First-Principles Study on the Stabilities, Electronic and Optical Properties of GexSn1-xSe Alloys


Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
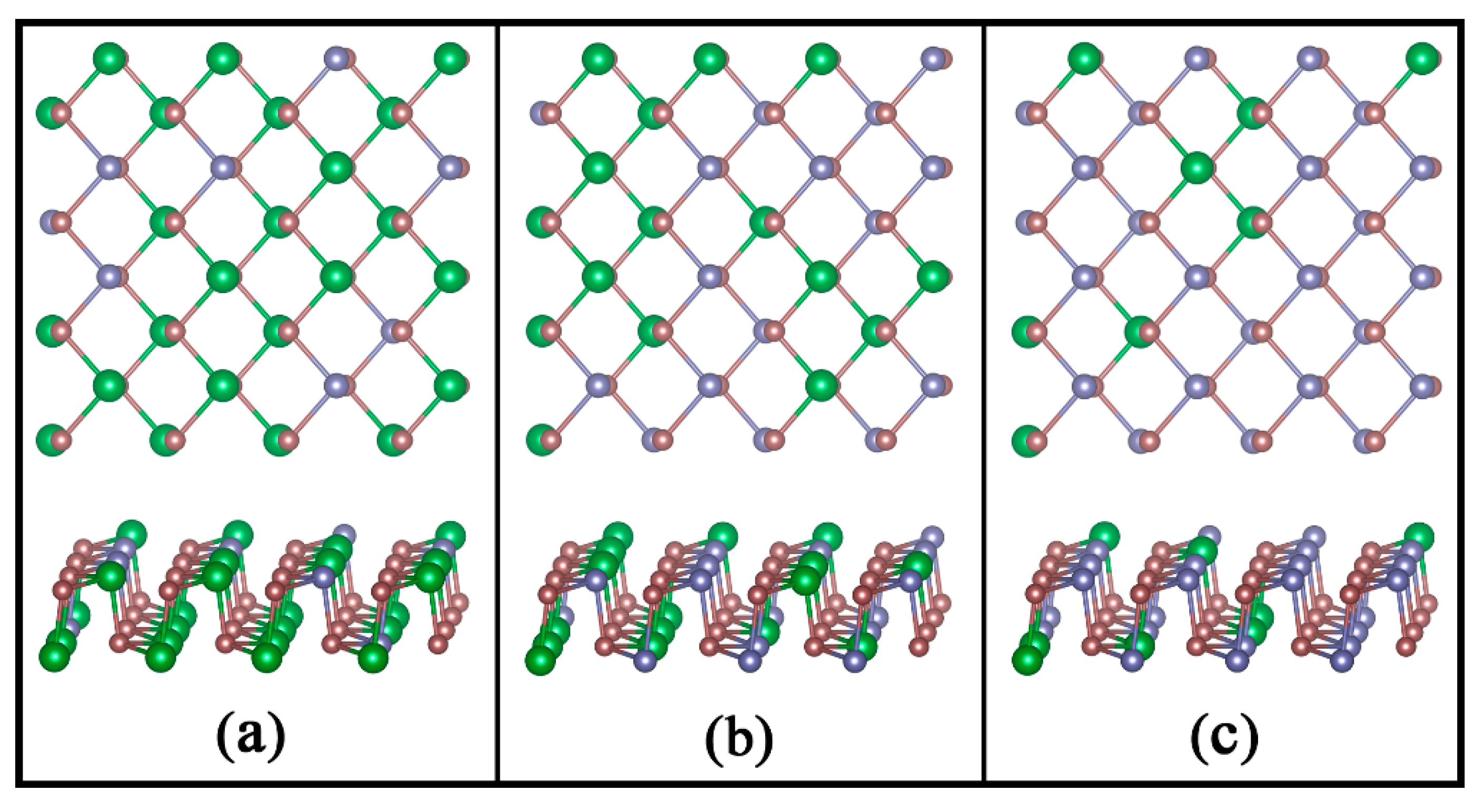
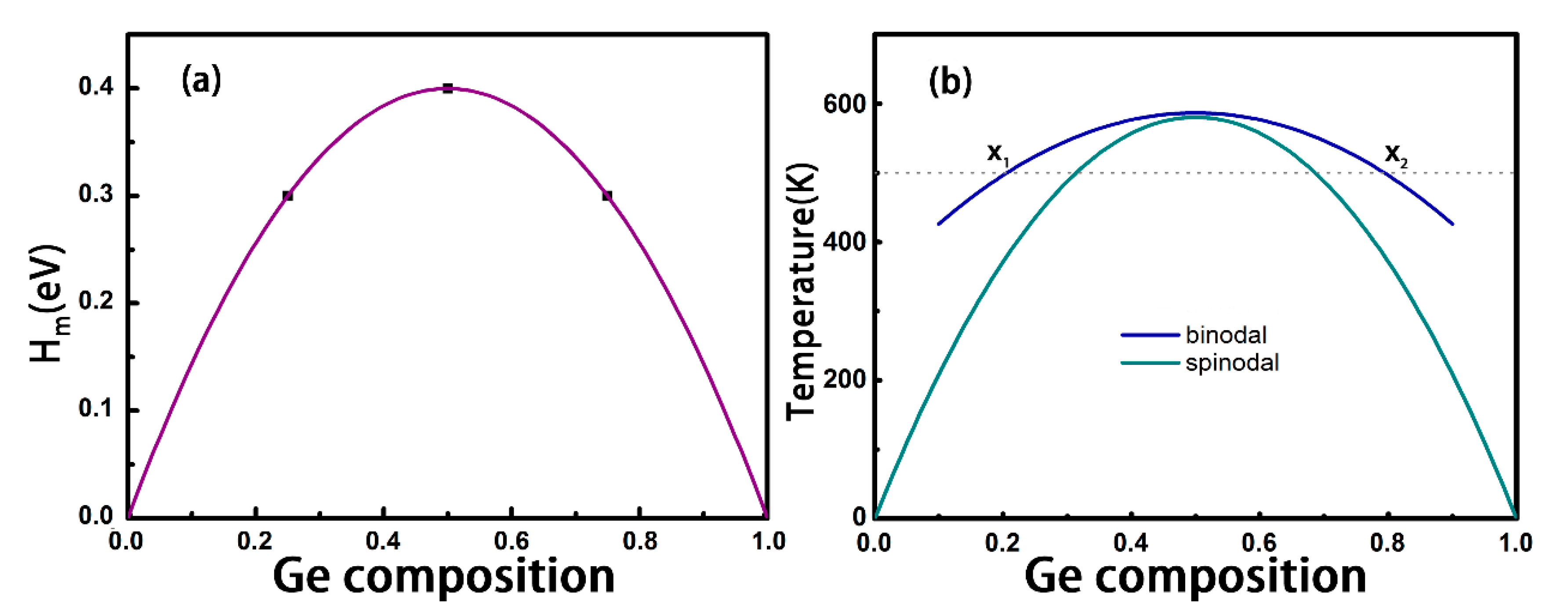
3.1. Structures and Stabilities of GexSn1-xSe Alloys
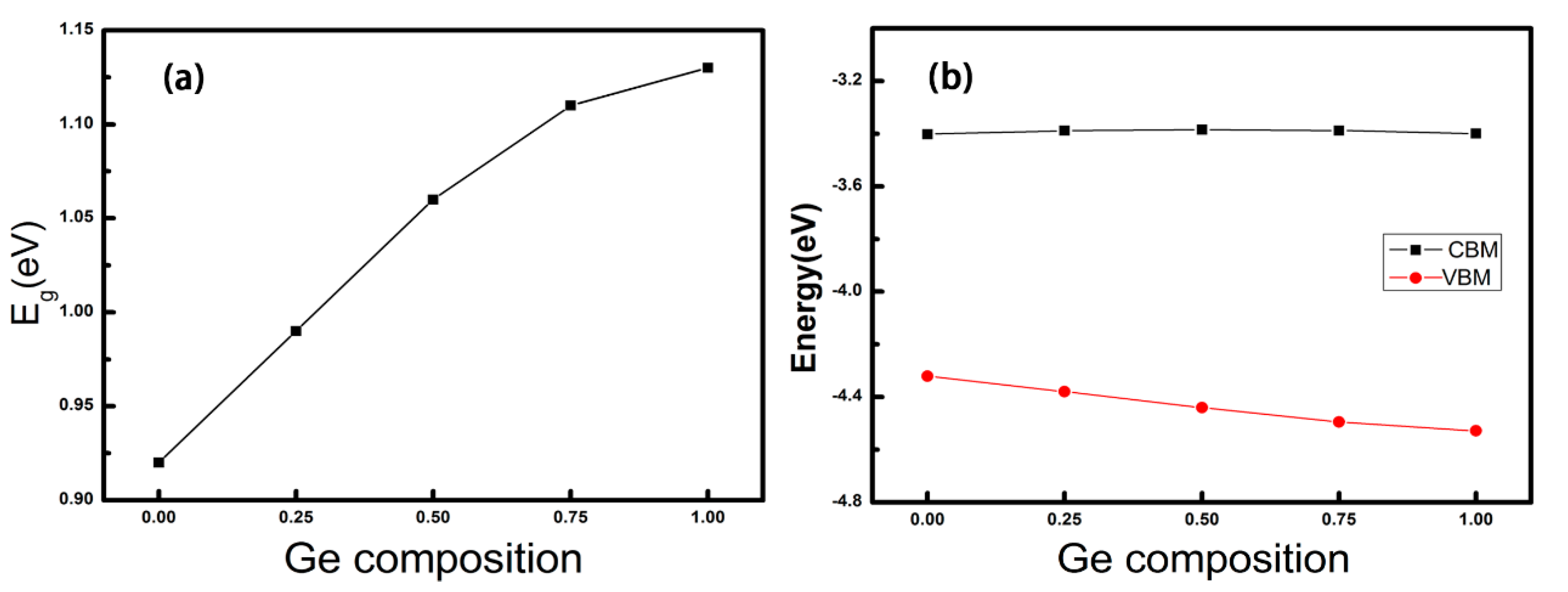
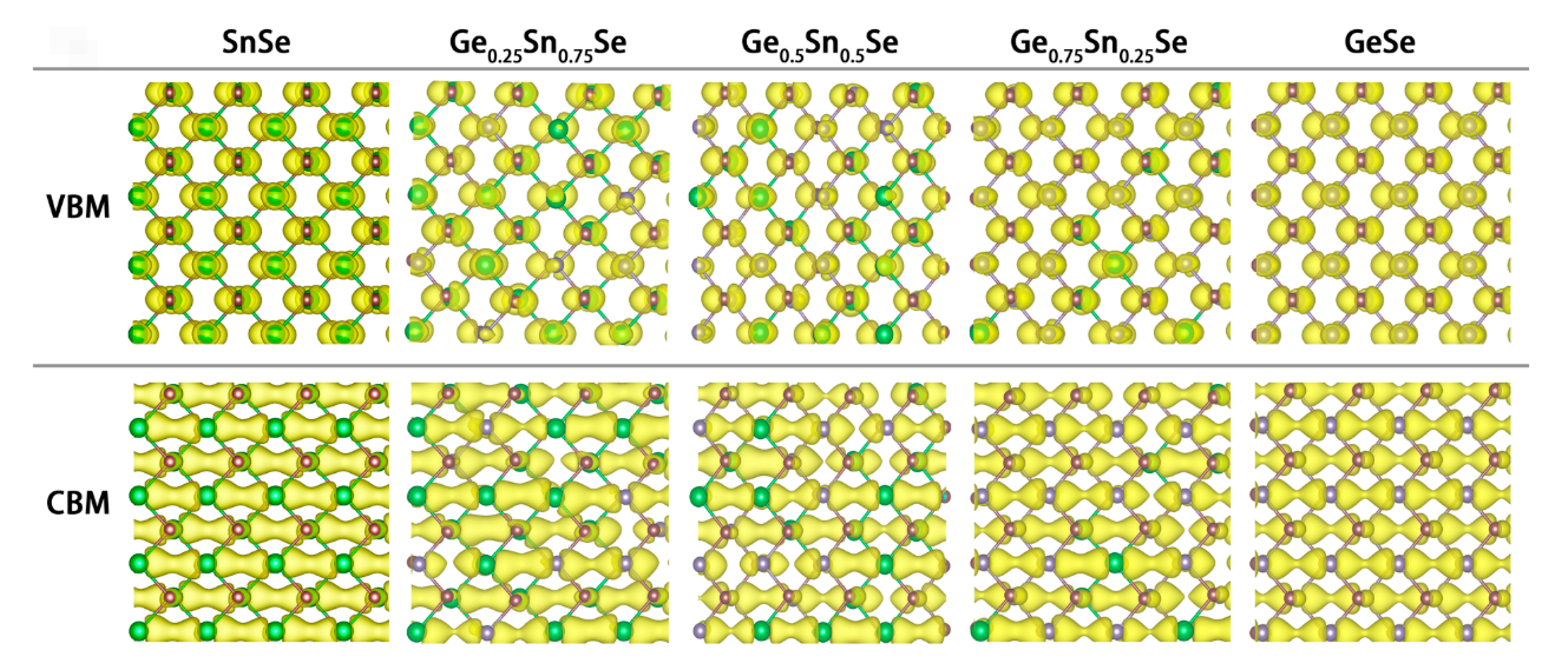
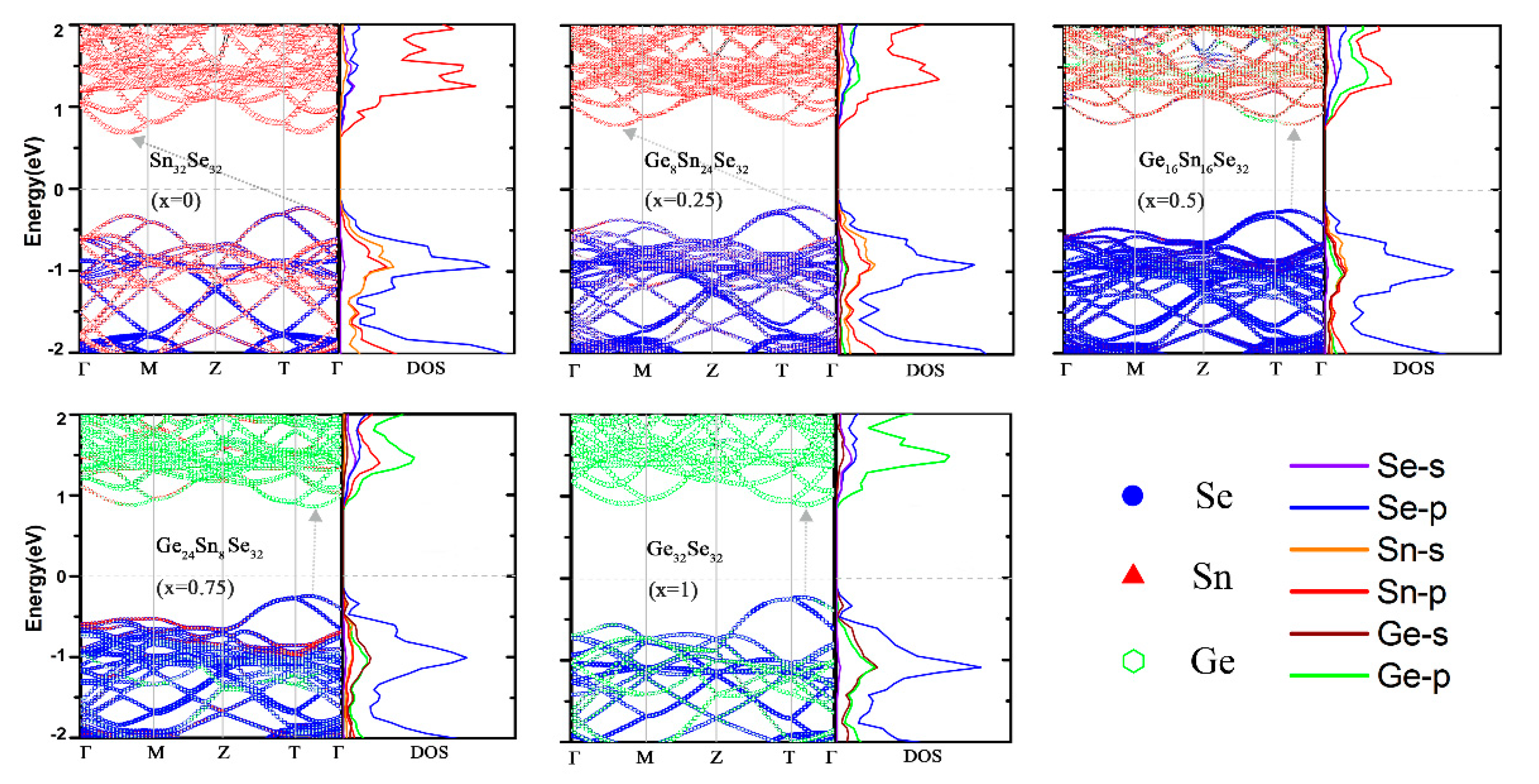
3.2. Electronic Properties of GexSn1-xSe Alloys
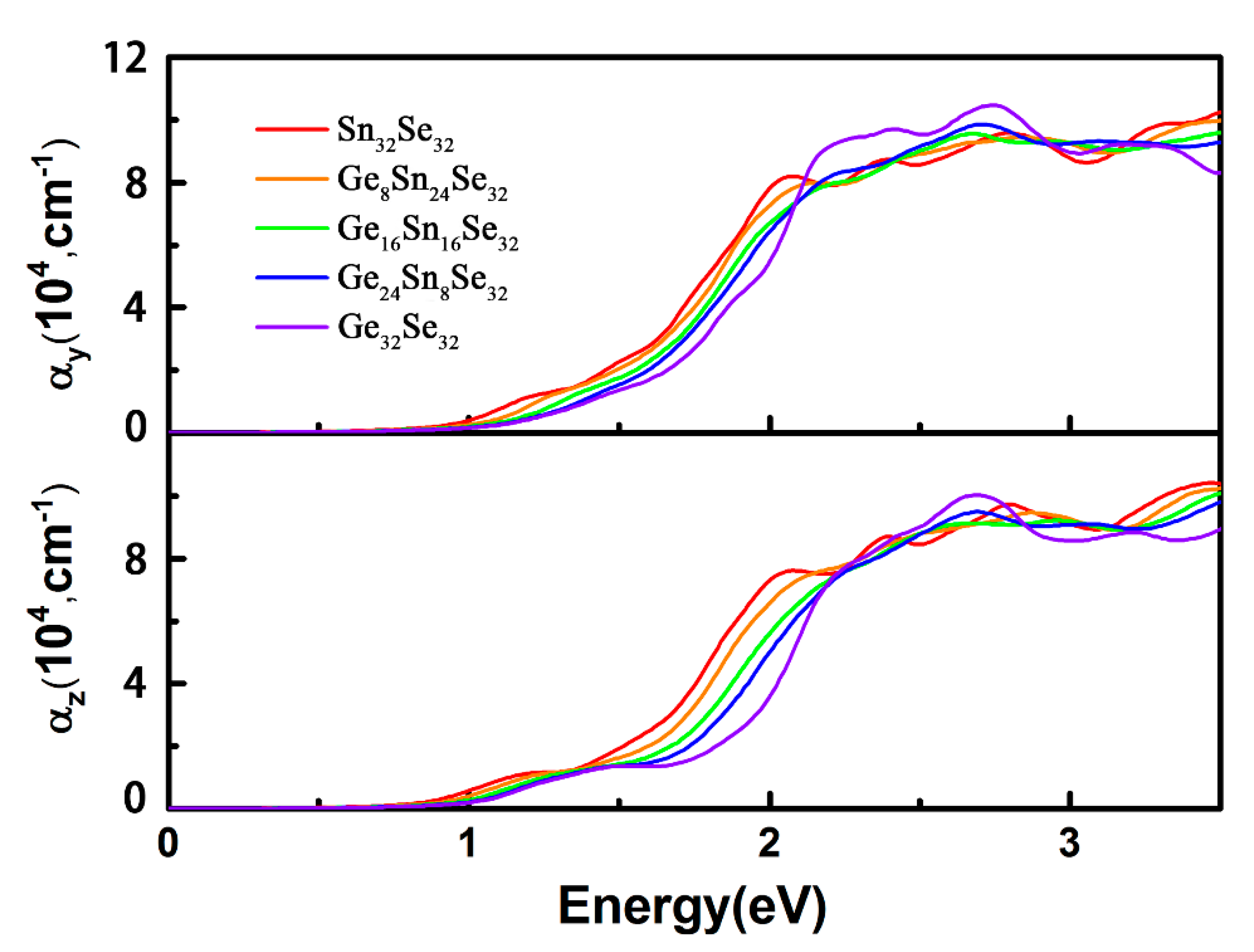
3.3. Optical Properties of GexSn1-xSe Alloys
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, K.I.; Sikes, K.J.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H.L. Ultrahigh Electron Mobility in Suspended Graphene. Solid State Commun. 2008, 146, 351–355. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.; Jiang, D.; Katsnelson, M.; Grigorieva, I.; Dubonos, S.; Firsov, A.A. Two-Dimensional Gas of Massless Dirac Fermions in Graphene. Nature 2005, 438, 197. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Jiang, D.; Schedin, F.; Booth, T.J.; Khotkevich, V.V.; Morozov, S.V.; Geim, A.K. Two-Dimensional Atomic Crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451–10453. [Google Scholar] [CrossRef] [PubMed]
- Fiori, G.; Bonaccorso, F.; Iannaccone, G.; Palacios, T.; Neumaier, D.; Seabaugh, A.; Banerjee, S.K.; Colombo, L. Electronics Based on Two-Dimensional Materials. Nat. Nanotechnol. 2014, 9, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.H.; Kalantarzadeh, K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and Optoelectronics of Two-Dimensional Transition Metal Dichalcogenides. Nat. Nanotechnol. 2012, 7, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Kong, X.; Hu, Z.X.; Yang, F.; Ji, W. High-Mobility Transport Anisotropy and Linear Dichroism in Few-Layer Black Phosphorus. Nat. Commun. 2014, 5, 4475. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hamsen, C.; Jia, X.; Kim, K.K.; Reina, A.; Hofmann, M.; Hsu, A.L.; Zhang, K.; Li, H.; Juang, Z.Y. Synthesis of Few-Layer Hexagonal Boron Nitride Thin Film by Chemical Vapor Deposition. Nano Lett. 2010, 10, 4134. [Google Scholar] [CrossRef] [PubMed]
- Naguib, M.; Mochalin, V.N.; Barsoum, M.W.; Gogotsi, Y. 25th Anniversary Article: Mxenes: A New Family of Two-Dimensional Materials. Adv. Mater. 2013, 26, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yan, Z.; Li, Y.; Chen, Z.; Zeng, H. Atomically Thin Arsenene and Antimonene: Semimetal–Semiconductor and Indirect–Direct Band-Gap Transitions. Angew. Chem. 2015, 127, 3155–3158. [Google Scholar] [CrossRef]
- Pumera, M.; Sofer, Z. 2d Monoelemental Arsenene, Antimonene, and Bismuthene: Beyond Black Phosphorus. Adv. Mater. 2017, 29, 1605299. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.; De Padova, P.; Quaresima, C.; Avila, J.; Frantzeskakis, E.; Asensio, M.C.; Resta, A.; Ealet, B.; Le Lay, G. Silicene: Compelling Experimental Evidence for Graphenelike Two-Dimensional Silicon. Phys. Rev. Lett. 2012, 108, 155501. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.-C.; Zhang, S.-F.; Ji, W.-X.; Zhang, C.-W.; Li, P.; Wang, P.-J. Tunable Electronic and Topological Properties of Germanene by Functional Group Modification. Nanomaterials 2018, 8, 145. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Cai, X.; Yi, S.; Chen, J.; Dai, Y.; Niu, C.; Guo, Z.; Xie, M.; Liu, F.; Cho, J.-H.; et al. Multivalency-Driven Formation of Te-Based Monolayer Materials: A Combined First-Principles and Experimental Study. Phys. Rev. Lett. 2017, 119, 106101. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-Layer MoS2 Transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Pan, H. Progress on the Theoretical Study of Two-Dimensional MoS2 Monolayer and Nanoribbon; Springer International Publishing: Cham, Switzerland, 2014; pp. 992–995. [Google Scholar]
- Wei, X.L.; Zhang, H.; Guo, G.C.; Li, X.B.; Lau, W.M.; Liu, L.M. Modulating the Atomic and Electronic Structures through Alloying and Heterostructure of Single-Layer MoS2. J. Mater. Chem. A 2014, 2, 2101–2109. [Google Scholar] [CrossRef]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Majumdar, K.; Liu, H.; Du, Y.; Wu, H.; Hatzistergos, M.; Hung, P.Y.; Tieckelmann, R.; Tsai, W.; Hobbs, C. Chloride Molecular Doping Technique on 2d Materials: WS2 and MoS2. Nano Lett. 2014, 14, 6275. [Google Scholar] [CrossRef] [PubMed]
- Low, T.; Rodin, A.S.; Carvalho, A.; Jiang, Y.; Wang, H.; Xia, F.; Castro Neto, A.H. Tunable Optical Properties of Multilayer Black Phosphorus Thin Films. Phys. Rev. B 2014, 90, 075434. [Google Scholar] [CrossRef]
- Xia, F.; Wang, H.; Jia, Y. Rediscovering Black Phosphorus as an Anisotropic Layered Material for Optoelectronics and Electronics. Nat. Commun. 2014, 5, 4458. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Fujisawa, K.; Lin, Z.; Lei, Y.; Mondschein, J.S.; Terrones, M.; Schaak, R.E. Low-Temperature Solution Synthesis of Transition Metal Dichalcogenide Alloys with Tunable Optical Properties. J. Am. Chem. Soc. 2017, 139, 11096–11105. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, T.; Zhang, Z.; Yang, L. Enhanced Doping Effect on Tuning Structural Phases of Monolayer Antimony. Appl. Phys. Lett. 2018, 112, 213104. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, Y.; Ito, Y.; Zhang, Y.; Du, J.; Fujita, T.; Hirata, A.; Tang, Z.; Chen, M. Synthesizing 1T-1H Two-Phase Mo1-xWxS2 Monolayers by Chemical Vapor Deposition. ACS Nano 2018, 12, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Darwish, A.A.A.; Rashad, M.; Bekheet, A.E.; El-Nahass, M.M. Linear and Nonlinear Optical Properties of GeSe2-xSnx (0 ≤ x ≤ 0.8) Thin Films for Optoelectronic Applications. J. Alloys Compd. 2017, 709, 640–645. [Google Scholar] [CrossRef]
- Li, X.; Dai, Y.; Ma, Y.; Han, S.; Huang, B. Graphene/G-C3N4 Bilayer: Considerable Band Gap Opening and Effective Band Structure Engineering. Phys. Chem. Chem. Phys. 2014, 16, 4230–4235. [Google Scholar] [CrossRef] [PubMed]
- Huo, N.; Kang, J.; Wei, Z.; Li, S.-S.; Li, J.; Wei, S.-H. Novel and Enhanced Optoelectronic Performances of Multilayer MoS2–WS2 Heterostructure Transistors. Adv. Funct. Mater. 2014, 24, 7025–7031. [Google Scholar] [CrossRef]
- Sapna, S.; Yuya, T.; Hisanori, S.; Ryo, K. Simple Fabrication of Air-Stable Black Phosphorus Heterostructures with Large-Area Hbn Sheets Grown by Chemical Vapor Deposition Method. 2D Materials 2016, 3, 035010. [Google Scholar]
- Huang, Y.; Chen, X.; Zhou, D.; Liu, H.; Wang, C.; Du, J.; Ning, L.; Wang, S. Stabilities, Electronic and Optical Properties of SnSe2(1–x)S2x Alloys: A First-Principles Study. J. Phys. Chem. C 2016, 120, 5839–5847. [Google Scholar] [CrossRef]
- Wang, Y.; Le Huang, L.H.; Li, B.; Shang, J.; Xia, C.; Fan, C.; Deng, H.-X.; Wei, Z.; Li, J. Composition-Tunable 2d SnSe2(1−x)S2x Alloys Towards Efficient Bandgap Engineering and High Performance (Opto)Electronics. J. Mater. Chem. C 2017, 5, 84–90. [Google Scholar] [CrossRef]
- Tongay, S.; Narang, D.S.; Kang, J.; Fan, W. Two-Dimensional Semiconductor Alloys: Monolayer Mo1−xWxSe2. Appl. Phys. Lett. 2014, 104, 012101. [Google Scholar] [CrossRef]
- Zheng, S.; Sun, L.; Yin, T.; Dubrovkin, A.M.; Liu, F.; Liu, Z.; Shen, Z.X.; Fan, H.J. Monolayers of WxMo1−xS2 Alloy Heterostructure with in-Plane Composition Variations. Appl. Phys. Lett. 2015, 106, 063113. [Google Scholar] [CrossRef]
- Tan, W.; Wei, Z.; Liu, X.; Liu, J.; Fang, X.; Fang, D.; Wang, X.; Wang, D.; Tang, J.; Fan, X. Ordered and Disordered Phases in Mo1−xWxS2 Monolayer. Sci. Rep. 2017, 7, 15124. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Yuan, L.; Liu, X.; Zhou, W.; Song, X.; Zhang, S. First-Principles Study of SO2 Sensors Based on Phosphorene and Its Isoelectronic Counterparts: GeS, GeSe, SnS, SnSe. Chem. Phys. Lett. 2017, 686, 83–87. [Google Scholar] [CrossRef]
- Peng, L.; Wang, C.; Qian, Q.; Bi, C.; Wang, S.; Huang, Y. Complete Separation of Carriers in the GeS/SnS Lateral Heterostructure by Uniaxial Tensile Strain. ACS. Appl. Mater. Interfaces 2017, 9, 40969–40977. [Google Scholar] [CrossRef] [PubMed]
- Shafique, A.; Shin, Y.H. Thermoelectric and Phonon Transport Properties of Two-Dimensional IV–VI Compounds. Sci. Rep. 2017, 7, 506. [Google Scholar] [CrossRef] [PubMed]
- Deb, A.K.; Kumar, V. Bandgap Engineering in Semiconducting One to Few Layers of SnS and SnSe. Phys. Status Solidi B Basic Res. 2017, 254, 1600379. [Google Scholar] [CrossRef]
- Buckley, J.J.; Rabuffetti, F.A.; Hinton, H.L.; Brutchey, R.L. Synthesis and Characterization of Ternary SnxGe1-xSe Nanocrystals. Chem. Mater. 2012, 24, 3514–3516. [Google Scholar] [CrossRef]
- Fu, Y.; Xu, J.; Liu, G.-Q.; Tan, X.; Liu, Z.; Wang, X.; Shao, H.; Jiang, H.; Liang, B.; Jiang, J. Study on Thermoelectric Properties of Polycrystalline SnSe by Ge Doping. J. Electron. Mater. 2017, 46, 3182–3186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys Rev. B Condens. Matter. 1993, 48, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- PE, B. Projector Augmented-Wave Method. Phys. Rev. B Condens. Matter. 1994, 50, 17953–17979. [Google Scholar]
- Perdew, J.P.; Wang, Y. Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy. Phys. Rev. B Condens. Matter. 1992, 45, 13244. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation. Phys. Rev. B Condens. Matter. 1993, 46, 6671–6687. [Google Scholar] [CrossRef]
- Monkhorst, H.J. Special Points for Brillouin-Zone Integrations. Phys. Rev. B Condens. Matter. 1976, 16, 1748–1749. [Google Scholar] [CrossRef]
- Zunger, A.; Wei, S.H.; Ferreira, L.G.; Bernard, J.E. Special Quasirandom Structures. Phys. Rev. Lett. 1990, 65, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Van de Walle, A.; Asta, M.; Ceder, G. The Alloy Theoretic Automated Toolkit: A User Guide. Calphad 2002, 26, 539–553. [Google Scholar] [CrossRef]
- Denton, A.R.; Ashcroft, N.W. Vegard’s Law. Phys. Rev. A 1991, 43, 3161. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.W. Alloy Phase Equilibria; Elsevier Pub. Co.: Amsterdam, The Netherlands, 1966; pp. 255–256. [Google Scholar]
- Gomes, L.C.; Carvalho, A. Phosphorene Analogues: Isoelectronic Two-Dimensional Group-IV Monochalcogenides with Orthorhombic Structure. Phys. Rev. B 2015, 92, 085406. [Google Scholar] [CrossRef]
- Fei, R.; Li, W.; Li, J.; Yang, L. Giant Piezoelectricity of Monolayer Group IV Monochalcogenides: SnSe, SnS, GeSe, and GeS. Appl. Phys. Lett. 2015, 107, 173104. [Google Scholar] [CrossRef]
- Shi, G.; Kioupakis, E. Anisotropic Spin Transport and Strong Visible-Light Absorbance in Few-Layer SnSe and GeSe. Nano Lett. 2015, 15, 6926–6931. [Google Scholar] [CrossRef] [PubMed]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Kim, Y.; Choi, I.-H. Optical and Electrical Properties of GeSe and SnSe Single Crystals. J. Korean Phys. Soc. 2018, 72, 238–242. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Cohen, M.H. Self-Consistent Field Approach to the Many-Electron Problem. Phy. Rev. 1959, 115, 786–790. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| x | 0.25 | 0.50 | 0.75 | 1.00 |
|---|---|---|---|---|
| Es | −3.77 | −7.77 | −11.87 | −16.21 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Q.; Peng, L.; Cui, Y.; Sun, L.; Du, J.; Huang, Y. First-Principles Study on the Stabilities, Electronic and Optical Properties of GexSn1-xSe Alloys. Nanomaterials 2018, 8, 876. https://doi.org/10.3390/nano8110876
Qian Q, Peng L, Cui Y, Sun L, Du J, Huang Y. First-Principles Study on the Stabilities, Electronic and Optical Properties of GexSn1-xSe Alloys. Nanomaterials. 2018; 8(11):876. https://doi.org/10.3390/nano8110876
Chicago/Turabian StyleQian, Qi, Lei Peng, Yu Cui, Liping Sun, Jinyan Du, and Yucheng Huang. 2018. "First-Principles Study on the Stabilities, Electronic and Optical Properties of GexSn1-xSe Alloys" Nanomaterials 8, no. 11: 876. https://doi.org/10.3390/nano8110876
APA StyleQian, Q., Peng, L., Cui, Y., Sun, L., Du, J., & Huang, Y. (2018). First-Principles Study on the Stabilities, Electronic and Optical Properties of GexSn1-xSe Alloys. Nanomaterials, 8(11), 876. https://doi.org/10.3390/nano8110876