Graphene Based Poly(Vinyl Alcohol) Nanocomposites Prepared by In Situ Green Reduction of Graphene Oxide by Ascorbic Acid: Influence of Graphene Content and Glycerol Plasticizer on Properties


Abstract
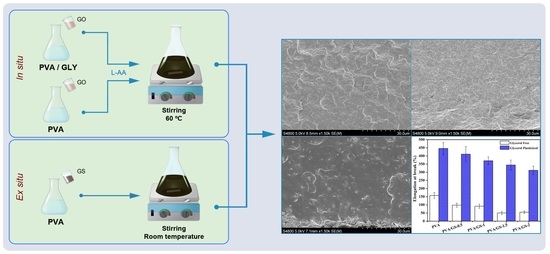
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Preparation of Graphite Oxide (GO) and Chemically Reduced Graphene Sheets (GS)
2.3. Synthesis of PVA/Graphene Sheets Nanocomposite Films (PVA/GS) by the in Situ Method
2.4. Preparation of PVA/Graphene Sheets Nanocomposite Film by the ex Situ Method
2.5. Characterization Techniques
3. Results and Discussion
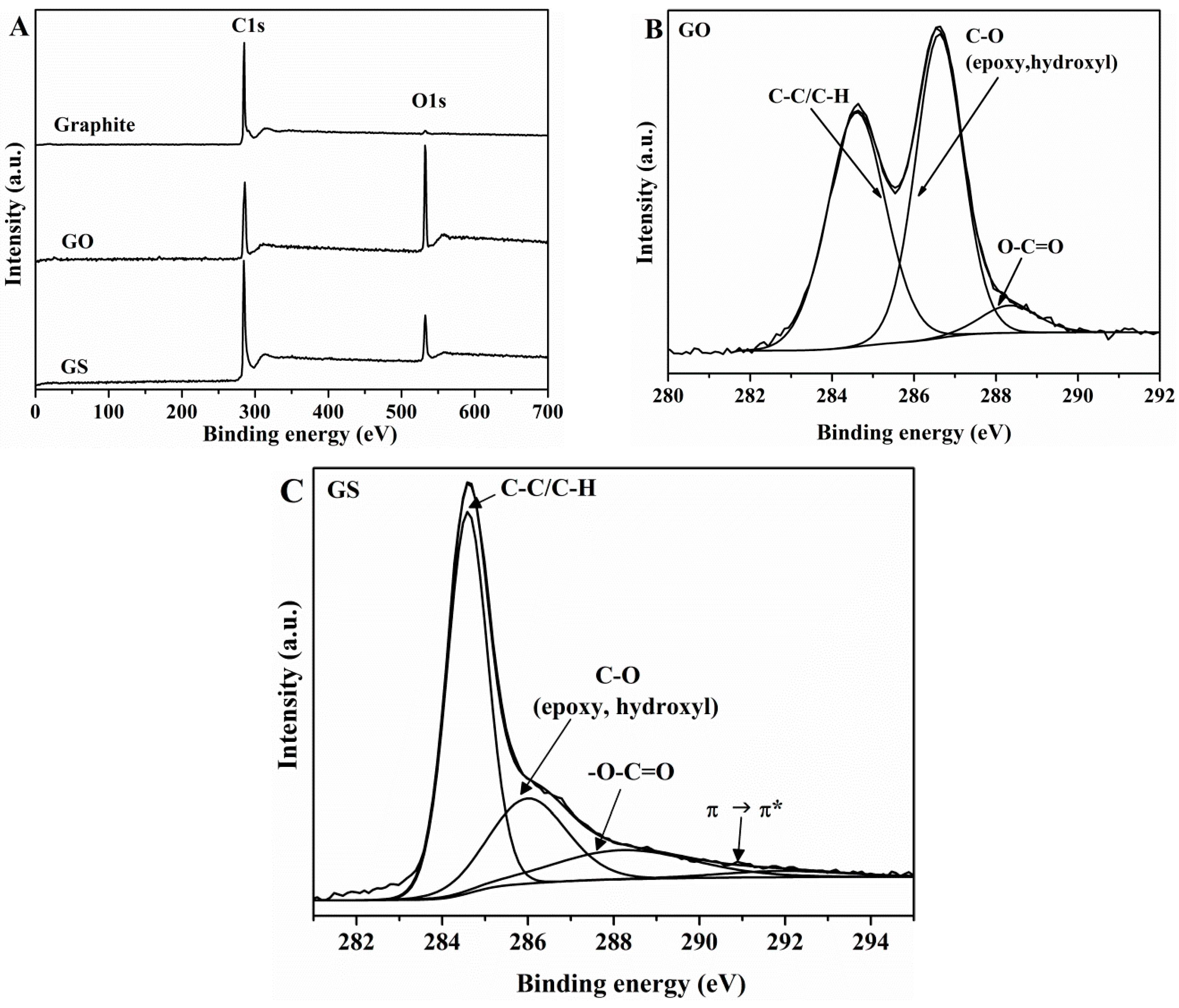
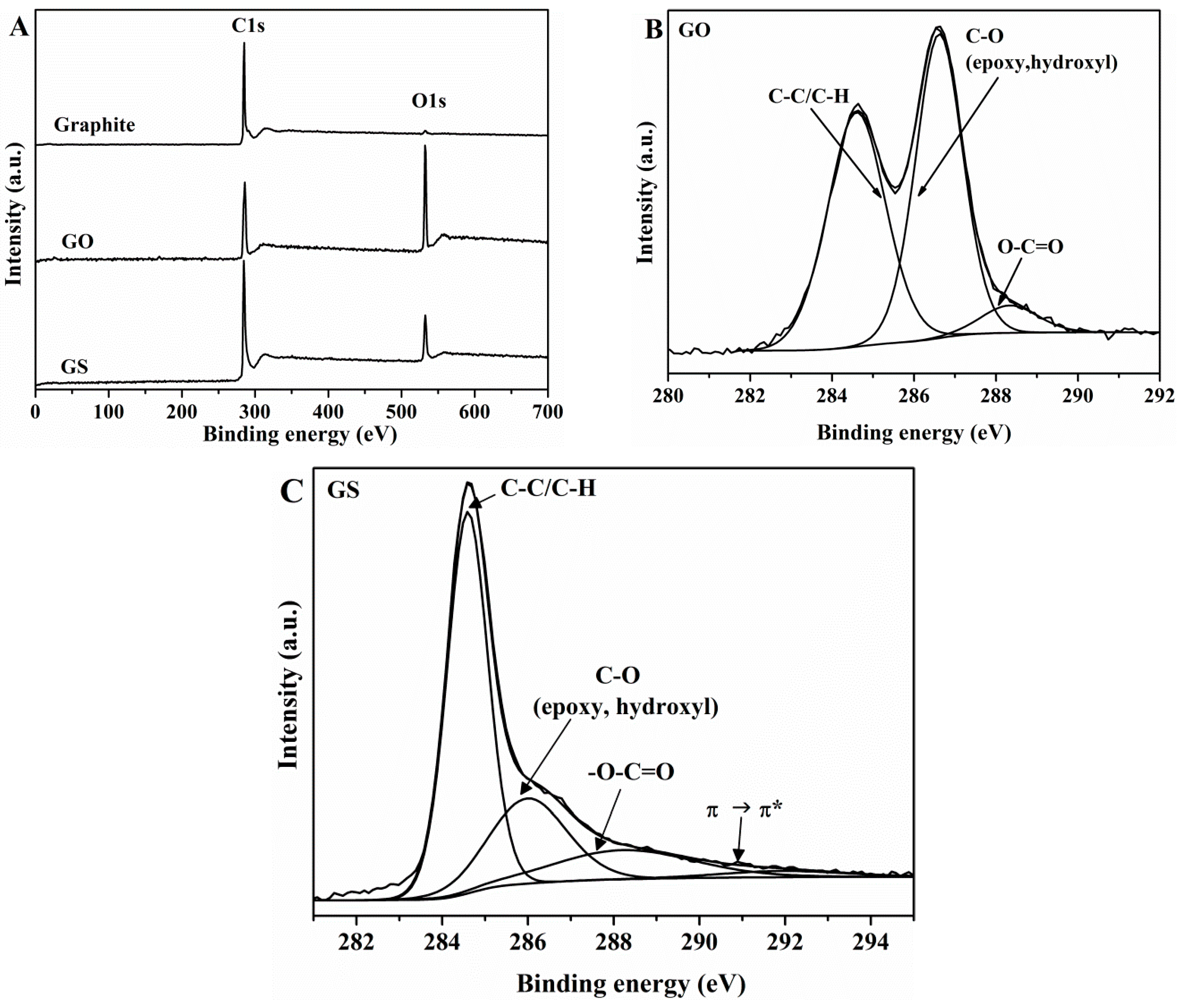
3.1. Characterization of GO and GS
3.2. Characterization of PVA/GS Nanocomposites
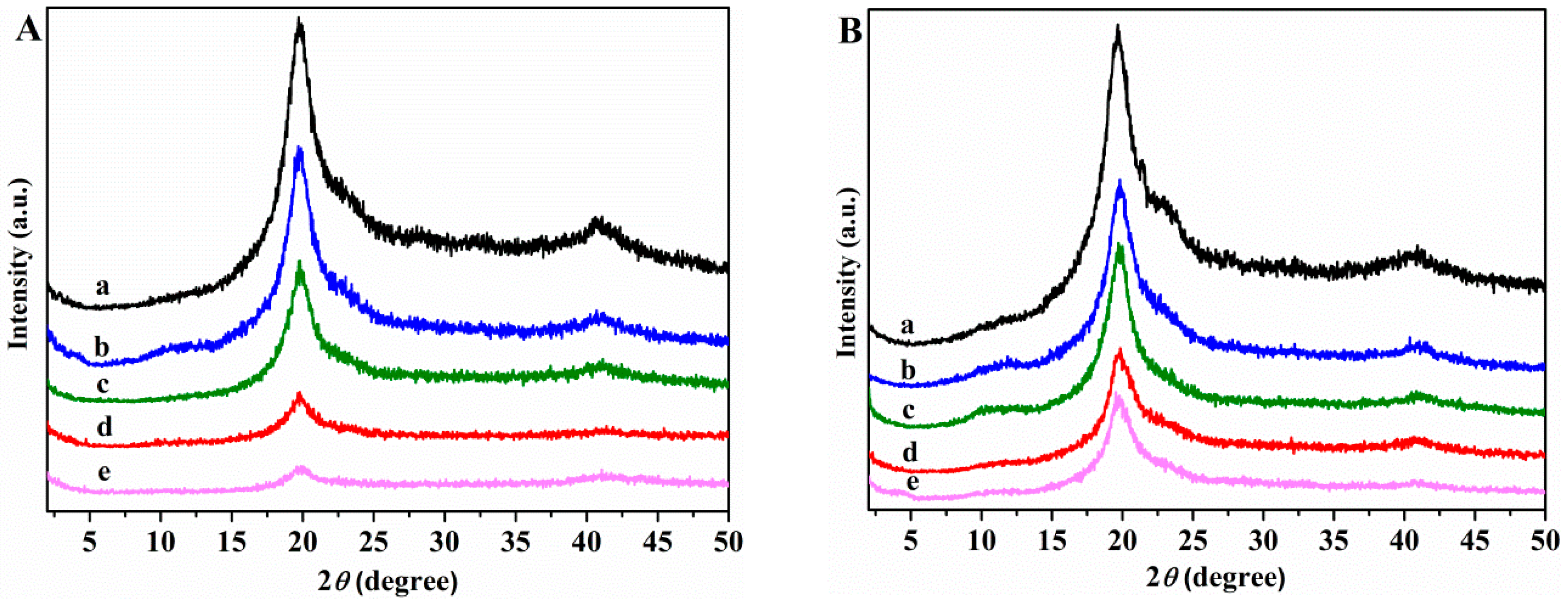
3.2.1. X-ray Diffraction
3.2.2. Nanostructure and Morphology
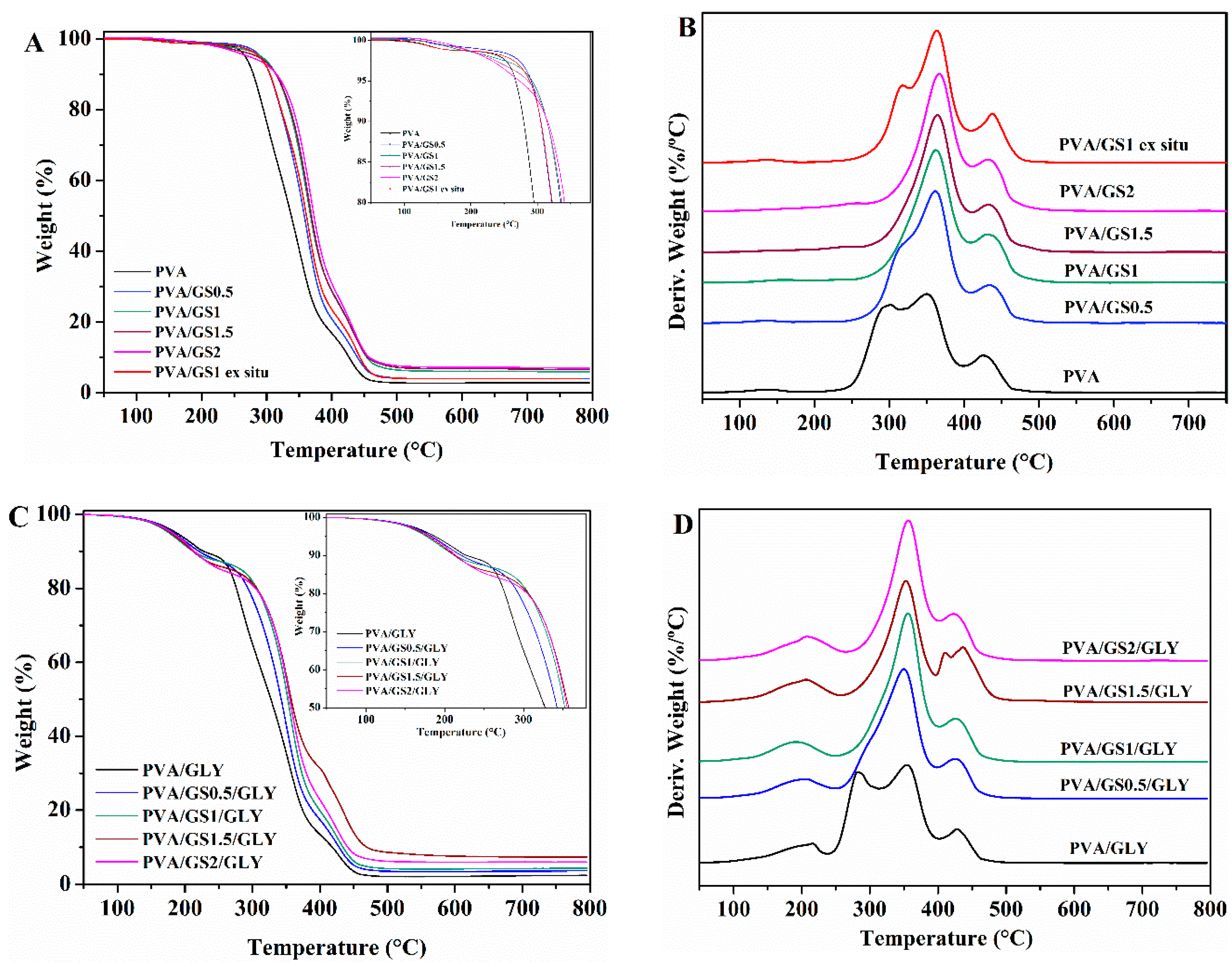
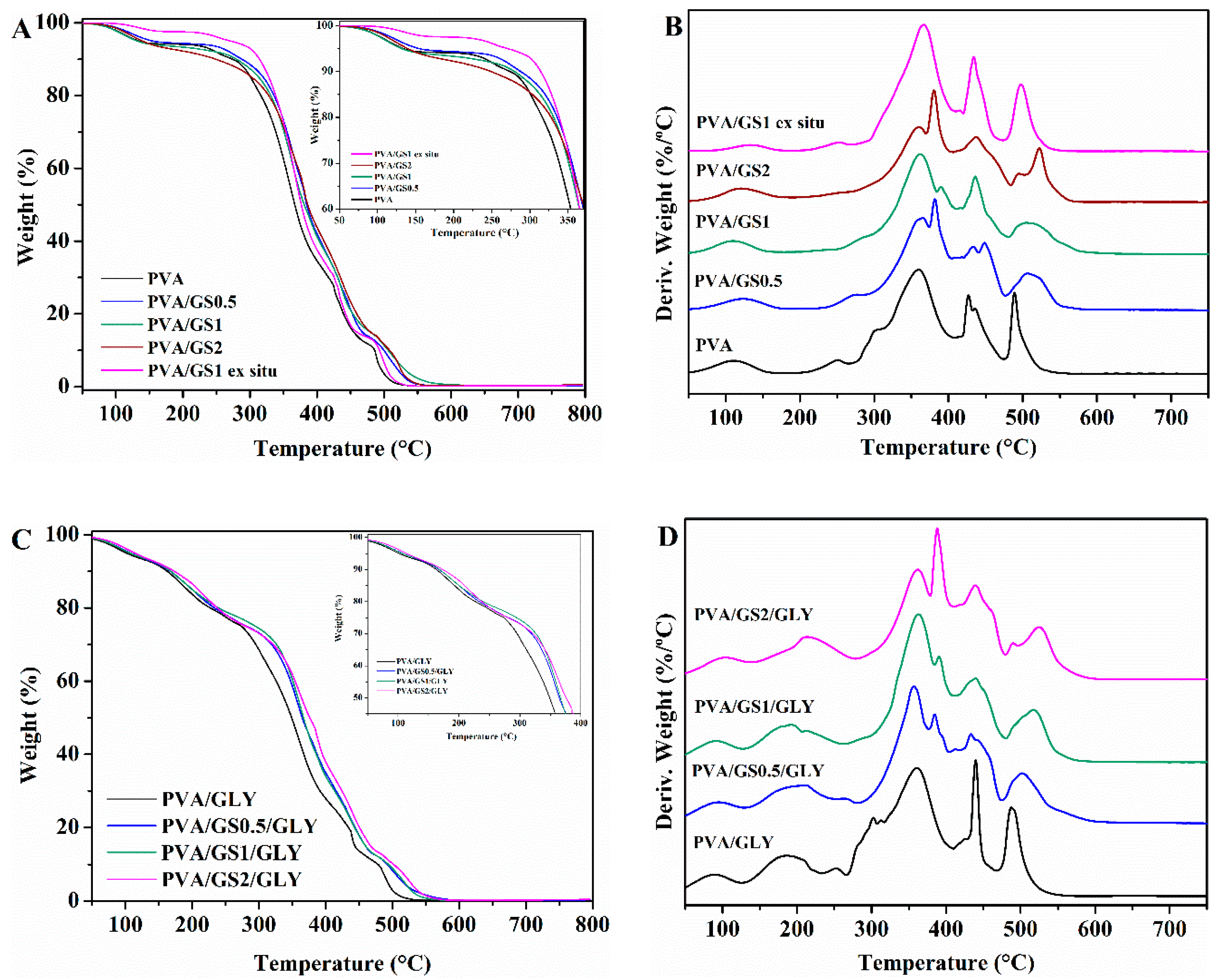
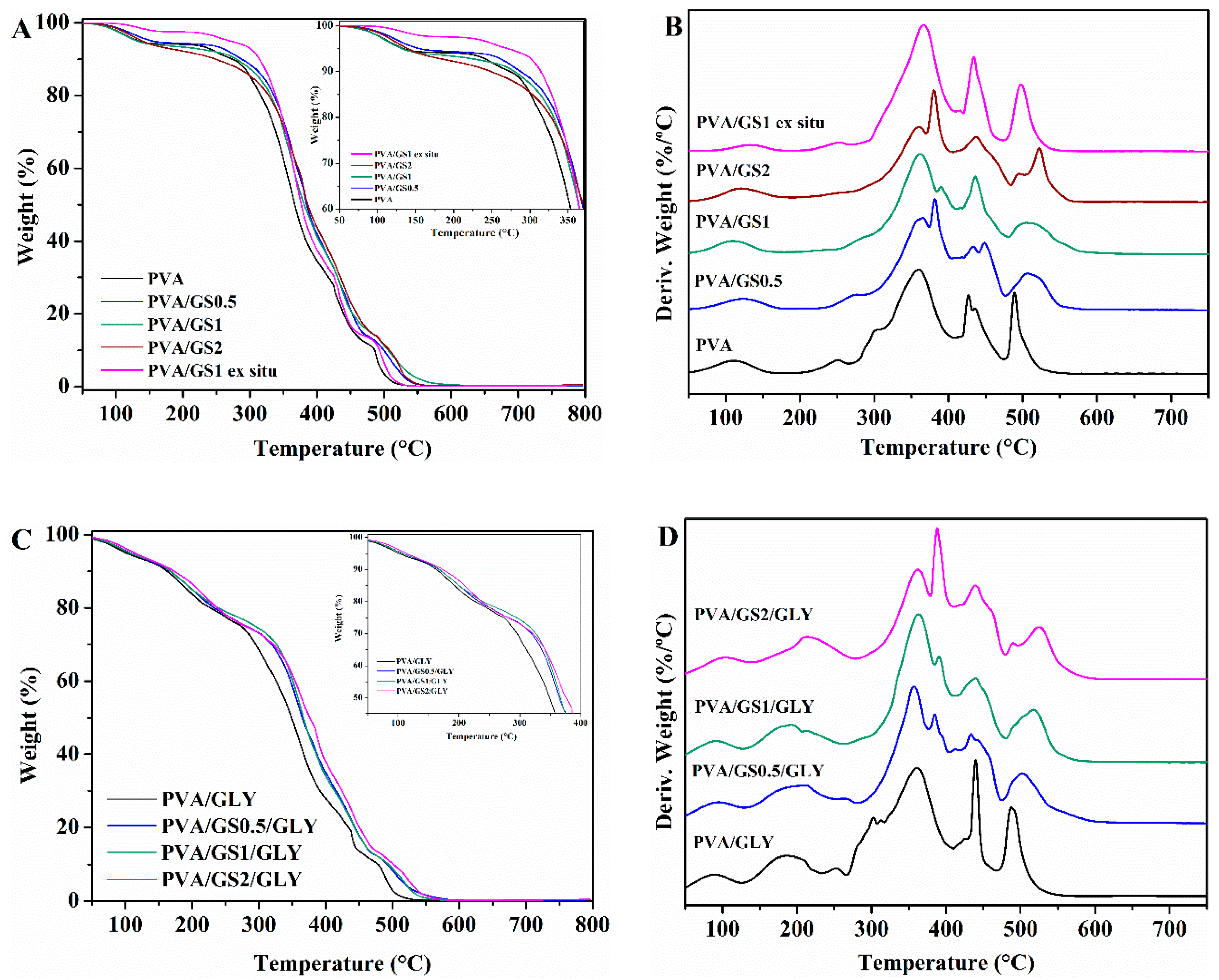
3.2.3. Thermal Properties
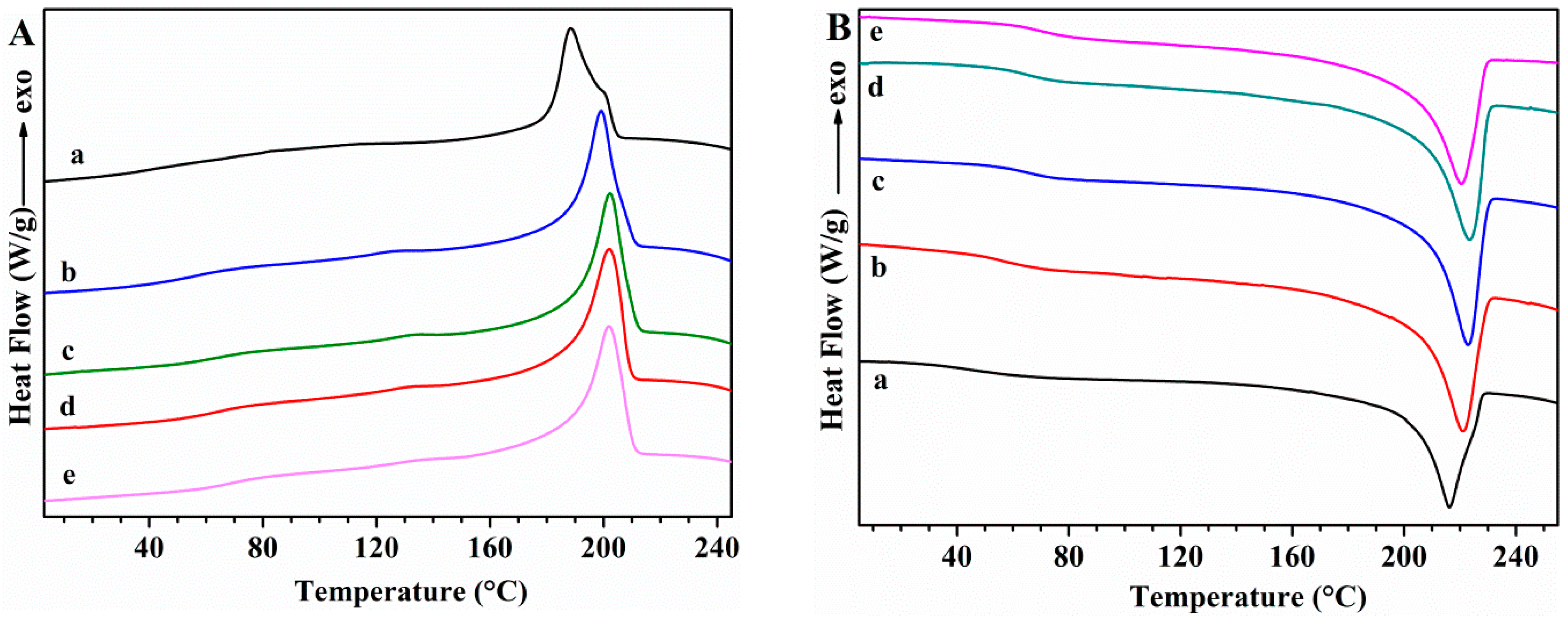
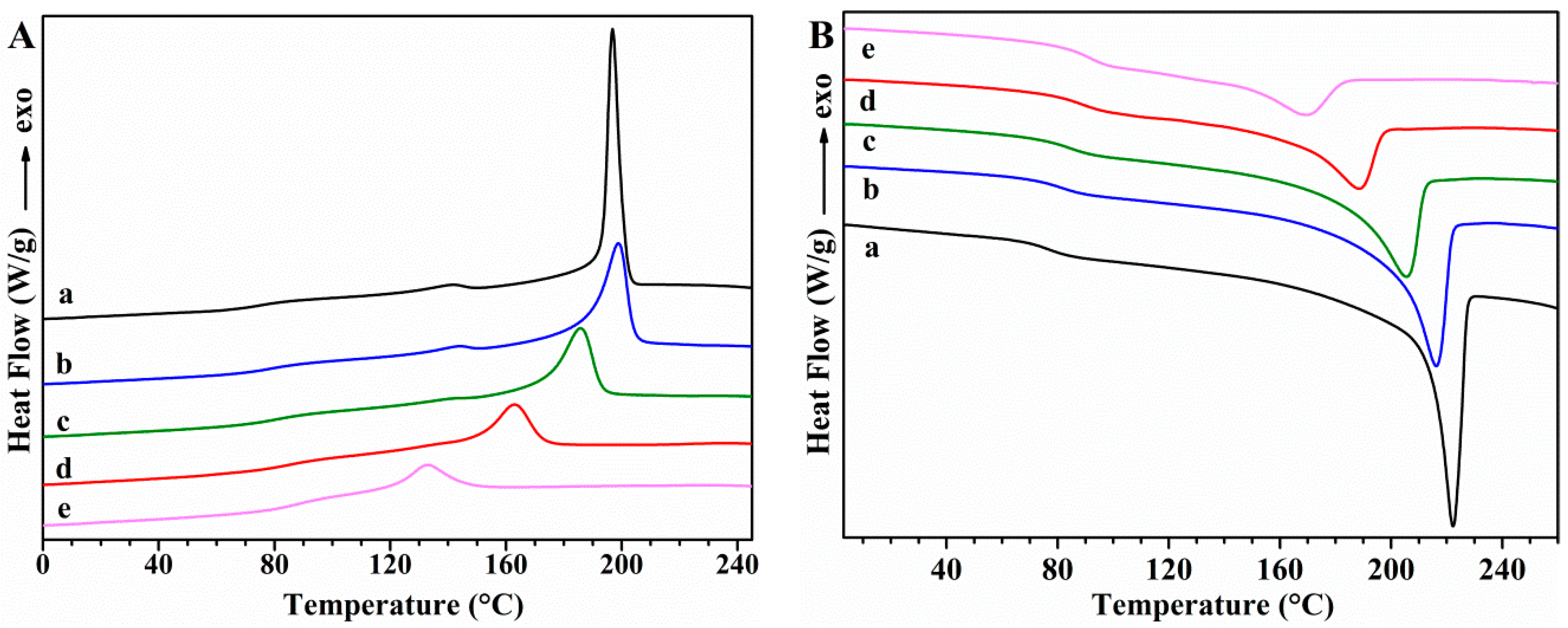
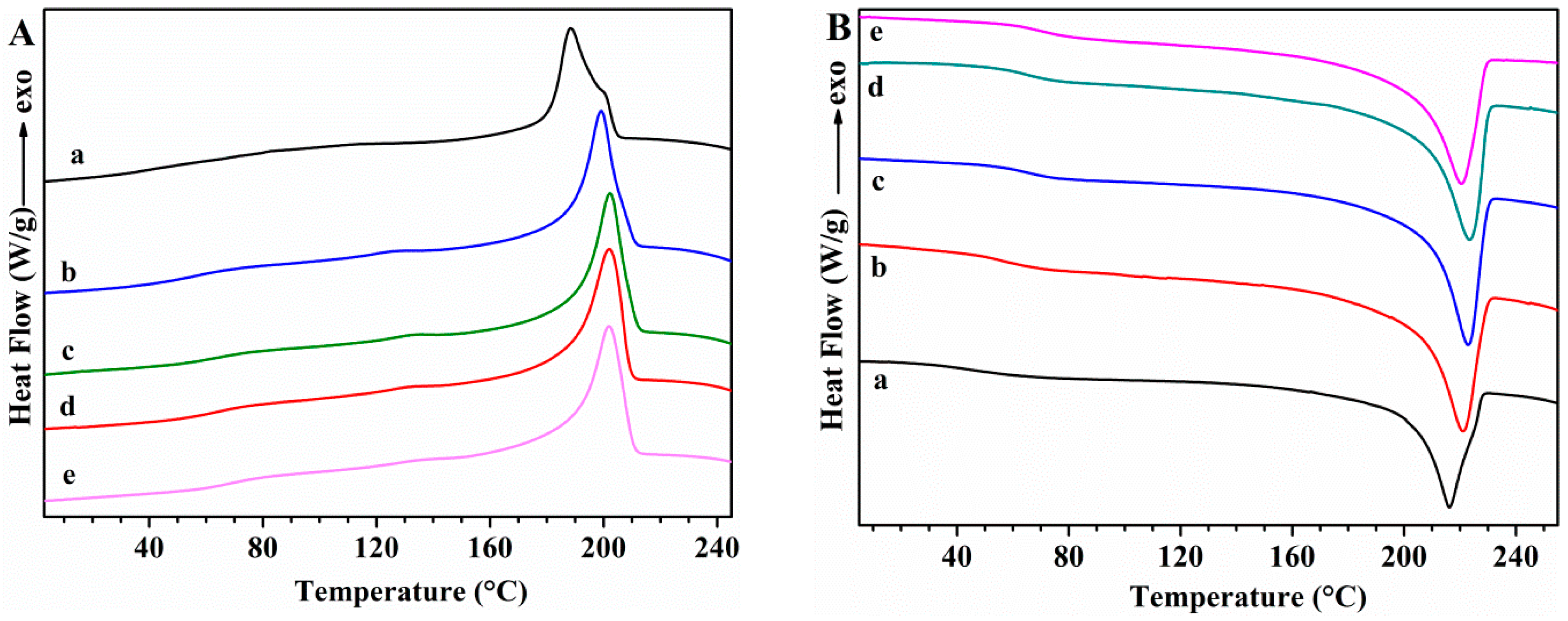
Differential Scanning Calorimetry (DSC)
Thermogravimetric Analysis (TGA)
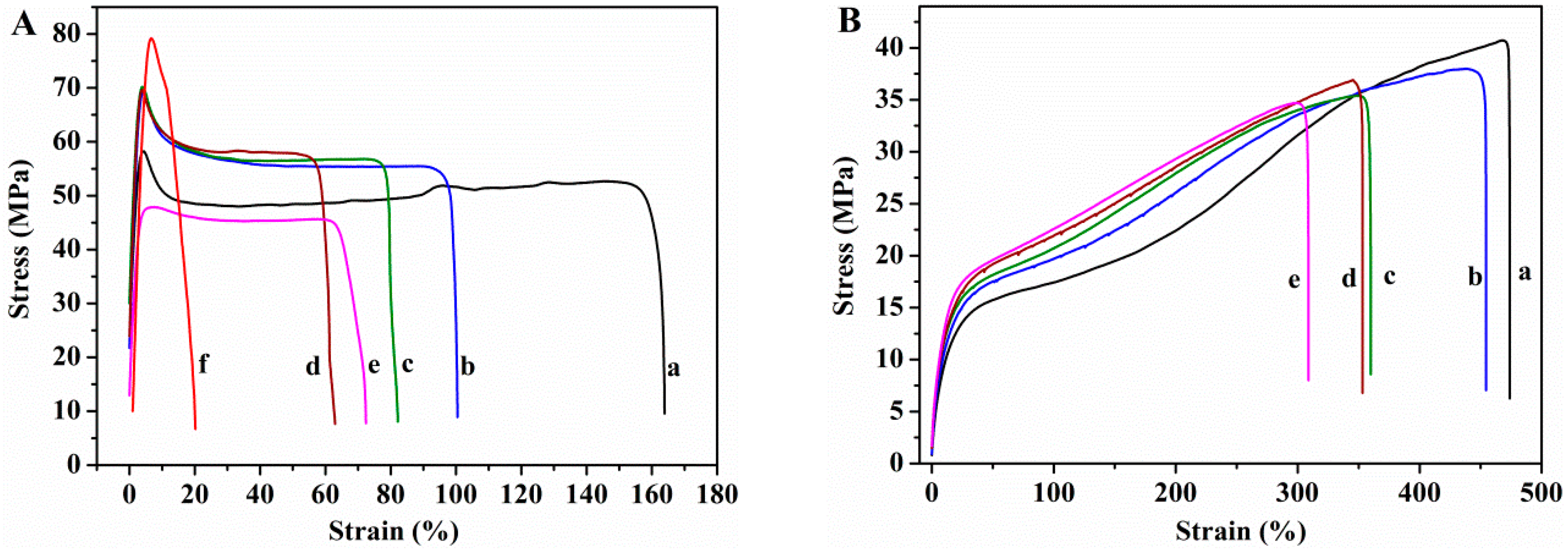
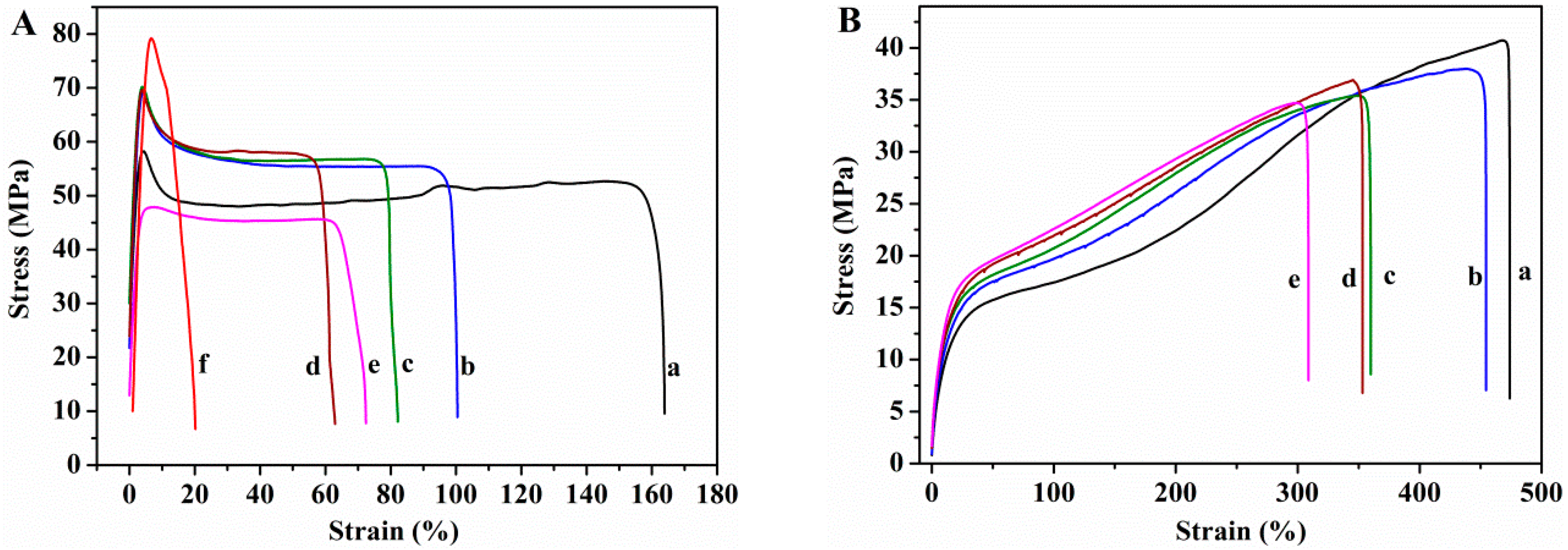
3.2.4. Mechanical Properties
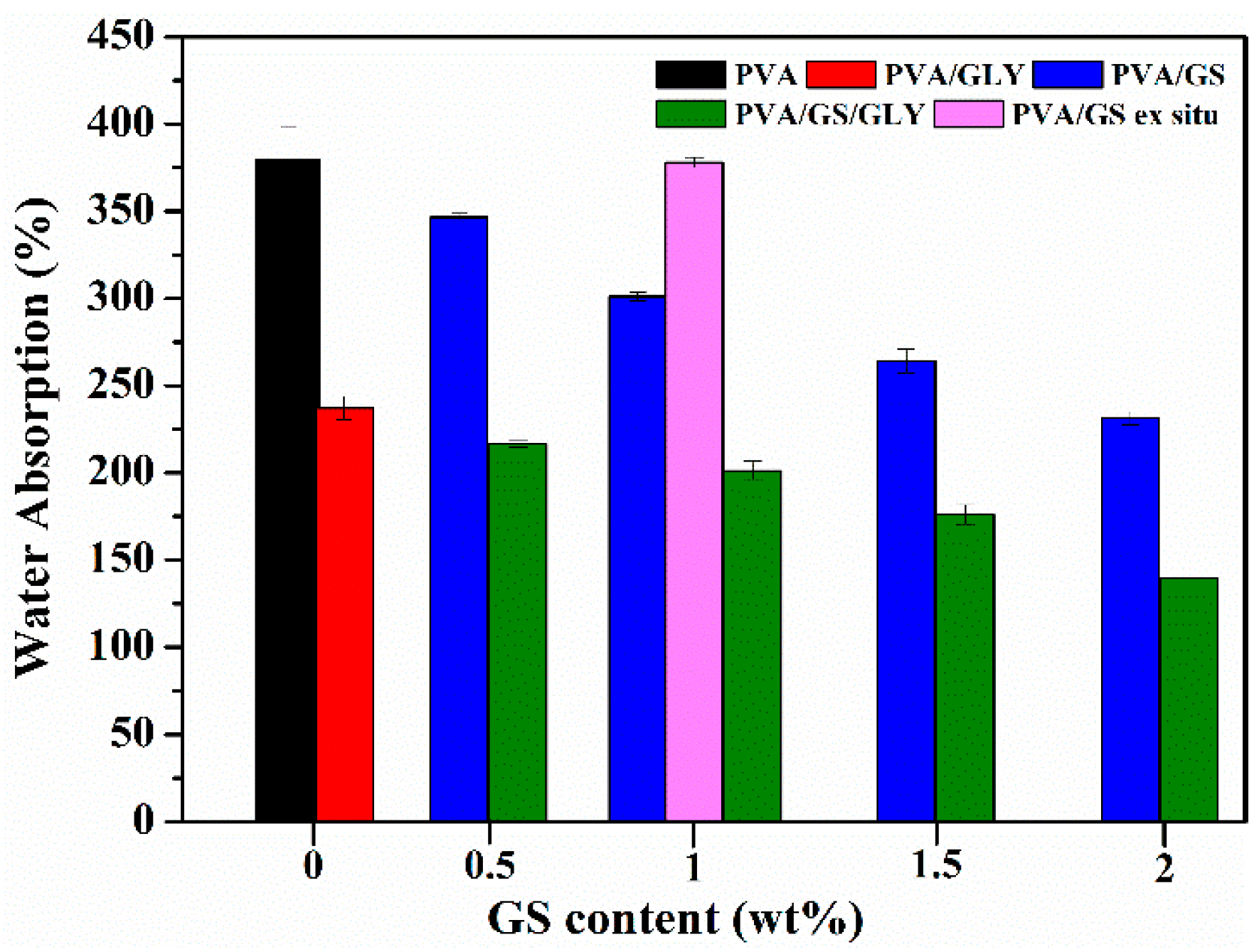
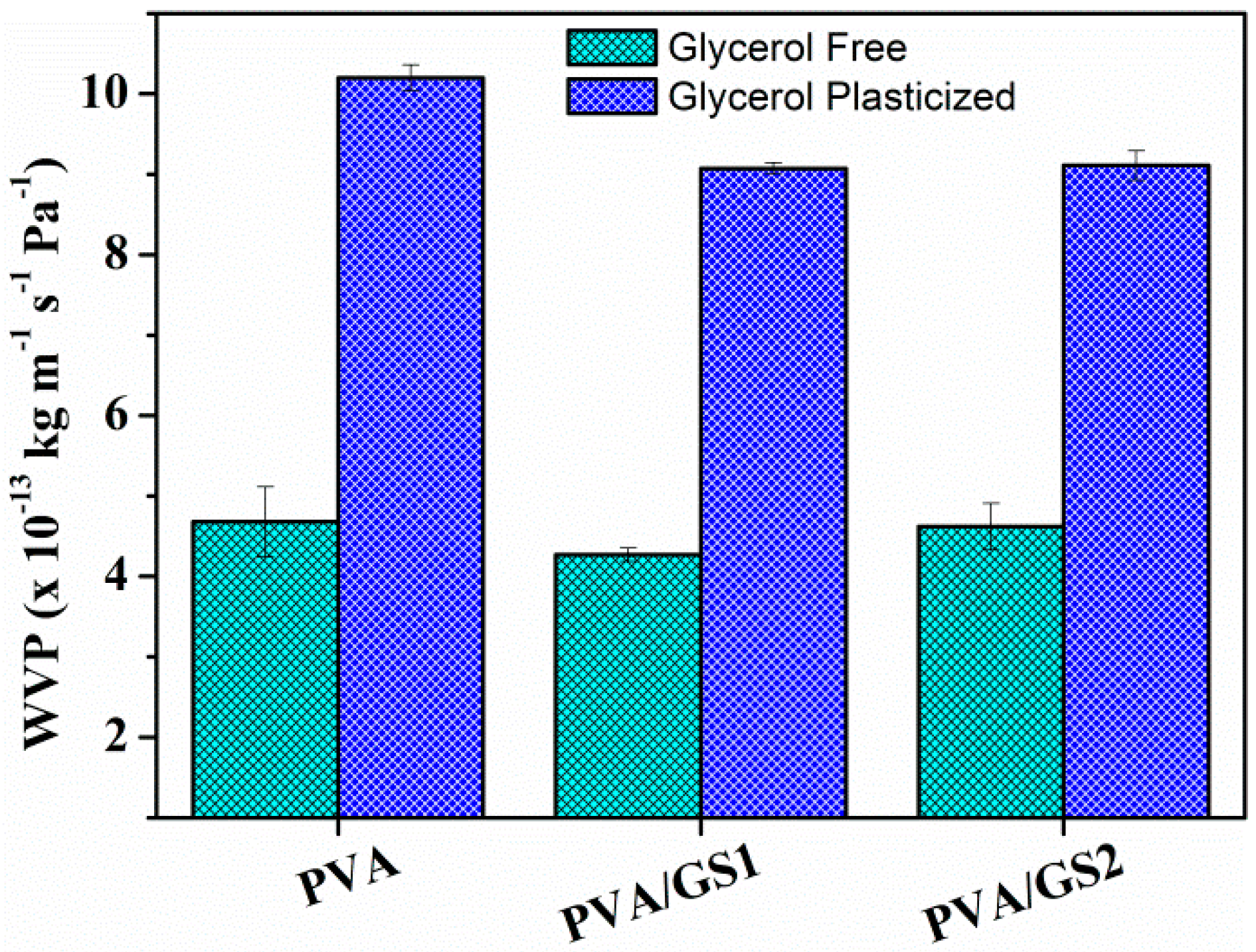
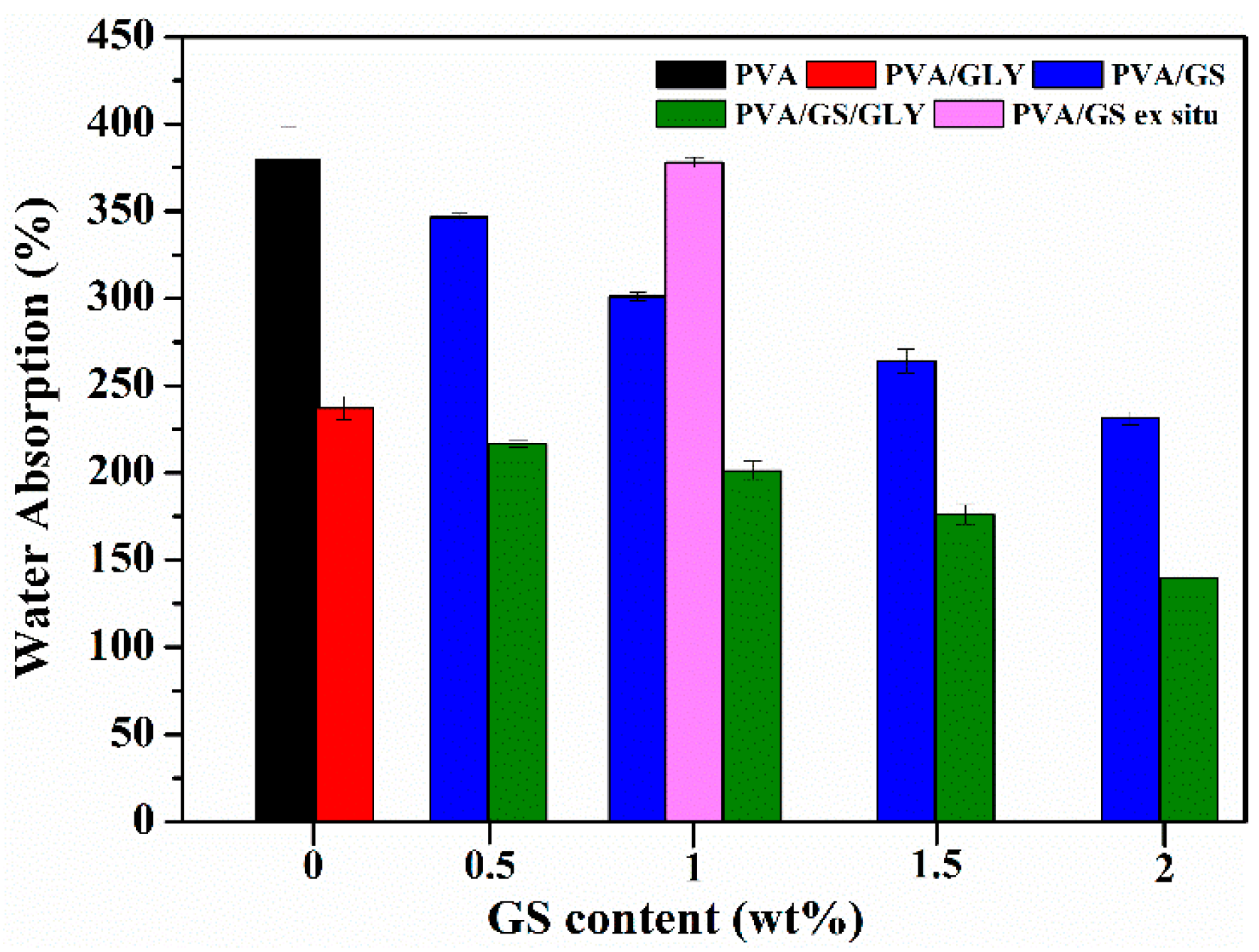
3.2.5. Water Absorption and Water Vapor Permeability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Finch, C.A. Poly(Vinyl Alcohol): Properties and Applications; John Wiley and Sons: London, UK, 1973. [Google Scholar]
- Chiellini, E.; Corti, A.; D’Antone, S.; Solaro, R. Biodegradation of poly(vinyl alcohol) based materials. Prog. Polym. Sci. 2003, 28, 963–1014. [Google Scholar] [CrossRef]
- DeMerlis, C.C.; Schoneker, D.R. Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem. Toxicol. 2003, 41, 319–326. [Google Scholar] [CrossRef]
- Mohsen-Nia, M.; Modarress, H. Viscometric study of aqueous poly(vinyl alcohol) (PVA) solutions as a binder in adhesive formulations. J. Adhes. Sci. Technol. 2006, 20, 1273–1280. [Google Scholar] [CrossRef]
- Paradossi, G.; Cavalieri, F.; Chiessi, E.; Spagnoli, C.; Cowman, M.K. Poly(vinyl alcohol) as versatile biomaterial for potential biomedical applications. J. Mater. Sci. Mater. Med. 2003, 14, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Musetti, A.; Paderni, K.; Fabbri, P.; Pulvirenti, A.; Al-Moghazy, M.; Fava, P. Poly(vinyl alcohol)-based film potentially suitable for antimicrobial packaging applications. J. Food Sci. 2014, 79, E577–E582. [Google Scholar] [CrossRef] [PubMed]
- Hyon, S.H.; Cha, W.I.; Ikada, Y.; Kita, M.; Ogura, Y.; Honda, Y. Poly (vinyl alcohol) hydrogels as soft contact lens material. J. Biomater. Sci. Polym. Ed. 1994, 5, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tian, H.; Xiang, A. Influence of polyol plasticizers on the properties of poly vinyl alcohol films fabricated by melt processing. J. Polym. Environ. 2012, 20, 63–69. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Q.; Li, L. Evaporation behaviour of water and its plasticizing effect in modified poly(vinyl alcohol) systems. Polym. Int. 2003, 52, 1820–1826. [Google Scholar] [CrossRef]
- Hodge, R.M.; Bastow, T.J.; Edward, G.H.; Simon, G.P.; Hill, A.J. Free volume and the mechanism of plasticization in water-swollen poly(vinyl alcohol). Macromolecules 1996, 29, 8137–8143. [Google Scholar] [CrossRef]
- Lim, L.Y.; Wan, L.S.C. The effect of plasticizers on the properties of poly(vinyl alcohol) films. Drug. Dev. Ind. Pharm. 1994, 20, 1007–1020. [Google Scholar] [CrossRef]
- Paul, D.R.; Robeson, L.M. Polymer nanotechnology: Nanocomposites. Polymer 2008, 49, 3187–3204. [Google Scholar] [CrossRef]
- Crosby, A.J.; Lee, J.Y. Polymer nanocomposites: The “nano” effect on mechanical properties. Polym. Rev. 2007, 47, 217–229. [Google Scholar] [CrossRef]
- Loh, K.P.; Boa, Q.; Ang, P.K.; Yang, J. The chemistry of graphene. J. Mater. Chem. 2010, 20, 2277–2289. [Google Scholar] [CrossRef]
- Kim, H.; Abdala, A.A.; Macosko, C.W. Graphene/polymer nanocomposites. Macromolecules 2010, 43, 6515–6530. [Google Scholar] [CrossRef]
- Kuilla, T.; Bhadra, S.; Yao, D.; Kim, N.H.; Bose, S.; Lee, J.H. Recent advances in graphene based polymer composites. Prog. Polym. Sci. 2010, 35, 1350–1375. [Google Scholar] [CrossRef]
- Hu, K.; Kulkarni, D.D.; Choi, I.; Tsukruk, V.V. Graphene-polymer nanocomposites for structural and functional applications. Prog. Polym. Sci. 2014, 39, 1934–1972. [Google Scholar] [CrossRef]
- Chee, W.K.; Lim, H.N.; Huang, N.M.; Harrison, I. Nanocomposites of graphene/polymers: A review. RSC Adv. 2015, 5, 68014–68051. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruof, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef]
- Park, S.; Ruoff, R. Chemical methods for the production of graphenes. Nat. Nanotechnol. 2009, 4, 217–224. [Google Scholar] [CrossRef]
- Stankovich, S.; Dikin, D.A.; Piner, R.D.; Kohlhaas, K.A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S.T.; Ruoff, R.S. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 2007, 45, 1558–1565. [Google Scholar] [CrossRef]
- Paredes, J.I.; Villar-Rodil, S.; Martínez-Alonso, A.; Tascón, J.M.D. Graphene oxide dispersions in organic solvents. Langmuir 2008, 24, 10560–10564. [Google Scholar] [CrossRef] [PubMed]
- Schniepp, H.C.; Li, J.L.; McAllister, M.J.; Sai, H.; Herrera-Alonso, M.; Adamson, D.H.; Prud’homme, R.K.; Car, R.; Saville, D.A.; Aksay, I.A. Functionalized single graphene sheets derived from splitting graphite oxide. J. Phys. Chem. B 2006, 110, 8535–8539. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.K.; Pumera, M. Chemical reduction of graphene oxide: A synthetic chemistry viewpoint. Chem. Soc. Rev. 2014, 43, 291–312. [Google Scholar] [CrossRef]
- Pei, S.; Cheng, H.M. The reduction of graphene oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- De Silva, K.K.H.; Huang, H.H.; Joshi, R.K.; Yoshimura, M. Chemical reduction of graphene oxide using green reductants. Carbon 2017, 119, 190–199. [Google Scholar] [CrossRef]
- Fernandez-Merino, M.J.; Guardia, L.; Paredes, J.I.; Villar-Rodil, S.; Solis-Fernandez, P.; Martinez-Alonso, A.; Tascon, J.M.D. Vitamin C is an ideal substitute for hydrazine in the reduction of graphene oxide suspensions. J. Phys. Chem. C 2010, 114, 6426–6432. [Google Scholar] [CrossRef]
- Mei, X.; Meng, X.; Wu, F. Hydrothermal method for the production of reduced graphene oxide. Physica E 2015, 68, 81–86. [Google Scholar] [CrossRef]
- Dubin, S.; Gilje, S.; Wang, K.; Tung, V.C.; Cha, K.; Hall, A.S.; Farrar, J.; Varshneya, R.; Yang, Y.; Kaner, R.B. A one-step, solvothermal reduction method for producing reduced graphene oxide dispersions in organic solvents. ACS Nano 2010, 4, 3845–3852. [Google Scholar] [CrossRef]
- Sasikala, S.P.; Poulin, P.; Aymonier, C. Advances in subcritical hydro-/solvothermal processing of graphene materials. Adv. Mater. 2017, 29, 1605473. [Google Scholar] [CrossRef]
- Liao, K.H.; Mittal, A.; Bose, S.; Leighton, C.; Mkhoyan, K.A.; Macosko, C.W. Aqueous only route toward graphene from graphite oxide. ACS Nano 2011, 5, 1253–1258. [Google Scholar] [CrossRef]
- Wang, H.; Robinson, J.T.; Li, X.; Dai, H. Solvothermal reduction of chemically exfoliated graphene sheets. J. Am. Chem. Soc. 2009, 131, 9910–9911. [Google Scholar] [CrossRef] [PubMed]
- Stankovich, S.; Dikin, D.A.; Dommett, D.; Kohlhaas, K.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Graphene-Based Composite Materials. Nature 2006, 442, 282–286. [Google Scholar] [CrossRef]
- Huang, X.; Yin, Z.; Wu, S.; Qi, X.; He, Q.; Zhang, Q.; Yan, Q.; Boey, F.; Zhang, H. Graphene-based materials: Synthesis, characterization, properties, and applications. Small 2011, 7, 1876–1902. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chen, F.; Tang, C.; Bai, H.; Zhang, Q.; Deng, H.; Fu, Q. The preparation of high performance and conductive poly(vinyl alcohol)/graphene nanocomposite via reducing graphite oxide with sodium hydrosulfite. Compos. Sci. Technol. 2011, 71, 1266–1270. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Q.; Chen, D. Enhanced Mechanical Properties of Graphene-Based Poly(vinyl alcohol) Composites. Macromolecules 2010, 43, 2357–2363. [Google Scholar] [CrossRef]
- Li, C.; Vongsvivut, J.; She, X.; Li, Y.; She, F.; Kong, L. New insight into non-isothermal crystallization of PVA–graphene composites. Phys. Chem. Chem. Phys. 2014, 16, 22145–22158. [Google Scholar] [CrossRef]
- Yang, X.; Li, L.; Shang, S.; Tao, X. Synthesis and characterization of layer-aligned poly(vinyl alcohol)/graphene nanocomposites. Polymer 2010, 51, 3431–3435. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; Xu, C.; Zhanga, M.; Shang, X. Preparation of graphene/poly(vinyl alcohol) nanocomposites with enhanced mechanical properties and water resistance. Polym. Int. 2011, 60, 816–822. [Google Scholar] [CrossRef]
- Salavagione, H.J.; Martinez, G.; Gomez, M.A. Synthesis of poly(vinyl alcohol)/reduced graphite oxide nanocomposites with improved thermal and electrical properties. J. Mater. Chem. 2009, 19, 5027–5032. [Google Scholar] [CrossRef]
- Bao, C.; Guo, Y.; Song, L.; Hu, Y. Poly(vinyl alcohol) nanocomposites based on graphene and graphite oxide: A comparative investigation of property and mechanism. J. Mater. Chem. 2011, 21, 13942–13950. [Google Scholar] [CrossRef]
- Ye, Y.S.; Cheng, M.Y.; Xie, X.L.; Rick, J.; Huang, Y.J.; Chang, F.C.; Hwang, B.J. Alkali doped polyvinyl alcohol/graphene electrolyte for direct methanol alkaline fuel cells. J. Power Sources 2013, 239, 424–432. [Google Scholar] [CrossRef]
- Kashyap, S.; Pratihar, S.K.; Behera, S.K. Strong and ductile graphene oxide reinforced PVA nanocomposites. J. Alloy. Compd. 2016, 684, 254–260. [Google Scholar] [CrossRef]
- Wang, N.; Chang, P.R.; Zheng, P.; Ma, X. Graphene–poly(vinyl alcohol) composites: Fabrication, adsorption and electrochemical properties. Appl. Surf. Sci. 2014, 314, 815–882. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, J.; Lv, W.; Song, Y.; Zheng, Q. Multifunctional graphene/poly(vinylalcohol) aerogels: In situ hydrothermal preparation and applications in broad-spectrum adsorption for dyes and oils. Carbon 2017, 123, 354–363. [Google Scholar] [CrossRef]
- Li, J.; Shao, L.; Yuan, L.; Wang, Y. A novel strategy for making poly(vinyl alcohol)/reduced graphite oxide nanocomposites by solvothermal reduction. Mater. Des. 2014, 54, 520–525. [Google Scholar] [CrossRef]
- Zhang, J.; He, Y.; Zhu, P.; Lin, S.; Ju, S.; Jiang, D. In situ reduction of graphene oxide in the poly(vinyl alcohol) matrix via microwave irradiation. Polym. Compos. 2017. [Google Scholar] [CrossRef]
- Xu, C.; Shi, X.; Ji, A.; Shi, L.; Zhou, C.; Cui, Y. Fabrication and characteristics of reduced graphene oxide produced with different green reductants. PLoS ONE 2015, 10, e0144842. [Google Scholar] [CrossRef]
- He, D.; Shen, L.; Zhang, X.; Wang, Y.; Bao, N.; Kung, H.H. An efficient and ecofriendly solution-chemical route for preparation of ultrastable reduced graphene oxide suspensions. AIChE J. 2014, 60, 2757–2764. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Cobos, M.; Gonzalez, B.; Fernandez, M.J.; Fernandez, M.D. Chitosan-graphene oxide nanocomposites: Effect of graphene oxide nanosheets and glycerol plasticizer on thermal and mechanical properties. J. Appl. Polym. Sci. 2017, 134, 45092–45106. [Google Scholar] [CrossRef]
- Cobos, M.; González, B.; Fernández, M.J.; Fernández, M.D. Study on the effect of graphene and glycerol plasticizer on the properties of chitosan-graphene nanocomposites via in situ green chemical reduction of graphene oxide. Int. J. Biol. Macromol. 2018, 114, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, K.; Veerapandian, M.; Yun, K.; Kim, S.J. The chemical and structural analysis of graphene oxide with different degrees of oxidation. Carbon 2013, 53, 38–49. [Google Scholar] [CrossRef]
- Tunistra, F.; Koenig, J.L. Raman spectrum of graphite. J. Chem. Phys. 1970, 53, 1126–1130. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef]
- Stankovich, S.; Piner, R.D.; Chen, X.; Wu, N.; Nguyen, S.T.; Ruoff, R.S. Stable aqueous dispersions of graphitic nanoplatelets via the reduction of exfoliated graphite oxide in the presence of poly(sodium 4-styrenesulfonate). J. Mater. Chem. 2006, 16, 155–158. [Google Scholar] [CrossRef]
- Zhan, D.; Ni, Z.; Chen, W.; Sun, L.; Luo, Z.; Lai, L.; Yu, T.; Wee, A.T.S.; Shen, Z. Electronic structure of graphite oxide and thermally reduced graphite oxide. Carbon 2011, 49, 1362–1366. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, L.; Li, F. Graphene Oxide: Physics and Applications; Springer: London, UK, 2015; pp. 20–22. [Google Scholar]
- Kaniyoor, A.; Ramaprabhu, S. A Raman spectroscopic investigation of graphite oxide derived graphene. AIP Adv. 2012, 2, 032183–13. [Google Scholar] [CrossRef]
- Ganguly, A.; Sharma, S.; Papakonstantinou, P.; Hamilton, J. Probing the thermal deoxygenation of graphene oxide using high-resolution in situ X-ray-based spectroscopies. J. Phys. Chem. C 2011, 115, 17009–17019. [Google Scholar]
- Assender, H.E.; Windle, A.H. Crystallinity in poly(vinyl alcohol). 1. An X-ray diffraction study of atactic PVOH. Polymer 1998, 39, 4295–4302. [Google Scholar] [CrossRef]
- Adame, D.; Beall, G.W. Direct measurement of the constrained polymer region in polyamide/clay nanocomposites and the implications for gas diffusion. Appl. Clay Sci. 2009, 42, 545–552. [Google Scholar] [CrossRef]
- Huang, H.D.; Xu, J.Z.; Fan, Y.; Xu, L.; Li, Z.M. Poly(L-lactic acid) Crystallization in a Confined Space Containing Graphene Oxide Nanosheets. J. Phys. Chem. B 2013, 117, 10641–10651. [Google Scholar] [CrossRef]
- Jang, J.; Lee, D.K. Plasticizer effect on the melting and crystallization behavior of polyvinyl alcohol. Polymer 2003, 44, 8139–8146. [Google Scholar] [CrossRef]
- Nishio, Y.; Haratani, T.; Takahashi, T.; Manley, R.S.J. Cellulose/poly(vinyl alcohol) blends: An estimation of thermodynamic polymer-polymer interaction by melting-point-depression analysis. Macromolecules 1989, 22, 2547–2549. [Google Scholar] [CrossRef]
- Liao, K.H.; Aoyama, S.; Abdala, A.A.; Macosko, C. Does graphene change Tg of nanocomposites? Macromolecules 2014, 47, 8311–8319. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Sumi, K. Thermal decomposition products of poly(vinyl alcohol). J. Polym. Sci. Part A-1 Polym. Chem. 1969, 17, 3151–3158. [Google Scholar] [CrossRef]
- Ballistreri, A.; Foti, S.; Montaudo, G.; Scamporrino, E. Evolution of aromatic compounds in the thermal decomposition of vinyl polymers. J. Polym. Sci. Polym. Chem. Ed. 1980, 18, 1147–1153. [Google Scholar] [CrossRef]
- Cao, Y.; Feng, J.; Wu, P. Preparation of organically dispersible graphene nanosheet powders through a lyophilization method and their poly(lactic acid) composites. Carbon 2010, 48, 3834–3839. [Google Scholar] [CrossRef]
- Kelen, T. Polymer Degradation; Van Nostrand Reinhold: New York, NY, USA, 1983. [Google Scholar]
- Budrugeac, P. Kinetics of the complex process of thermo-oxidative degradation of poly(vinyl alcohol). J. Therm. Anal. Calorim. 2008, 92, 291–296. [Google Scholar] [CrossRef]
- Xianda, Y.; Anlai, W.; Suqin, C. Water-vapor permeability of polyvinyl alcohol films. Desalination 1987, 62, 293–297. [Google Scholar] [CrossRef]
- Klopffer, M.H.; Flaconnèche, B. Transport Properties of Gases in Polymers: Bibliographic Review. Oil Gas Sci. Technol. 2001, 56, 223–244. [Google Scholar] [CrossRef]
- Wan, L.S.C.; Lim, L.Y. Drug release from heat treated polyvinyl alcohol films. Drug Dev. Ind. Pharm. 1992, 18, 1895–1906. [Google Scholar] [CrossRef]
- Kwon, J.S.; Kim, D.Y.; Seo, H.W.; Jeong, S.H.; Kim, J.H.; Kim, M.S. Preparation of erythromycin-loaded poly(vinylalcohol) film and investigation of its feasibility as a transdermal delivery carrier. Tissue Eng. Regen. Med. 2014, 11, 211–216. [Google Scholar] [CrossRef]
- Malipeddi, V.R.; Awasthi, R.; Ghisleni, D.M.; Souza-Braga, M.; Kikuchi, I.S.; Andreoli-Pinto, T.J.; Dua, K. Preparation and characterization of metoprolol tartrate containing matrix type transdermal drug delivery system. Drug Deliv. Transl. Res. 2017, 7, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Minghetti, P.; Cilurzo, F.; Liberti, V.; Montanari, L. Dermal therapeutic systems permeable to water vapour. Int. J. Pharmaceut. 1997, 158, 165–172. [Google Scholar] [CrossRef]
- Novoselov, S.; Fal’ko, V.I.; Colombo, L.; Gellert, P.R.; Schwab, M.G.; Kim, K. A roadmap for graphene. Nature 2012, 490, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Kim, Y.K.; Shin, D.; Ryoo, S.R.; Hong, B.H.; Min, D.H. Biomedical applications of graphene and graphene oxide. Acc. Chem. Res. 2013, 46, 2211–2224. [Google Scholar] [CrossRef]
- Feng, L.; Liu, Z. Graphene in biomedicine: Opportunities and challenges. Nanomedicine 2011, 6, 317–324. [Google Scholar] [CrossRef]
- McCallion, C.; Burthem, J.; Rees-Unwin, K.; Golovanov, A.; Pluen, A. Graphene in therapeutics delivery: Problems, solutions and future opportunities. Eur. J. Pharm. Biopharm. 2016, 104, 235–250. [Google Scholar] [CrossRef]
- Liu, J.; Cui, L.; Losic, D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013, 9, 9243–9257. [Google Scholar] [CrossRef]
- Liao, K.H.; Lin, Y.S.; Macosko, C.W.; Haynes, C.L. Cytotoxicity of graphene oxide and graphene in human erythrocytes and skin fibroblasts. ACS Appl. Mater. Interfaces 2011, 3, 2607–2615. [Google Scholar] [CrossRef]
- Sanchez, C.; Jachak, A.; Hurt, R.H.; Kane, A.B. Biological interactions of graphene-family nanomaterials: An interdisciplinary review. Chem. Res. Toxicol. 2012, 25, 15–34. [Google Scholar] [CrossRef]
- Li, Y.Q.; Yu, T.; Yang, T.Y.; Zheng, L.X.; Liao, K. Bio-inspired nacre-like composite films based on graphene with superior mechanical, electrical, and biocompatible properties. Adv. Mater. 2012, 24, 3426–3431. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Glycerin; GRAS status as a direct human food ingredient. Fed. Regist. 1979, 48, 5759–5760. [Google Scholar]
- Güngör, S.; Erdal, M.; Özsoy, Y. Plasticizers in transdermal drug delivery systems. In Recent Advances in Plasticizers; Luqman, M., Ed.; InTech: Rijeka, Croatia, 2012; pp. 91–112. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tg (°C) | Tm (°C) | ΔHm (J/g) | Tc (°C) | ΔHc(J/g) | Xc (%) |
|---|---|---|---|---|---|---|
| PVA | 76.1 | 221.7 | 76.2 | 197.7 | 66.5 | 53.7 |
| PVA/GS-0.5 | 80.3 | 215.8 | 67.9 | 199.2 | 51.6 | 48.1 |
| PVA/GS-1 | 84.4 | 205.2 | 53.8 | 185.9 | 39.8 | 38.3 |
| PVA/GS-1.5 | 88.0 | 188.2 | 34.1 | 163.0 | 28.3 | 24.4 |
| PVA/GS-2 | 91.9 | 169.2 | 21.0 | 133.0 | 21.6 | 15.1 |
| PVA/GS-1 ex situ | 75.0 | 221.9 | 75.7 | 200.1 | 60.1 | 53.9 |
| PVA/GLY | 42.5 | 216.0 | 49.8 | 188.7 | 47.7 | 35.1 |
| PVA/GS-0.5/GLY | 72.0 | 218.3 | 58.8 | 199.6 | 50.1 | 41.6 |
| PVA/GS-1/GLY | 77.0 | 205.5 | 48.8 | 189.5 | 39.2 | 34.7 |
| PVA/GS-1.5/GLY | 73.6 | 196.6 | 33.8 | 176.8 | 28.2 | 24.2 |
| PVA/GS-2/GLY | 83.6 | 187.9 | 31.7 | 167.6 | 21.1 | 22.8 |
| Sample | T5% (°C) | T50% (°C) | Residue (%) | ||
|---|---|---|---|---|---|
| N2 | O2 | N2 | O2 | N2 | |
| PVA | 271 | 281 | 338 | 367 | 2.9 |
| PVA/GS-0.5 | 293 | 296 | 358 | 384 | 4.1 |
| PVA/GS-1 | 300 | 291 | 367 | 381 | 6.0 |
| PVA/GS-1.5 | 295 | 368 | 6.6 | ||
| PVA/GS-2 | 287 | 284 | 372 | 386 | 7.1 |
| PVA/GS-1 ex situ | 295 | 303 | 361 | 376 | 4.1 |
| PVA/GLY | 266 | 272 | 327 | 349 | 2.5 |
| PVA/GS-0.5/GLY | 286 | 317 | 342 | 365 | 3.7 |
| PVA/GS-1/GLY | 298 | 291 | 352 | 367 | 4.3 |
| PVA/GS-1.5/GLY | 302 | 358 | 7.3 | ||
| PVA/GS-2/GLY | 310 | 311 | 355 | 377 | 6.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cobos, M.; Fernández, M.J.; Fernández, M.D. Graphene Based Poly(Vinyl Alcohol) Nanocomposites Prepared by In Situ Green Reduction of Graphene Oxide by Ascorbic Acid: Influence of Graphene Content and Glycerol Plasticizer on Properties. Nanomaterials 2018, 8, 1013. https://doi.org/10.3390/nano8121013
Cobos M, Fernández MJ, Fernández MD. Graphene Based Poly(Vinyl Alcohol) Nanocomposites Prepared by In Situ Green Reduction of Graphene Oxide by Ascorbic Acid: Influence of Graphene Content and Glycerol Plasticizer on Properties. Nanomaterials. 2018; 8(12):1013. https://doi.org/10.3390/nano8121013
Chicago/Turabian StyleCobos, Mónica, M. Jesús Fernández, and M. Dolores Fernández. 2018. "Graphene Based Poly(Vinyl Alcohol) Nanocomposites Prepared by In Situ Green Reduction of Graphene Oxide by Ascorbic Acid: Influence of Graphene Content and Glycerol Plasticizer on Properties" Nanomaterials 8, no. 12: 1013. https://doi.org/10.3390/nano8121013
APA StyleCobos, M., Fernández, M. J., & Fernández, M. D. (2018). Graphene Based Poly(Vinyl Alcohol) Nanocomposites Prepared by In Situ Green Reduction of Graphene Oxide by Ascorbic Acid: Influence of Graphene Content and Glycerol Plasticizer on Properties. Nanomaterials, 8(12), 1013. https://doi.org/10.3390/nano8121013