; Demchenko, A.V.; Stine, K.J. Electrochemical impedance spectroscopy study of carbohydrate-terminated alkanethiol monolayers on nanoporous gold: Implications for pore wetting. J. Electroanal. Chem. 2016, 782, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Li, H.; Wang, M.e.; Zhang, K.; Si, P. Examining the effects of self-assembled monolayers on nanoporous gold based amperometric glucose biosensors. Analyst 2014, 139, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Wittstock, A.; Zielasek, V.; Biener, J.; Friend, C.M.; Baeumer, M. Nanoporous gold catalysts for selective gas-phase oxidative coupling of methanol at low temperature. Science 2010, 327, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Daggumati, P.; Matharu, Z.; Seker, E. Effect of nanoporous gold thin film morphology on electrochemical DNA sensing. Anal. Chem. 2015, 87, 8149–8156. [Google Scholar] [CrossRef] [PubMed]
- Pandey, B.; Bhattarai, J.K.; Pornsuriyasak, P.; Fujikawa, K.; Catania, R.; Demchenko, A.V.; Stine, K.J. Square-wave voltammetry assays for glycoproteins on nanoporous gold. J. Electroanal. Chem. 2014, 717–718, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Huang, W.; Zheng, J.; Niu, Z.; Li, Z. Nonenzymatic amperometric response of glucose on a nanoporous gold film electrode fabricated by a rapid and simple electrochemical method. Biosens. Bioelectron. 2011, 26, 3555–3561. [Google Scholar] [CrossRef] [PubMed]
- Seker, E.; Berdichevsky, Y.; Staley, K.J.; Yarmush, M.L. Microfabrication-compatible nanoporous gold foams as biomaterials for drug delivery. Adv. Healthc. Mater. 2012, 1, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Pornsuriyasak, P.; Ranade, S.C.; Li, A.; Parlato, M.C.; Sims, C.R.; Shulga, O.V.; Stine, K.J.; Demchenko, A.V. STICS: Surface-tethered iterative carbohydrate synthesis. Chem. Commun. 2009, 1834–1836. [Google Scholar] [CrossRef] [PubMed]
- Daggumati, P.; Appelt, S.; Matharu, Z.; Marco, M.L.; Seker, E. Sequence-specific electrical purification of nucleic acids with nanoporous gold electrodes. J. Am. Chem. Soc. 2016, 138, 7711–7717. [Google Scholar] [CrossRef] [PubMed]
- Alla, A.J.; FB, D.A.; Bhattarai, J.K.; Cooper, J.A.; Tan, Y.H.; Demchenko, A.V.; Stine, K.J. Selective capture of glycoproteins using lectin-modified nanoporous gold monolith. J. Chromatogr. A 2015, 1423, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Meng, F.; Xie, Y.; Liu, J.; Ding, Y. Direct N2H4/H2O2 fuel cells powered by nanoporous gold leaves. Sci. Rep. 2012, 2, 941. [Google Scholar] [CrossRef] [PubMed]
- Biener, J.; Wittstock, A.; Zepeda-Ruiz, L.A.; Biener, M.M.; Zielasek, V.; Kramer, D.; Viswanath, R.N.; Weissmuller, J.; Baumer, M.; Hamza, A.V. Surface-chemistry-driven actuation in nanoporous gold. Nat. Mater. 2009, 8, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.Y.; Yuan, H.T.; Iwasa, Y.; Chen, M.W. Three-dimensional nanoporous gold for electrochemical supercapacitors. Scr. Mater. 2011, 64, 923–926. [Google Scholar] [CrossRef]
- Bhattarai, J.K. Electrochemical Synthesis of Nanostructured Noble Metal Films for Biosensing. Ph.D. Dissertation, University of Missouri, St. Louis, MO, USA, 2014. [Google Scholar]
- Bhattarai, J.K.; Sharma, A.; Fujikawa, K.; Demchenko, A.V.; Stine, K.J. Electrochemical synthesis of nanostructured gold film for the study of carbohydrate–lectin interactions using localized surface plasmon resonance spectroscopy. Carbohydr. Res. 2015, 405, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Cobley, C.M.; Xia, Y. Gold and nanotechnology. Elements 2009, 5, 309–313. [Google Scholar] [CrossRef]
- Cobley, C.M.; Chen, J.; Cho, E.C.; Wang, L.V.; Xia, Y. Gold nanostructures: A class of multifunctional materials for biomedical applications. Chem. Soc. Rev. 2011, 40, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, N.V.; Fujikawa, K.; Tan, Y.H.; Nigudkar, S.S.; Stine, K.J.; Demchenko, A.V. Surface-tethered iterative carbohydrate synthesis: A spacer study. J. Org. Chem. 2013, 78, 6849–6857. [Google Scholar] [CrossRef] [PubMed]
- Biener, J.; Nyce, G.W.; Hodge, A.M.; Biener, M.M.; Hamza, A.V.; Maier, S.A. Nanoporous plasmonic metamaterials. Adv. Mater. 2008, 20, 1211–1217. [Google Scholar] [CrossRef]
- Xiao, X.; Si, P.; Magner, E. An overview of dealloyed nanoporous gold in bioelectrochemistry. Bioelectrochemistry 2016, 109, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Daggumati, P.; Matharu, Z.; Wang, L.; Seker, E. Biofouling-resilient nanoporous gold electrodes for DNA sensing. Anal. Chem. 2015, 87, 8618–8622. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Li, Y.; Ji, G.; Zhou, G.; Huang, X.; Qu, Y.; Gao, P. Immobilization of lignin peroxidase on nanoporous gold: Enzymatic properties and in situ release of H2O2 by co-immobilized glucose oxidase. Bioresour. Technol. 2009, 100, 3837–3842. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Xue, L.; Ji, G.; Zhou, G.; Huang, X.; Qu, Y.; Gao, P. Enzyme-modified nanoporous gold-based electrochemical biosensors. Biosens. Bioelectron. 2009, 24, 3014–3018. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-B.; Liu, Y.-L.; Zhang, H.-W.; Xiao, C.; Qin, Y.; Duo, H.-H.; Xu, J.-Q.; Guo, S.; Pang, D.-W.; Huang, W.-H. Electrochemical monitoring of hydrogen sulfide release from single cells. ChemElectroChem 2016, 3, 1998–2002. [Google Scholar] [CrossRef]
- Jia, F.; Yu, C.; Ai, Z.; Zhang, L. Fabrication of nanoporous gold film electrodes with ultrahigh surface area and electrochemical activity. Chem. Mater. 2007, 19, 3648–3653. [Google Scholar] [CrossRef]
- Sukeri, A.; Saravia, L.P.H.; Bertotti, M. A facile electrochemical approach to fabricate a nanoporous gold film electrode and its electrocatalytic activity towards dissolved oxygen reduction. Phys. Chem. Chem. Phys. 2015, 17, 28510–28514. [Google Scholar] [CrossRef] [PubMed]
- Nishio, K.; Masuda, H. Anodization of gold in oxalate solution to form a nanoporous black film. Angew. Chem. Int. Ed. 2011, 50, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Kim, J. Fabrication of nanoporous Au films with ultra-high surface area for sensitive electrochemical detection of glucose in the presence of Cl. Appl. Surf. Sci. 2014, 297, 84–88. [Google Scholar] [CrossRef]
- Deng, Y.; Huang, W.; Chen, X.; Li, Z. Facile fabrication of nanoporous gold film electrodes. Electrochem. Commun. 2008, 10, 810–813. [Google Scholar] [CrossRef]
- Zhou, C.; Xia, Y.; Huang, W.; Li, Z. A rapid anodic fabrication of nanoporous gold in NH4Cl solution for nonenzymatic glucose detection. J. Electrochem. Soc. 2014, 161, H802–H808. [Google Scholar] [CrossRef]
- Yang, S.; Zheng, Y.; Zhang, X.; Ding, S.; Li, L.; Zha, W. Molecularly imprinted electrochemical sensor based on the synergic effect of nanoporous gold and copper nanoparticles for the determination of cysteine. J. Solid State Electrochem. 2016, 20, 2037–2044. [Google Scholar] [CrossRef]
- Cherevko, S.; Chung, C.-H. Direct electrodeposition of nanoporous gold with controlled multimodal pore size distribution. Electrochem. Commun. 2011, 13, 16–19. [Google Scholar] [CrossRef]
- Huang, J.F.; Sun, I.W. Fabrication and surface functionalization of nanoporous gold by electrochemical alloying/dealloying of Au–Zn in an ionic liquid, and the self-assembly of l-cysteine monolayers. Adv. Funct. Mater. 2005, 15, 989–994. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, L.; Zhang, L.; Ma, H.; Ding, Y. Electrocatalytic oxidation of d-glucose at nanoporous Au and Au-Ag alloy electrodes in alkaline aqueous solutions. Electrochim. Acta 2009, 54, 7286–7293. [Google Scholar] [CrossRef]
- Chen-Wiegart, Y.-C.K.; Wang, S.; McNulty, I.; Dunand, D.C. Effect of Ag–Au composition and acid concentration on dealloying front velocity and cracking during nanoporous gold formation. Acta Mater. 2013, 61, 5561–5570. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, X.; Qi, Z.; Wang, Y.; Zhang, Z. A benign route to fabricate nanoporous gold through electrochemical dealloying of Al–Au alloys in a neutral solution. Electrochim. Acta 2009, 54, 6190–6198. [Google Scholar] [CrossRef]
- McCurry, D.A.; Kamundi, M.; Fayette, M.; Wafula, F.; Dimitrov, N. All electrochemical fabrication of a platinized nanoporous Au thin-film catalyst. ACS Appl. Mater. Interfaces 2011, 3, 4459–4468. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, J. Effect of pH on anodic formation of nanoporous gold films in chloride solutions: Optimization of anodization for ultrahigh porous structures. Langmuir 2014, 30, 4844–4851. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Searson, P.C. Single nanoporous gold nanowire sensors. J. Phys. Chem. B 2006, 110, 4318–4322. [Google Scholar] [CrossRef] [PubMed]
- Bok, H.-M.; Kim, S.; Yoo, S.-H.; Kim, S.K.; Park, S. Synthesis of perpendicular nanorod arrays with hierarchical architecture and water slipping superhydrophobic properties. Langmuir 2008, 24, 4168–4173. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Searson, P.C. Synthesis and characterization of nanoporous gold nanowires. J. Phys. Chem. B 2003, 107, 4494–4499. [Google Scholar] [CrossRef]
- Ke, X.; Xu, Y.; Yu, C.; Zhao, J.; Cui, G.; Higgins, D.; Li, Q.; Wu, G. Nanoporous gold on three-dimensional nickel foam: An efficient hybrid electrode for hydrogen peroxide electroreduction in acid media. J. Power Sources 2014, 269, 461–465. [Google Scholar] [CrossRef]
- Ji, C.; Searson, P.C. Fabrication of nanoporous gold nanowires. Appl. Phys. Lett. 2002, 81, 4437. [Google Scholar] [CrossRef]
- Chauvin, A.; Delacote, C.; Molina-Luna, L.; Duerrschnabel, M.; Boujtita, M.; Thiry, D.; Du, K.; Ding, J.; Choi, C.-H.; Tessier, P.-Y.; et al. Planar arrays of nanoporous gold nanowires: When electrochemical dealloying meets nanopatterning. ACS Appl. Mater. Interfaces 2016, 8, 6611–6620. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gradilla, V.; Sattayasamitsathit, S.; Soto, F.; Kuralay, F.; Yardimci, C.; Wiitala, D.; Galarnyk, M.; Wang, J. Ultrasound-propelled nanoporous gold wire for efficient drug loading and release. Small 2014, 10, 4154–4159. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, F.; Zeng, J.; Qi, J.; Lu, J.; Shih, W.-C. Microfluidic surface-enhanced Raman scattering sensor with monolithically integrated nanoporous gold disk arrays for rapid and label-free biomolecular detection. J. Biomed. Opt. 2014, 19, 111611–111618. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Zhao, F.; Zenasni, O.; Li, J.; Shih, W.C. Nanoporous gold disks functionalized with stabilized G-quadruplex moieties for sensing small molecules. ACS Appl. Mater. Interfaces 2016, 8, 29968–29976. [Google Scholar] [CrossRef] [PubMed]
- Pedireddy, S.; Lee, H.K.; Tjiu, W.W.; Phang, I.Y.; Tan, H.R.; Chua, S.Q.; Troadec, C.; Ling, X.Y. One-step synthesis of zero-dimensional hollow nanoporous gold nanoparticles with enhanced methanol electrooxidation performance. Nat. Commun. 2014, 5, 4947. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.S.; Pedireddy, S.; Lee, Y.H.; Tjiu, W.W.; Liu, Y.; Yang, Z.; Ling, X.Y. Nanoporous gold nanoframes with minimalistic architectures: Lower porosity generates stronger surface-enhanced Raman scattering capabilities. Chem. Mater. 2015, 27, 7827–7834. [Google Scholar] [CrossRef]
- Pedireddy, S.; Lee, H.K.; Koh, C.S.L.; Tan, J.M.R.; Tjiu, W.W.; Ling, X.Y. Nanoporous gold bowls: A kinetic approach to control open shell structures and size-tunable lattice strain for electrocatalytic applications. Small 2016, 12, 4531–4540. [Google Scholar] [CrossRef] [PubMed]
- Nyce, G.W.; Hayes, J.R.; Hamza, A.V.; Satcher, J.H. Synthesis and characterization of hierarchical porous gold materials. Chem. Mater. 2007, 19, 344–346. [Google Scholar] [CrossRef]
- Zhao, F.; Zeng, J.; Parvez Arnob, M.M.; Sun, P.; Qi, J.; Motwani, P.; Gheewala, M.; Li, C.-H.; Paterson, A.; Strych, U.; et al. Monolithic NPG nanoparticles with large surface area, tunable plasmonics, and high-density internal hot-spots. Nanoscale 2014, 6, 8199–8207. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Schaaf, P. Nanoporous gold nanoparticles. J. Mater. Chem. 2012, 22, 5344–5348. [Google Scholar] [CrossRef]
- Khristosov, M.K.; Bloch, L.; Burghammer, M.; Kauffmann, Y.; Katsman, A.; Pokroy, B. Sponge-like nanoporous single crystals of gold. Nat. Commun. 2015, 6, 8841. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Altomare, M.; Yoo, J.E.; Schmuki, P. Efficient photocatalytic H2 evolution: Controlled dewetting-dealloying to fabricate site-selective high-activity nanoporous Au particles on highly ordered TiO2 nanotube arrays. Adv. Mater. 2015, 27, 3208–3215. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ji, R.; Albrecht, A.; Schaaf, P. Ordered arrays of nanoporous gold nanoparticles. Beilstein J. Nanotechnol. 2012, 3, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Cattarin, S.; Kramer, D.; Lui, A.; Musiani, M.M. Preparation and characterization of gold nanostructures of controlled dimension by electrochemical techniques. J. Phys. Chem. C 2007, 111, 12643–12649. [Google Scholar] [CrossRef]
- Dursun, A.; Pugh, D.V.; Corcoran, S.G. Dealloying of Ag–Au alloys in halide-containing electrolytes: Affect on critical potential and pore size. J. Electrochem. Soc. 2003, 150, B355–B360. [Google Scholar] [CrossRef]
- Sieradzki, K.; Dimitrov, N.; Movrin, D.; McCall, C.; Vasiljevic, N.; Erlebacher, J. The dealloying critical potential. J. Electrochem. Soc. 2002, 149, B370. [Google Scholar] [CrossRef]
- Kamundi, M.; Bromberg, L.; Fey, E.; Mitchell, C.; Fayette, M.; Dimitrov, N. Impact of structure and composition on the dealloying of AuxAg(1−x) alloys on the nanoscale. J. Phys. Chem. C 2012, 116, 14123–14133. [Google Scholar] [CrossRef]
- Seker, E.; Reed, M.; Begley, M. A thermal treatment approach to reduce microscale void formation in blanket nanoporous gold films. Scr. Mater. 2009, 60, 435–438. [Google Scholar] [CrossRef]
- Kim, M.; Ha, W.-J.; Anh, J.-W.; Kim, H.-S.; Park, S.-W.; Lee, D. Fabrication of nanoporous gold thin films on silicon substrate by multilayer deposition of Au and Ag. J. Alloys Compd. 2009, 484, 28–32. [Google Scholar] [CrossRef]
- Snyder, J.; Livi, K.; Erlebacher, J. Dealloying silver/gold alloys in neutral silver nitrate solution: Porosity evolution, surface composition, and surface oxides. J. Electrochem. Soc. 2008, 155, C464–C473. [Google Scholar] [CrossRef]
- Chen, H.; Jiang, C.; Yu, C.; Zhang, S.; Liu, B.; Kong, J. Protein chips and nanomaterials for application in tumor marker immunoassays. Biosens. Bioelectron. 2009, 24, 3399–3411. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.H.; Davis, J.A.; Fujikawa, K.; Ganesh, N.V.; Demchenko, A.V.; Stine, K.J. Surface area and pore size characteristics of nanoporous gold subjected to thermal, mechanical, or surface modification studied using gas adsorption isotherms, cyclic voltammetry, thermogravimetric analysis, and scanning electron microscopy. J. Mater. Chem. 2012, 22, 6733–6745. [Google Scholar] [CrossRef] [PubMed]
- Seker, E.; Gaskins, J.T.; Bart-Smith, H.; Zhu, J.; Reed, M.L.; Zangari, G.; Kelly, R.; Begley, M.R. The effects of post-fabrication annealing on the mechanical properties of freestanding nanoporous gold structures. Acta Mater. 2007, 55, 4593–4602. [Google Scholar] [CrossRef]
- Arnob, M.M.P.; Zhao, F.; Zeng, J.; Santos, G.M.; Li, M.; Shih, W.-C. Laser rapid thermal annealing enables tunable plasmonics in nanoporous gold nanoparticles. Nanoscale 2014, 6, 12470–12475. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.A.R.; Wang, L.; Biener, J.; Seker, E.; Biener, M.M.; Matthews, M.J. Engineering on-chip nanoporous gold material libraries via precision photothermal treatment. Nanoscale 2016, 8, 785–795. [Google Scholar] [CrossRef] [PubMed]
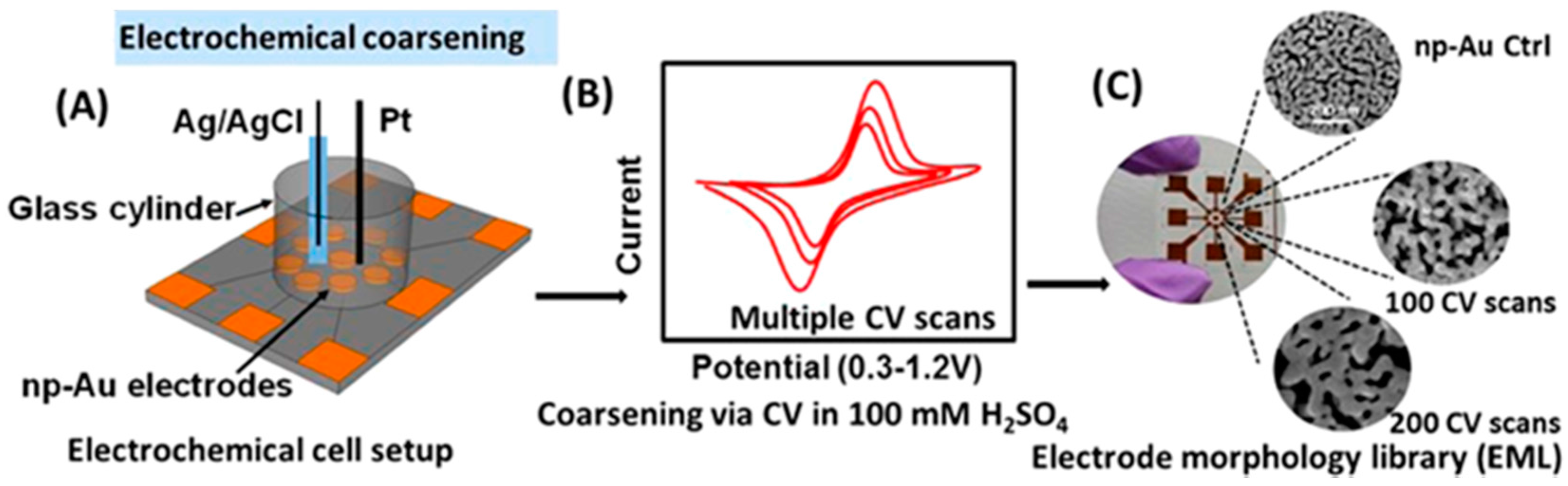
- Dorofeeva, T.S.; Seker, E. Electrically tunable pore morphology in nanoporous gold thin films. Nano Res. 2015, 8, 2188–2198. [Google Scholar] [CrossRef]
- Dorofeeva, T.S.; Seker, E. In situ electrical modulation and monitoring of nanoporous gold morphology. Nanoscale 2016, 8, 19551–19556. [Google Scholar] [CrossRef] [PubMed]
- Dorofeeva, T.S.; Matharu, Z.; Daggumati, P.; Seker, E. Electrochemically triggered pore expansion in nanoporous gold thin films. J. Phys. Chem. C 2016, 120, 4080–4086. [Google Scholar] [CrossRef]
- Zeng, G.; Zhang, C.; Huang, D.; Lai, C.; Tang, L.; Zhou, Y.; Xu, P.; Wang, H.; Qin, L.; Cheng, M. Practical and regenerable electrochemical aptasensor based on nanoporous gold and thymine-Hg2+-thymine base pairs for Hg2+ detection. Biosens. Bioelectron. 2017, 90, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski, P.N.; Scott, A.M.; Faulkner, C.J.; Berron, B.J.; Cliffel, D.E.; Jennings, G.K. Functionalized nanoporous gold leaf electrode films for the immobilization of photosystem I. ACS Nano 2008, 2, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Wang, L.; Liu, Z.; Ding, Y. Nanoporous gold leaf for amperometric determination of nitrite. Electroanalysis 2010, 23, 381–386. [Google Scholar] [CrossRef]
- Chen, A.Y.; Shi, S.S.; Qiu, Y.D.; Xie, X.F.; Ruan, H.H.; Gu, J.F.; Pan, D. Pore-size tuning and optical performances of nanoporous gold films. Microporous Mesoporous Mater. 2015, 202, 50–56. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Guo, Y.-G.; Zhang, H.-M.; Hu, J.-S.; Wan, L.-J.; Bai, C.-L. Nanoarchitectured metal film electrodes with high electroactive surface areas. Thin Solid Films 2005, 484, 341–345. [Google Scholar] [CrossRef]
- Quynh, B.T.P.; Byun, J.Y.; Kim, S.H. Non-enzymatic amperometric detection of phenol and catechol using nanoporous gold. Sens. Actuators B 2015, 221, 191–200. [Google Scholar] [CrossRef]
- Lu, L.; Huang, X.; Dong, Y.; Huang, Y.; Pan, X.; Wang, X.; Feng, M.; Luo, Y.; Fang, D. Facile method for fabrication of self-supporting nanoporous gold electrodes via cyclic voltammetry in ethylene glycol, and their application to the electrooxidative determination of catechol. Microchim. Acta 2015, 182, 1509–1517. [Google Scholar] [CrossRef]
- Yang, M.; Chen, X.; Liu, J.-H.; Huang, X.-J. Enhanced anti-interference on electrochemical detection of arsenite with nanoporous gold in mild condition. Sens. Actuators B 2016, 234, 404–411. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Huang, P.-J.J.; Ding, J.; Liu, J. Aptamer-based biosensors for biomedical diagnostics. Analyst 2014, 139, 2627–2640. [Google Scholar] [CrossRef] [PubMed]
- Zamay, G.S.; Zamay, T.N.; Kolovskii, V.A.; Shabanov, A.V.; Glazyrin, Y.E.; Veprintsev, D.V.; Krat, A.V.; Zamay, S.S.; Kolovskaya, O.S.; Gargaun, A. Electrochemical aptasensor for lung cancer-related protein detection in crude blood plasma samples. Sci. Rep. 2016, 6, 34350. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Sun, Y.; Huang, X.; Qu, Y. A sensitive nanoporous gold-based electrochemical aptasensor for thrombin detection. Colloids Surf. B 2010, 79, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Kashefi-Kheyrabadi, L.; Mehrgardi, M.A. Aptamer-based electrochemical biosensor for detection of adenosine triphosphate using a nanoporous gold platform. Bioelectrochemistry 2013, 94, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhou, C.; Yan, X.; Yan, Y.; Wang, Q. Aptamer-functionalized nanoporous gold film for high-performance direct electrochemical detection of bisphenol A in human serum. Anal. Chim. Acta 2015, 883, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lai, C.; Zeng, G.; Huang, D.; Tang, L.; Yang, C.; Zhou, Y.; Qin, L.; Cheng, M. Nanoporous Au-based chronocoulometric aptasensor for amplified detection of Pb2+ using DNAzyme modified with Au nanoparticles. Biosens. Bioelectron. 2016, 81, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, S.; Zhang, L.; Wang, L.; Wu, H.; Pan, D.; Fan, C. Sequence-specific detection of femtomolar DNA via a chronocoulometric DNA sensor (CDS): Effects of nanoparticle-mediated amplification and nanoscale control of DNA assembly at electrodes. J. Am. Chem. Soc. 2006, 128, 8575–8580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, S.; Wang, L.; Pan, D.; Fan, C. A gold nanoparticle-based chronocoulometric DNA sensor for amplified detection of DNA. Nat. Protoc. 2007, 2, 2888–2895. [Google Scholar] [CrossRef] [PubMed]
- Steel, A.B.; Herne, T.M.; Tarlov, M.J. Electrochemical quantitation of DNA immobilized on gold. Anal. Chem. 1998, 70, 4670–4677. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Liu, A.; Xu, X.; Sun, Z.; Chen, J.; Wang, K.; Liu, Q.; Lin, X.; Lin, J. Detection of femtomolar level osteosarcoma-related gene via a chronocoulometric DNA biosensor based on nanostructure gold electrode. Int. J. Nanomed. 2012, 7, 527–536. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, K.; Lan, D.; Li, X.; Zhang, S. Electrochemical DNA biosensor based on nanoporous gold electrode and multifunctional encoded DNA−Au bio bar codes. Anal. Chem. 2008, 80, 9124–9130. [Google Scholar] [CrossRef] [PubMed]
- Ahangar, L.E.; Mehrgardi, M.A. Nanoporous gold electrode as a platform for the construction of an electrochemical DNA hybridization biosensor. Biosens. Bioelectron. 2012, 38, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Huang, J.; Shi, G.; Zhang, W.; Jin, L. A sensitive nanoporous gold-based electrochemical DNA biosensor for Escherichia coli detection. Anal. Lett. 2011, 44, 2559–2570. [Google Scholar] [CrossRef]
- Zhong, G.; Liu, A.; Chen, X.; Wang, K.; Lian, Z.; Liu, Q.; Chen, Y.; Du, M.; Lin, X. Electrochemical biosensor based on nanoporous gold electrode for detection of PML/RARα fusion gene. Biosens. Bioelectron. 2011, 26, 3812–3817. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Liu, P.; Ye, S.; Zhang, S. Ultrasensitive electrochemical detection of DNA based on PbS nanoparticle tags and nanoporous gold electrode. Biosens. Bioelectron. 2009, 24, 3113–3119. [Google Scholar] [CrossRef] [PubMed]
- Milton, R.D.; Minteer, S.D. Direct enzymatic bioelectrocatalysis: Differentiating between myth and reality. J. R. Soc. Interface 2017, 14, 20170253. [Google Scholar] [CrossRef] [PubMed]
- Du Toit, H.; Di Lorenzo, M. Glucose oxidase directly immobilized onto highly porous gold electrodes for sensing and fuel cell applications. Electrochim. Acta 2014, 138, 86–92. [Google Scholar] [CrossRef]
- Gregg, B.A.; Heller, A. Cross-linked redox gels containing glucose oxidase for amperometric biosensor applications. Anal. Chem. 1990, 62, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Dong, Y.; Wang, J.; Li, Q.; Wu, X. Direct electrochemistry and bioelectrocatalysis of horseradish peroxidase entrapped in a self-supporting nanoporous gold electrode: A new strategy to improve the orientation of immobilized enzymes. Anal. Methods 2015, 7, 6686–6694. [Google Scholar] [CrossRef]
- Mie, Y.; Ikegami, M.; Komatsu, Y. Nanoporous structure of gold electrode fabricated by anodization and its efficacy for direct electrochemistry of human cytochrome P450. Chem. Lett. 2016, 45, 640–642. [Google Scholar] [CrossRef]
- Salaj-Kosla, U.; Poller, S.; Schuhmann, W.; Shleev, S.; Magner, E. Direct electron transfer of Trametes hirsuta laccase adsorbed at unmodified nanoporous gold electrodes. Bioelectrochemistry 2013, 91, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Salaj-Kosla, U.; Poeller, S.; Beyl, Y.; Scanlon, M.D.; Beloshapkin, S.; Shleev, S.; Schuhmann, W.; Magner, E. Direct electron transfer of bilirubin oxidase (Myrothecium verrucaria) at an unmodified nanoporous gold biocathode. Electrochem. Commun. 2012, 16, 92–95. [Google Scholar] [CrossRef]
- Chen, L.; Fujita, T.; Chen, M. Biofunctionalized nanoporous gold for electrochemical biosensors. Electrochim. Acta 2012, 67, 1–5. [Google Scholar] [CrossRef]
- Yang, X.N.; Huang, X.B.; Hang, R.Q.; Zhang, X.Y.; Qin, L.; Tang, B. Improved catalytic performance of porcine pancreas lipase immobilized onto nanoporous gold via covalent coupling. J. Mater. Sci. 2016, 51, 6428–6435. [Google Scholar] [CrossRef]
- Du, X.; Liu, X.; Li, Y.; Wu, C.; Wang, X.; Xu, P. Efficient biocatalyst by encapsulating lipase into nanoporous gold. Nanoscale Res. Lett. 2013, 8, 180. [Google Scholar] [CrossRef] [PubMed]
- Hakamada, M.; Takahashi, M.; Mabuchi, M. Enzyme electrodes stabilized by monolayer-modified nanoporous Au for biofuel cells. Gold Bull. 2012, 45, 9–15. [Google Scholar] [CrossRef]
- Hsiao, M.W. Electrochemical oxidation of glucose on single crystal and polycrystalline gold surfaces in phosphate buffer. J. Electrochem. Soc. 1996, 143, 759–767. [Google Scholar] [CrossRef]
- Park, S.; Boo, H.; Chung, T.D. Electrochemical non-enzymatic glucose sensors. Anal. Chim. Acta 2006, 556, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Toghill, K.E.; Compton, R.G. Electrochemical non-enzymatic glucose sensors: A perspective and an evaluation. Int. J. Electrochem. Sci. 2010, 5, 1246–1301. [Google Scholar]
- Seo, B.; Kim, J. Electrooxidation of glucose at nanoporous gold surfaces: Structure dependent electrocatalysis and its application to amperometric detection. Electroanalysis 2010, 22, 939–945. [Google Scholar] [CrossRef]
- Lang, X.; Qian, L.; Guan, P.; Zi, J.; Chen, M. Localized surface plasmon resonance of nanoporous gold. Appl. Phys. Lett. 2011, 98, 093701. [Google Scholar] [CrossRef]
- Ahmadalinezhad, A.; Kafi, A.K.M.; Chen, A. Glucose biosensing based on the highly efficient immobilization of glucose oxidase on a Prussian blue modified nanostructured Au surface. Electrochem. Commun. 2009, 11, 2048–2051. [Google Scholar] [CrossRef]
- Li, T.; Jia, F.; Fan, Y.; Ding, Z.; Yang, J. Fabrication of nanoporous thin-film working electrodes and their biosensing applications. Biosens. Bioelectron. 2013, 42, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Salaj-Kosla, U.; Scanlon, M.D.; Baumeister, T.; Zahma, K.; Ludwig, R.; O’Conghaile, P.; MacAodha, D.; Leech, D.; Magner, E. Mediated electron transfer of cellobiose dehydrogenase and glucose oxidase at osmium polymer-modified nanoporous gold electrodes. Anal. Bioanal. Chem. 2013, 405, 3823–3830. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, M.-E.; Li, H.; Si, P. One-step fabrication of bio-functionalized nanoporous gold/poly(3,4-ethylenedioxythiophene) hybrid electrodes for amperometric glucose sensing. Talanta 2013, 116, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.T.Y.; Kinkead, B.; Gates, B.D. Ordered porous gold electrodes to enhance the sensitivity of enzyme-based glucose sensors. J. Electrochem. Soc. 2014, 161, B3103–B3106. [Google Scholar] [CrossRef]
- Xiao, X.; Ulstrup, J.; Li, H.; Wang, M.E.; Zhang, J.; Si, P. Nanoporous gold assembly of glucose oxidase for electrochemical biosensing. Electrochim. Acta 2014, 130, 559–567. [Google Scholar] [CrossRef]
- Wu, C.; Sun, H.; Li, Y.; Liu, X.; Du, X.; Wang, X.; Xu, P. Biosensor based on glucose oxidase-nanoporous gold co-catalysis for glucose detection. Biosens. Bioelectron. 2015, 66, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Heller, A.; Feldman, B. Electrochemical glucose sensors and their applications in diabetes management. Chem. Rev. 2008, 108, 2482–2505. [Google Scholar] [CrossRef] [PubMed]
- Ferri, S.; Kojima, K.; Sode, K. Review of glucose oxidases and glucose dehydrogenases: A bird’s eye view of glucose sensing enzymes. J. Diabetes Sci. Technol. 2011, 5, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Si, P.; Huang, Y.; Wang, T.; Ma, J. Nanomaterials for electrochemical non-enzymatic glucose biosensors. RSC Adv. 2013, 3, 3487–3502. [Google Scholar] [CrossRef]
- Qiu, H.; Huang, X. Effects of Pt decoration on the electrocatalytic activity of nanoporous gold electrode toward glucose and its potential application for constructing a nonenzymatic glucose sensor. J. Electroanal. Chem. 2010, 643, 39–45. [Google Scholar] [CrossRef]
- Huang, J.F. Facile preparation of an ultrathin nickel film coated nanoporous gold electrode with the unique catalytic activity to oxidation of glucose. Chem. Commun. 2009, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Cha, A.; Lee, Y.; Lee, C. Nonenzymatic amperometric glucose sensor based on nanoporous gold/ruthenium electrode. Electroanalysis 2011, 23, 2057–2062. [Google Scholar] [CrossRef]
- Tavakkoli, N.; Nasrollahi, S. Non-enzymatic glucose sensor based on palladium coated nanoporous gold film electrode. Aust. J. Chem. 2013, 66, 1097–1104. [Google Scholar] [CrossRef]
- Guo, M.-M.; Wang, P.-S.; Zhou, C.-H.; Xia, Y.; Huang, W.; Li, Z. An ultrasensitive non-enzymatic amperometric glucose sensor based on a Cu-coated nanoporous gold film involving co-mediating. Sens. Actuators B 2014, 203, 388–395. [Google Scholar] [CrossRef]
- Lang, X.-Y.; Fu, H.-Y.; Hou, C.; Han, G.-F.; Yang, P.; Liu, Y.-B.; Jiang, Q. Nanoporous gold supported cobalt oxide microelectrodes as high-performance electrochemical biosensors. Nat. Commun. 2013, 4, 2169. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.-M.; Yin, X.-L.; Zhou, C.-H.; Xia, Y.; Huang, W.; Li, Z. Ultrasensitive nonenzymatic sensing of glucose on Ni(OH)2-coated nanoporous gold film with two pairs of electron mediators. Electrochim. Acta 2014, 142, 351–358. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, M.e.; Li, H.; Pan, Y.; Si, P. Non-enzymatic glucose sensors based on controllable nanoporous gold/copper oxide nanohybrids. Talanta 2014, 125, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.; Zhang, Z.; Zhang, P.; Xiao, S.; Wang, L.; Dong, Y.; Yuan, H.; Li, P.; Sun, Y.; Jiang, X.; et al. 3D nanoporous gold scaffold supported on graphene paper: Freestanding and flexible electrode with high loading of ultrafine PtCo alloy nanoparticles for electrochemical glucose sensing. Anal. Chim. Acta 2016, 938, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Li, Z.; Gan, L.; Zhao, J.; Cui, G.; Kellogg, W.; Matera, D.; Higgins, D.; Wu, G. Three-dimensional nanoporous Au films as high-efficiency enzyme-free electrochemical sensors. Electrochim. Acta 2015, 170, 337–342. [Google Scholar] [CrossRef]
- Xu, H.; Zheng, Q.L.; Yang, P.; Liu, J.S.; Xing, S.J.; Jin, L.T. Electrochemical synthesis of silver nanoparticles-coated gold nanoporous film electrode and its application to amperometric detection for trace Cr(VI). Sci. China Chem. 2011, 54, 1004–1010. [Google Scholar] [CrossRef]
- Siepenkoetter, T.; Salaj-Kosla, U.; Magner, E. The immobilization of fructose dehydrogenase on nanoporous gold electrodes for the detection of fructose. ChemElectroChem 2017, 4, 905–912. [Google Scholar] [CrossRef]
- Ahmadalinezhad, A.; Chen, A. High-performance electrochemical biosensor for the detection of total cholesterol. Biosens. Bioelectron. 2011, 26, 4508–4513. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Tian, Y.; Liu, H.; Luo, Y. Nanoporous gold film encapsulating cytochrome c for the fabrication of a H2O2 biosensor. Biomaterials 2009, 30, 3183–3188. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, X.; Li, Y.; Du, X.; Wang, X.; Xu, P. Lipase-nanoporous gold biocomposite modified electrode for reliable detection of triglycerides. Biosens. Bioelectron. 2014, 53, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhang, H.; Jia, F.; Qin, W.; Du, D. Assembly of carbon nanotubes on a nanoporous gold electrode for acetylcholinesterase biosensor design. Sens. Actuators B 2014, 199, 284–290. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Z.; Sun, H.; Wang, X.; Xu, P. Selective determination of phenols and aromatic amines based on horseradish peroxidase-nanoporous gold co-catalytic strategy. Biosens. Bioelectron. 2016, 79, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Liu, Z.; Wu, C.; Xu, P.; Wang, X. Amperometric inhibitive biosensor based on horseradish peroxidase-nanoporous gold for sulfide determination. Sci. Rep. 2016, 6, 30905. [Google Scholar] [CrossRef] [PubMed]
- Shulga, O.V.; Zhou, D.; Demchenko, A.V.; Stine, K.J. Detection of free prostate specific antigen (fPSA) on a nanoporous gold platform. Analyst 2008, 133, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Yan, X.; Zhu, C.; Du, D.; Lin, Y. Recent advances in electrochemical immunosensors. Anal. Chem. 2017, 89, 138–156. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Y.; Zhou, J.; Yan, W.; Li, X.; Zhu, J.-J. Electrochemical impedance immunosensor based on three-dimensionally ordered macroporous gold film. Anal. Chem. 2008, 80, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Bertok, T.; Sediva, A.; Katrlik, J.; Gemeiner, P.; Mikula, M.; Nosko, M.; Tkac, J. Label-free detection of glycoproteins by the lectin biosensor down to attomolar level using gold nanoparticles. Talanta 2013, 108, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Jiang, J.; Shen, G.; Yu, R. Impedance immunosensor based on receptor protein adsorbed directly on porous gold film. Anal. Chim. Acta 2005, 553, 190–195. [Google Scholar] [CrossRef]
- Dawan, S.; Wannapob, R.; Kanatharana, P.; Limbut, W.; Numnuam, A.; Samanman, S.; Thavarungkul, P. One-step porous gold fabricated electrode for electrochemical impedance spectroscopy immunosensor detection. Electrochim. Acta 2013, 111, 374–383. [Google Scholar] [CrossRef]
- Li, R.; Wu, D.; Li, H.; Xu, C.; Wang, H.; Zhao, Y.; Cai, Y.; Wei, Q.; Du, B. Label-free amperometric immunosensor for the detection of human serum chorionic gonadotropin based on nanoporous gold and graphene. Anal. Biochem. 2011, 414, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhao, Y.; Xu, C.; Wu, D.; Cai, Y.; He, J.; Li, H.; Du, B.; Yang, M. Nanoporous gold film based immunosensor for label-free detection of cancer biomarker. Biosens. Bioelectron. 2011, 26, 3714–3718. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Zhang, Y.; Li, H.; Wu, D.; Xin, X.; Zhang, S.; Yu, H.; Wei, Q.; Du, B. Ultrasensitive electrochemical immunosensor for zeranol detection based on signal amplification strategy of nanoporous gold films and nano-montmorillonite as labels. Anal. Chim. Acta 2013, 758, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.Y.; Wei, Q.; Xu, C.; Li, H.; Wu, D.; Cai, Y.; Mao, K.; Cui, Z.; Du, B. Label-free electrochemical immunosensor for sensitive detection of kanamycin. Sens. Actuators B 2011, 155, 618–625. [Google Scholar] [CrossRef]
- Pandey, B.; Demchenko, A.; Stine, K. Nanoporous gold as a solid support for protein immobilization and development of an electrochemical immunoassay for prostate specific antigen and carcinoembryonic antigen. Microchim. Acta 2012, 179, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Li, H.; Hu, K.; Lin, J.M. Electrochemical immunoassay of hepatitis B surface antigen by the amplification of gold nanoparticles based on the nanoporous gold electrode. Talanta 2010, 80, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Jiao, X.; Chen, D. Ultrasensitive electrochemical immunosensor for CA 15-3 using thionine-nanoporous gold-graphene as a platform and horseradish peroxidase-encapsulated liposomes as signal amplification. Analyst 2012, 137, 4440–4447. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ma, Z. Electrochemical immunosensor based on nanoporpus gold loading thionine for carcinoembryonic antigen. Anal. Chim. Acta 2013, 780, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Guo, Z.; Gao, L.; Zhang, Y.; Fan, D.; Ji, G.; Du, B.; Wei, Q. Ultrasensitive electrochemical immunosensor for carbohydrate antigen 72-4 based on dual signal amplification strategy of nanoporous gold and polyaniline-Au asymmetric multicomponent nanoparticles. Biosens. Bioelectron. 2015, 64, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Xu, L.; Zhang, H.; Yu, A.; Lai, G. Enzymatically catalytic signal tracing by a glucose oxidase and ferrocene dually functionalized nanoporous gold nanoprobe for ultrasensitive electrochemical measurement of a tumor biomarker. Analyst 2016, 141, 4381–4387. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, L.; Ge, S.; Song, X.; Ge, L.; Yan, M.; Yu, J. A 3D origami multiple electrochemiluminescence immunodevice based on a porous silver-paper electrode and multi-labeled nanoporous gold-carbon spheres. Chem. Commun. 2013, 49, 7687–7689. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, R.; Zhang, X. Electrochemiluminescence immunoassay at a nanoporous gold leaf electrode and using CdTe quantum dots as labels. Microchim. Acta 2011, 172, 285–290. [Google Scholar] [CrossRef]
- Li, L.; Li, W.; Ma, C.; Yang, H.; Ge, S.; Yu, J. Paper-based electrochemiluminescence immunodevice for carcinoembryonic antigen using nanoporous gold-chitosan hybrids and graphene quantum dots functionalized Au@Pt. Sens. Actuators B 2014, 202, 314–322. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Advantages | Disadvantages |
|---|---|---|
| Electrochemical etching of Au electrode | One-step process No need to prepare alloy beforehand No need of highly concentrated corrosive chemicals Low chances of impurity on surface | Difficult to control size of pores and ligaments Can be time consuming |
| Electrodeposition | One-step process No need to prepare alloy beforehand Stable and highly pure structure can be formed | Difficult to create thicker structure Difficult to control size of pores and ligaments |
| Dealloying (a) Chemical (b) Electrochemical | Easy and no need of instrumentation Large number of samples can be prepared at the same time in a batch Size of pores and ligament can be tuned easily Optimal for self-supported np-Au structures Better control over pores and ligaments size when an alloy is a thin layer No need of highly corrosive solvents | Use of corrosive solvents May contain impurities from less noble metals Once dealloyed (self-supported structures), difficult to use as a working electrode because of fragile nature and connection problem Time consuming if thicker and large number of electrodes have to be prepared Electrolyte gets contaminated after dealloying and may need to be changed after each dealloying |
| Tech | Sensing Method | Analyte | Probe/Label | Linear Range | LOD | Ref. |
|---|---|---|---|---|---|---|
| CC | Hybridization | DNA | [Ru(NH3)6]3+ | 50–250 fM | 5.6 fM | [97] |
| CC | Hybridization | DNA | AuNP/[Ru(NH3)6]3+ | 0.08–1600 fM | 28 aM | [98] |
| CC | DNAzyme | Pb2+ | [Ru(NH3)6]3+ | 0.05–100 nM | 12 pM | [93] |
| CC | Aptasensing | Thrombin | AuNP/[Ru(NH3)6]3+ | 0.01–22 nM | 30 fM | [90] |
| DPV | Hybridization | Hg2+ | Ferrocene | 0.01–5000 nM | 3.6 pM | [78] |
| DPV | Aptasensing | Bisphenol A | - | 0.1–100 nM | 0.056 nM | [92] |
| DPV | Aptasensing | ATP | DABA | 0.1–3000 µM | 0.1 µM | [91] |
| DPV | Hybridization | DNA | Methylene blue | 60–220 pM | 6.7 pM | [101] |
| DPV | Hybridization | E. coli | Methylene blue | 50–50000 cfu·µL−1 | 50 cfu·µL−1 | [100] |
| DPASV | Hybridization | DNA | PbS-AuNP | 0.9–70 fM | 0.26 fM | [102] |
| SWV | Hybridization | DNA | [Fe(CN)6]3−/4− | 10–200 nM | 10 nM | [28] |
| Electrode | Rf | Potential a | Mediator | Linear (mM) | LOD (µM) | Sensitivity (µA·cm−2·mM−1) | Ref. |
|---|---|---|---|---|---|---|---|
| GOx-Chi/PB/np-Au/Ti | NA | −1.0 V b | PB | 0.1–2.0 | 2.5 | 177 c | [119] |
| Naf/GOx/np-Au/GC | NA | 0.4 V | - | 1–18 | 196 | 0.049 c | [30] |
| GOx/SAM/np-Au/GC | NA | −0.2 V b | - | 3–8 | 10 | 8.6 | [110] |
| Naf/GOx/PB/np-Au/Cr/Si | 40 | 0 V b | PB | 2–30 | 300 | 50 | [120] |
| GOx/Os(bpy)2P/np-Au/SiO2 | NA | NA | Os(bpy)2P | NA | 2 | 75 | [121] |
| GOx/PEDOT/np-Au/GC | NA | 0.2 V | BQ | 0.1–15 | 10 | 7.3 | [122] |
| GOx/np-Au/Au/Si | 36 | NA | - | 0.1–0.5 | 73 | 21.14 | [123] |
| GOx/GA/CA/np-Au/GC | 7 | 0.2 V 0.3 V | BQ FCA | 1–10 1–10 | NA NA | 3.53 1.35 | [124] |
| GOx/DTDPA/np-Au/GC | 8 | 0.2 V | BQ | 1–10 | NA | 2.187 | [9] |
| GOx/np-Au/GC | 8 | 0.3 V | - | 0.05–10 | 1.02 | 12.1 | [125] |
| Tech | Electrode | Analyte | Linear (mM) | LOD (µM) | Sensitivity (µA·cm−2·mM−1) | Ref. |
|---|---|---|---|---|---|---|
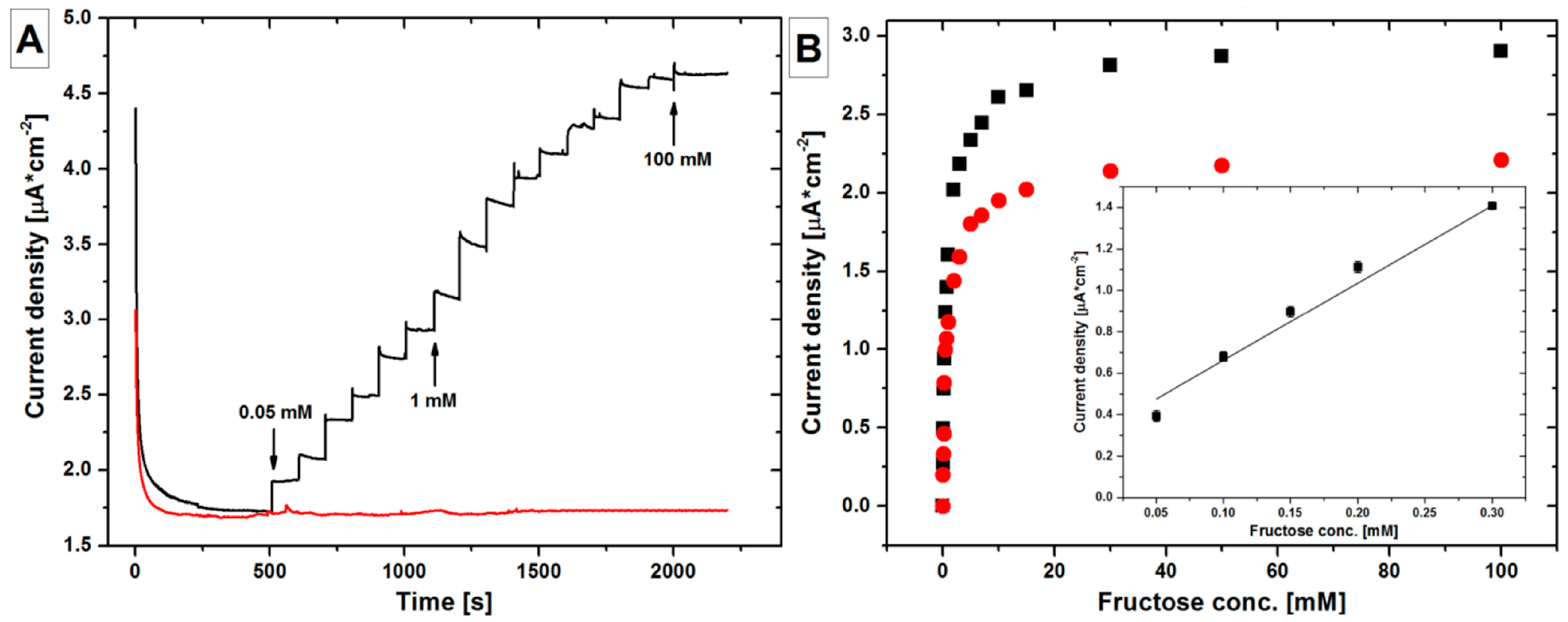
| CA | FDH/ND/np-Au/glass | Fructose | 0.05–0.3 | 1.2 | 3.7 | [140] |
| Nafion/ADH/np-Au/GC | Alcohol | 1.0–8.0 | 120 | 0.19 a | [30] | |
| Cyt c/np-Au/ITO | H2O2 | 0.010–12 | 6.3 | 2.8 | [142] | |
| HRP/np-Au/Au | H2O2 | 0.010–0.380 | 2.6 | 21 | [106] | |
| CV | ChOx+ChE+HRP/np-Au/Ti | Cholesterol | 0.97–7.8 | 12.9 | 29.33 | [141] |
| Lipase/np-Au/GC | Tributyrin | 1.65–8.27 | 88.6 | 0.009 | [143] | |
| AChE/MWCNT/Ci/np-Au/Au | Malathion | 0.003–0.150 | 0.0015 | NA | [144] | |
| DPV | HRP/np-Au/GC | Catechol | 7–150 | 0.66 | 31.8 | [145] |
| HRP/np-Au/GC | Sulfide | 0.1–40 | 0.027 | 1720 | [146] |
| Tech | Ab/Electrode | Antigen | Label/Probe | Linear | LOD | Ref. |
|---|---|---|---|---|---|---|
| EIS | Ab1/np-Au/GC | human IgG | Ab2-HRP | 0.011–11 ng·mL−1 | 0.009 ng·mL−1 | [151] |
| EIS | Ab/11-MUA/np-Au/Au | HSA | Label-free | 0.010–10,000 pM | 10 fM | [152] |
| CV | Ab/np-Au/GS/GC | hCG | Label-free | 0.5–40.00 ng·mL−1 | 0.034 ng·mL−1 | [153] |
| CV | Ab/np-Au/GC | PSA | Label-free | 0.05–26 ng·mL−1 | 3 pg·mL−1 | [154] |
| CV | Ab1/np-Au/GC | zeranol | Ab2/HRP/TH/SM | 0.01–12 ng·mL−1 | 3 pg·mL−1 | [155] |
| SWV | Ab1/np-Au/PB-C/GS /GC | kanamycin | Label-free | 0.02–14 ng·mL−1 | 6.31 pg·mL−1 | [156] |
| SWV | Ab1-ALP/LA/np-Au | PSA CEA | ALP | 1–30 ng·mL−1 0.2–10 ng·mL−1 | 0.75 ng·mL−1 0.015 ng·mL−1 | [157] |
| DPV | Ab1/DTSP/np-Au/GC | HBsAg | Ab2-HRP/AuNPs | 0.01–1.0 ng·mL−1 | 2.3 pg·mL−1 | [158] |
| DPV | Ab1/TH/np-Au/GS/GC | CA 15-3 | Ab2/HRP@Lip | 2 × 10−5–40 U·mL−1 | 2 × 10−6 U·mL−1 | [159] |
| DPV | Ab1/AuNP/TH/np-Au/GC | CEA | Label-free | 0.01–100 ng·mL−1 | 3 pg·mL−1 | [160] |
| CA | Ab1/np-Au/GC | CA 72-4 | PANI/Au AMNPs | 2–200 U·mL−1 | 0.10 U·mL−1 | [161] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattarai, J.K.; Neupane, D.; Nepal, B.; Mikhaylov, V.; Demchenko, A.V.; Stine, K.J. Preparation, Modification, Characterization, and Biosensing Application of Nanoporous Gold Using Electrochemical Techniques. Nanomaterials 2018, 8, 171. https://doi.org/10.3390/nano8030171
Bhattarai JK, Neupane D, Nepal B, Mikhaylov V, Demchenko AV, Stine KJ. Preparation, Modification, Characterization, and Biosensing Application of Nanoporous Gold Using Electrochemical Techniques. Nanomaterials. 2018; 8(3):171. https://doi.org/10.3390/nano8030171
Chicago/Turabian StyleBhattarai, Jay K., Dharmendra Neupane, Bishal Nepal, Vasilii Mikhaylov, Alexei V. Demchenko, and Keith J. Stine. 2018. "Preparation, Modification, Characterization, and Biosensing Application of Nanoporous Gold Using Electrochemical Techniques" Nanomaterials 8, no. 3: 171. https://doi.org/10.3390/nano8030171
APA StyleBhattarai, J. K., Neupane, D., Nepal, B., Mikhaylov, V., Demchenko, A. V., & Stine, K. J. (2018). Preparation, Modification, Characterization, and Biosensing Application of Nanoporous Gold Using Electrochemical Techniques. Nanomaterials, 8(3), 171. https://doi.org/10.3390/nano8030171