Physical Properties and Photovoltaic Application of Semiconducting Pd2Se3 Monolayer



Abstract
1. Introduction
2. Computational Methods
3. Results and Discussion
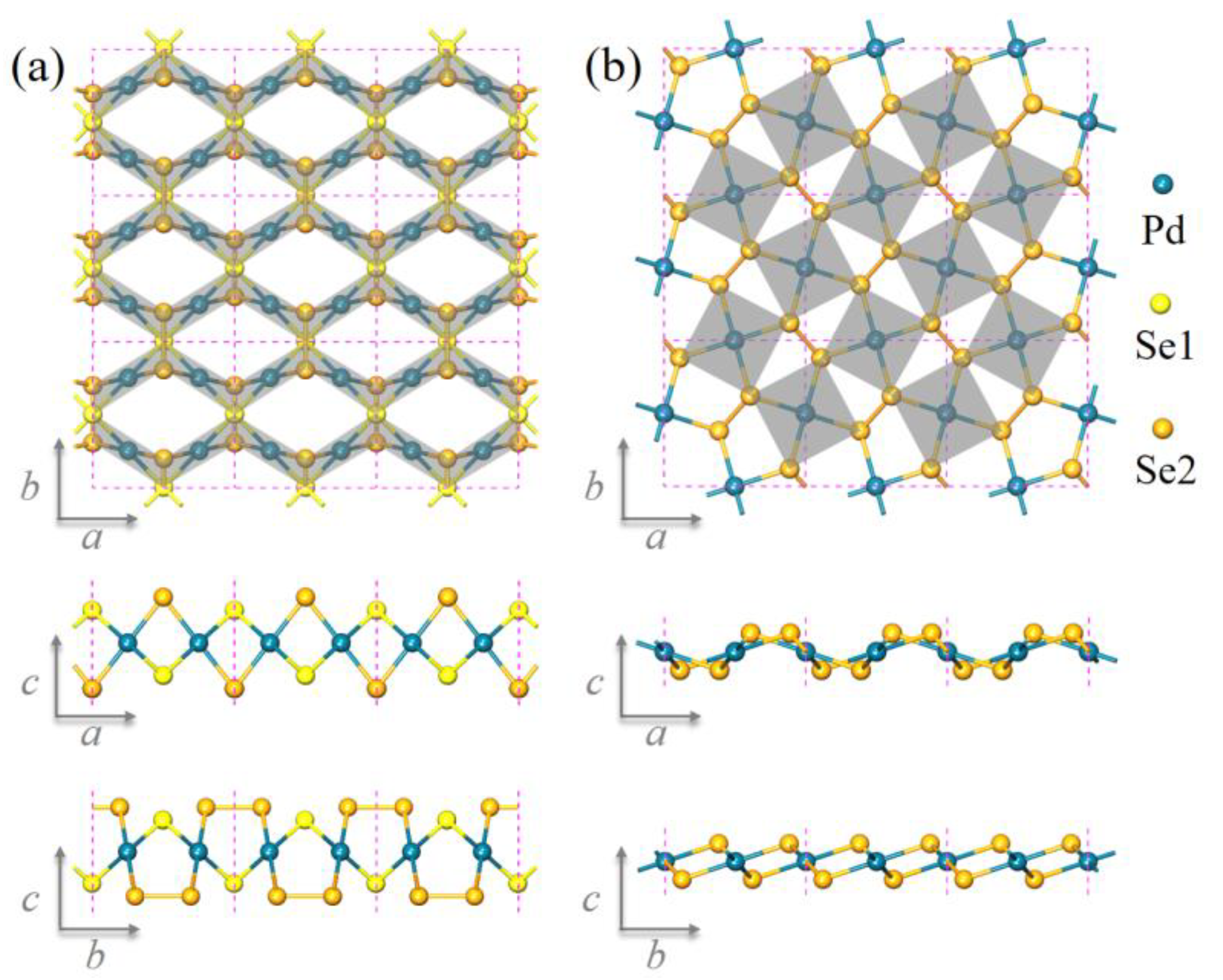
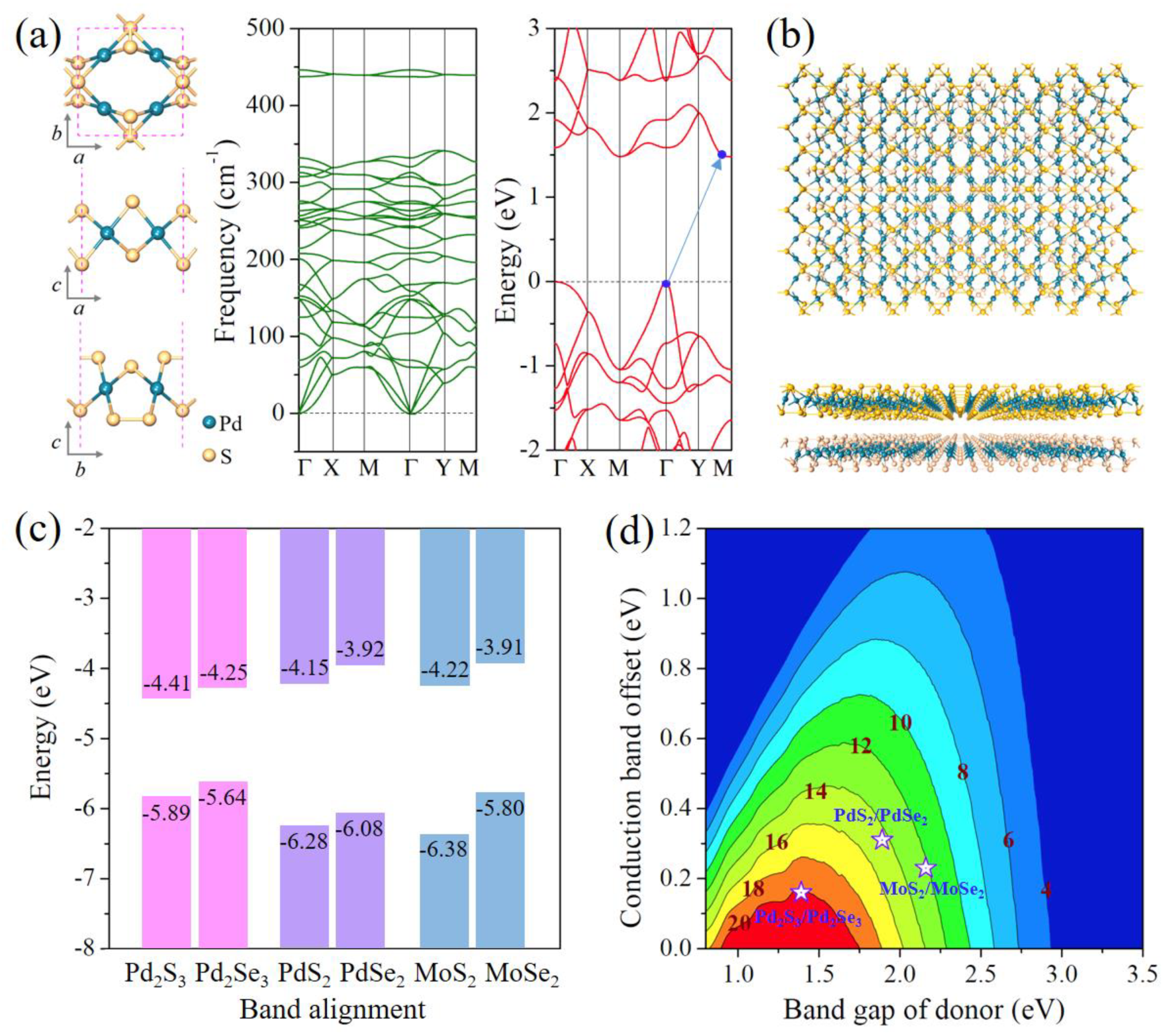
3.1. Geometric Structure of Pd2Se3
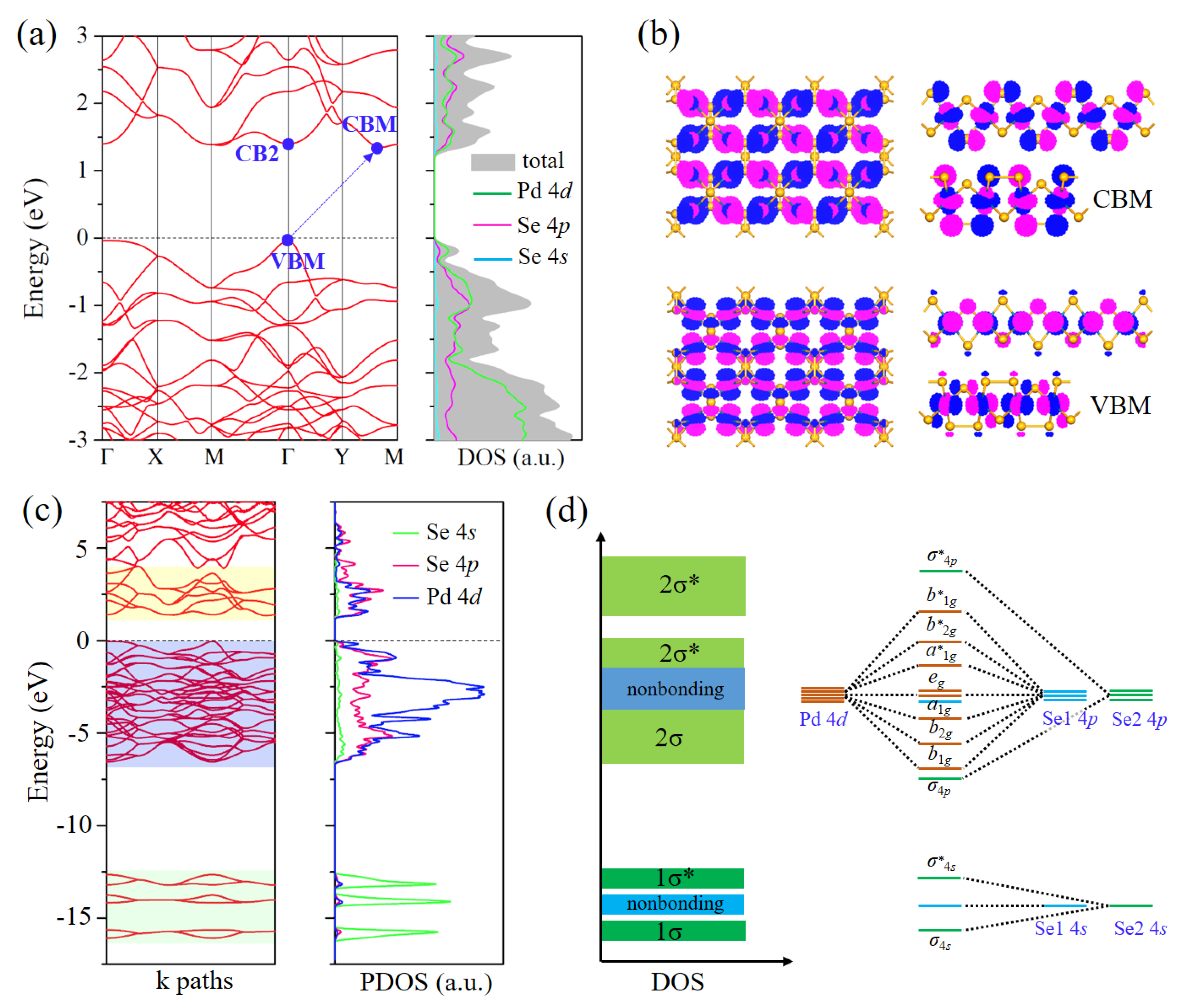
3.2. Electronic Properties of Pd2Se3
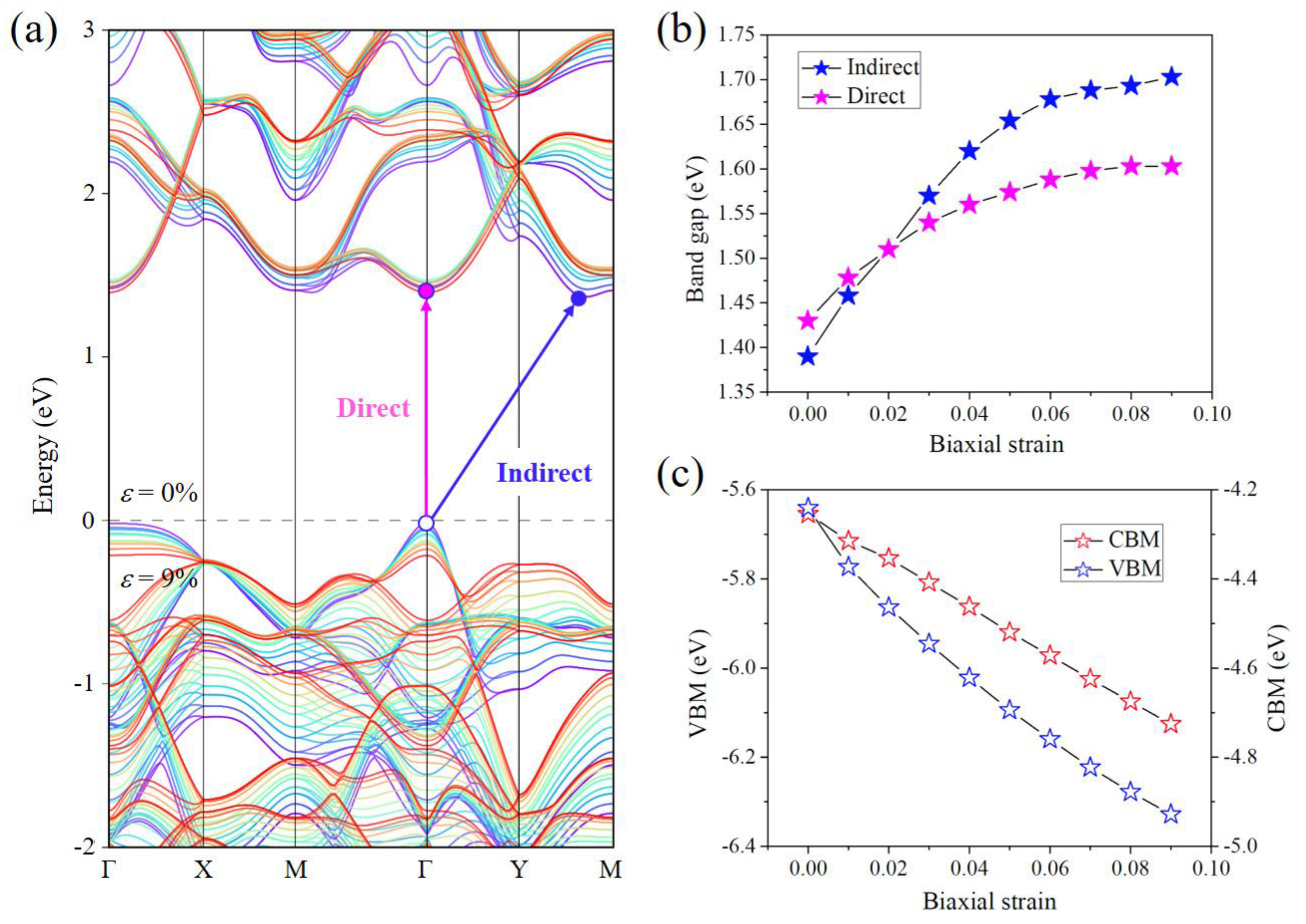
3.3. Strain Engineering of Electronic Band Structure of Pd2Se3
3.4. Transport Properties of Pd2Se3
3.5. Optical Properties of Pd2Se3
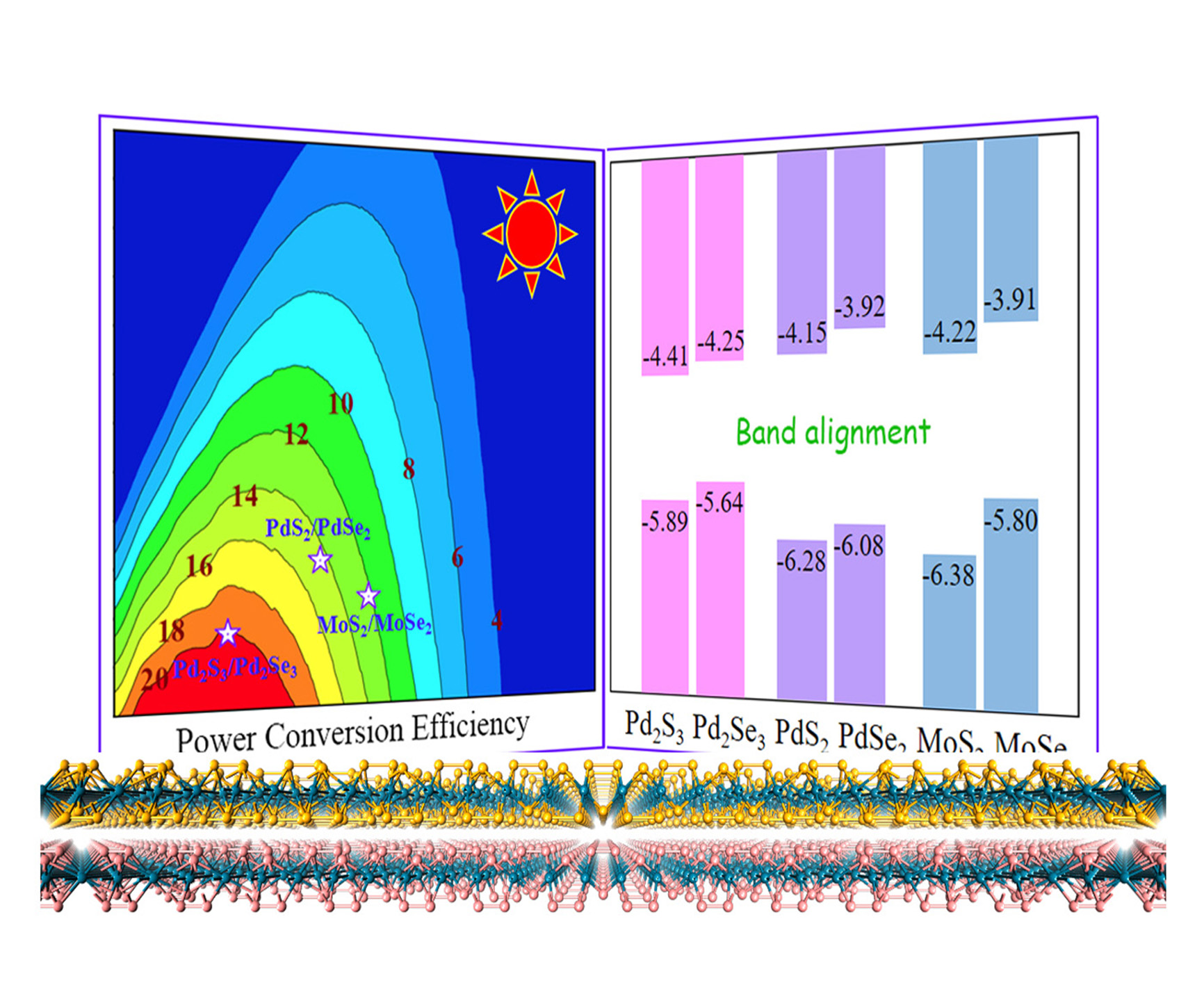
3.6. Extension and Photovoltaic Application of Pd2Se3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, Y.H.; Zhang, X.Q.; Zhang, W.; Chang, M.T.; Lin, C.T.; Chang, K.D.; Yu, Y.C.; Wang Jacob, T.W.; Chang, C.S.; Li, L.J.; et al. Synthesis of Large-Area MoS2 Atomic Layers with Chemical Vapor Deposition. Adv. Mater. 2012, 24, 2320–2325. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Utama, M.I.B.; Lin, J.; Gong, X.; Zhang, J.; Zhao, Y.; Pantelides, S.T.; Wang, J.; Dong, Z.; Liu, Z.; et al. Large-Area Synthesis of Monolayer and Few-Layer MoSe2 Films on SiO2 Substrates. Nano Lett. 2014, 14, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-Q.; Zhang, Y.; Lin, S.; Zheng, C.; Zhong, Y.L.; Xia, X.; Li, Z.; Sophia, P.J.; Fuhrer, M.S.; Cheng, Y.-B.; et al. Synthesis and Transfer of Large-Area Monolayer WS2 Crystals: Moving toward the Recyclable Use of Sapphire Substrates. ACS Nano 2015, 9, 6178–6187. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-K.; Pu, J.; Hsu, C.-L.; Chiu, M.-H.; Juang, Z.-Y.; Chang, Y.-H.; Chang, W.-H.; Iwasa, Y.; Takenobu, T.; Li, L.-J. Large-Area Synthesis of Highly Crystalline WSe2 Monolayers and Device Applications. ACS Nano 2014, 8, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Komsa, H.-P.; Yeh, C.-H.; Björkman, T.; Liang, Z.-Y.; Ho, C.-H.; Huang, Y.-S.; Chiu, P.-W.; Krasheninnikov, A.V.; Suenaga, K. Single-Layer ReS2: Two-Dimensional Semiconductor with Tunable in-Plane Anisotropy. ACS Nano 2015, 9, 11249–11257. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qiao, J.; Yu, P.; Hu, Z.; Lin, Z.; Lau, S.P.; Liu, Z.; Ji, W.; Chai, Y. Extraordinarily Strong Interlayer Interaction in 2D Layered PtS2. Adv. Mater. 2016, 28, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shi, H.; Siegrist, T.; Singh, D.J. Electronic, Transport, and Optical Properties of Bulk and Mono-Layer PdSe2. Appl. Phys. Lett. 2015, 107, 153902. [Google Scholar] [CrossRef]
- Oyedele, A.D.; Yang, S.; Liang, L.; Puretzky, A.A.; Wang, K.; Zhang, J.; Yu, P.; Pudasaini, P.R.; Ghosh, A.W.; Liu, Z.; et al. PdSe2: Pentagonal Two-Dimensional Layers with High Air Stability for Electronics. J. Am. Chem. Soc. 2017, 139, 14090–14097. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Sun, X.; Wu, C.; Peng, L.; Lin, C.; Hu, S.; Yang, J.; Xie, Y. Metallic Few-Layered VS2 Ultrathin Nanosheets: High Two-Dimensional Conductivity for in-Plane Supercapacitors. J. Am. Chem. Soc. 2011, 133, 17832–17838. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Hotta, T.; Koretsune, T.; Watanabe, K.; Taniguchi, T.; Sugawara, K.; Takahashi, T.; Shinohara, H.; Kitaura, R. Two-Dimensional Metallic NbS2: Growth, Optical Identification and Transport Properties. 2D Mater. 2016, 3, 025027. [Google Scholar] [CrossRef]
- Voiry, D.; Yang, J.; Chhowalla, M. Recent Strategies for Improving the Catalytic Activity of 2D TMD Nanosheets toward the Hydrogen Evolution Reaction. Adv. Mater. 2016, 28, 6197–6206. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-Layer MoS2 Transistors. Nat. Nanotechnol. 2011, 6, 147. [Google Scholar] [CrossRef] [PubMed]
- Akinwande, D.; Petrone, N.; Hone, J. Two-Dimensional Flexible Nanoelectronics. Nat. Commun. 2014, 5, 5678. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, L.; Lee, Y.-H.; Shi, Y.; Hsu, A.; Chin, M.L.; Li, L.-J.; Dubey, M.; Kong, J.; Palacios, T. Integrated Circuits Based on Bilayer MoS2 Transistors. Nano Lett. 2012, 12, 4674–4680. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Li, H.; Li, H.; Jiang, L.; Shi, Y.; Sun, Y.; Lu, G.; Zhang, Q.; Chen, X.; Zhang, H. Single-Layer MoS2 Phototransistors. ACS Nano 2012, 6, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanchez, O.; Lembke, D.; Kayci, M.; Radenovic, A.; Kis, A. Ultrasensitive Photodetectors Based on Monolayer MoS2. Nat. Nanotechnol. 2013, 8, 497. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.F.; He, K.; Shan, J.; Heinz, T.F. Control of Valley Polarization in Monolayer MoS2 by Optical Helicity. Nat. Nanotechnol. 2012, 7, 494. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Wang, G.; Han, W.; Ye, H.; Zhu, C.; Shi, J.; Niu, Q.; Tan, P.; Wang, E.; Liu, B.; et al. Valley-Selective Circular Dichroism of Monolayer Molybdenum Disulphide. Nat. Commun. 2012, 3, 887. [Google Scholar] [CrossRef] [PubMed]
- Soulard, C.; Rocquefelte, X.; Petit, P.E.; Evain, M.; Jobic, S.; Itié, J.P.; Munsch, P.; Koo, H.J.; Whangbo, M.H. Experimental and Theoretical Investigation on the Relative Stability of the PdS2- and Pyrite-Type Structures of PdSe2. Inorg. Chem. 2004, 43, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.L.; Yu, P.; Liu, F.; Hong, J.; Wang, X.; Zeng, Q.; Hsu, C.H.; Zhu, C.; Zhou, J.; Wang, X.; et al. High Mobility 2D Palladium Diselenide Field-Effect Transistors with Tunable Ambipolar Characteristics. Adv. Mater. 2017, 29, 1602969. [Google Scholar] [CrossRef] [PubMed]
- Lebègue, S.; Björkman, T.; Klintenberg, M.; Nieminen, R.M.; Eriksson, O. Two-Dimensional Materials from Data Filtering and Ab Initio Calculations. Phys. Rev. X 2013, 3, 031002. [Google Scholar]
- Wang, Y.; Li, Y.; Chen, Z. Not Your Familiar Two Dimensional Transition Metal Disulfide: Structural and Electronic Properties of the PdS2 Monolayer. J. Mater. Chem. C 2015, 3, 9603–9608. [Google Scholar] [CrossRef]
- Lin, J.; Zuluaga, S.; Yu, P.; Liu, Z.; Pantelides, S.T.; Suenaga, K. Novel Pd2Se3 Two-Dimensional Phase Driven by Interlayer Fusion in Layered PdSe2. Phys. Rev. Lett. 2017, 119, 016101. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, F.; Wang, Y.; Qiao, M.; Li, Y. Pd2Se3 Monolayer: A Novel Two-Dimensional Material with Excellent Electronic, Transport, and Optical Properties. J. Mater. Chem. C 2018, 6, 4494–4500. [Google Scholar] [CrossRef]
- Naghavi, S.S.; He, J.; Xia, Y.; Wolverton, C. Pd2Se3 Monolayer: A Promising Two-Dimensional Thermoelectric Material with Ultralow Lattice Thermal Conductivity and High Power Factor. Chem. Mater. 2018, 30, 5639–5647. [Google Scholar] [CrossRef]
- Sebastian, Z.; Junhao, L.; Kazu, S.; Sokrates, T.P. Two-Dimensional PdSe2-Pd2Se3 Junctions Can Serve as Nanowires. 2D Mater. 2018, 5, 035025. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Erratum: “Hybrid Functionals Based on a Screened Coulomb Potential” [J. Chem. Phys. 118, 8207 (2003)]. J. Chem. Phys. 2006, 124, 219906. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Parlinski, K.; Li, Z.Q.; Kawazoe, Y. First-Principles Determination of the Soft Mode in Cubic ZrO2. Phys. Rev. Lett. 1997, 78, 4063–4066. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First Principles Phonon Calculations in Materials Science. Scripta Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Bardeen, J.; Shockley, W. Deformation Potentials and Mobilities in Non-Polar Crystals. Phys. Rev. 1950, 80, 72–80. [Google Scholar] [CrossRef]
- Qiao, J.; Kong, X.; Hu, Z.-X.; Yang, F.; Ji, W. High-Mobility Transport Anisotropy and Linear Dichroism in Few-Layer Black Phosphorus. Nat. Commun. 2014, 5, 4475. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, G.; Zhang, Y.-W. Polarity-Reversed Robust Carrier Mobility in Monolayer MoS2 Nanoribbons. J. Am. Chem. Soc. 2014, 136, 6269–6275. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Zeng, X.C. Titanium Trisulfide Monolayer: Theoretical Prediction of a New Direct-Gap Semiconductor with High and Anisotropic Carrier Mobility. Angew. Chem. Int. Ed. 2015, 54, 7572–7576. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Ma, Y.; Li, Y.; Heine, T. GeP3: A Small Indirect Band Gap 2d Crystal with High Carrier Mobility and Strong Interlayer Quantum Confinement. Nano Lett. 2017, 17, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Sinha, T.P.; Mookerjee, A. Electronic Structure, Chemical Bonding, and Optical Properties of Paraelectric BaTiO3. Phys. Rev. B 2000, 62, 8828–8834. [Google Scholar] [CrossRef]
- Miao, N.; Xu, B.; Bristowe, N.C.; Zhou, J.; Sun, Z. Tunable Magnetism and Extraordinary Sunlight Absorbance in Indium Triphosphide Monolayer. J. Am. Chem. Soc. 2017, 139, 11125–11131. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Niu, X.; Ouyang, Y.; Zhou, Q.; Wang, J. Ultrathin Semiconducting Bi2Te2S and Bi2Te2Se with High Electron Mobilities. J. Phys. Chem. Lett. 2018, 9, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Paier, J.; Marsman, M.; Kresse, G. Dielectric Properties and Excitons for Extended Systems from Hybrid Functionals. Phys. Rev. B 2008, 78, 121201. [Google Scholar] [CrossRef]
- Scharber, M.C.; Mühlbacher, D.; Koppe, M.; Denk, P.; Waldauf, C.; Heeger, A.J.; Brabec, C.J. Design Rules for Donors in Bulk-Heterojunction Solar Cells-Towards 10 % Energy-Conversion Efficiency. Adv. Mater. 2006, 18, 789–794. [Google Scholar] [CrossRef]
- Bernardi, M.; Palummo, M.; Grossman, J.C. Semiconducting Monolayer Materials as a Tunable Platform for Excitonic Solar Cells. ACS Nano 2012, 6, 10082–10089. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Zeng, X.C. Bilayer Phosphorene: Effect of Stacking Order on Bandgap and Its Potential Applications in Thin-Film Solar Cells. J. Phys. Chem. Lett. 2014, 5, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, V.D.S.O.; Linghu, J.; Zhang, C.; Feng, Y.P.; Shen, L. Heterostructures of Phosphorene and Transition Metal Dichalcogenides for Excitonic Solar Cells: A First-Principles Study. Appl. Phys. Lett. 2016, 108, 122105. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, S.; Cai, B.; Huang, Y.; Zou, Y.; Guo, B.; Gu, Y.; Zeng, H. A Promising Two-Dimensional Solar Cell Donor: Black Arsenic–Phosphorus Monolayer with 1.54 eV Direct Bandgap and Mobility Exceeding 14,000 cm2v−1s−1. Nano Energy 2016, 28, 433–439. [Google Scholar] [CrossRef]
- Lee, J.; Huang, J.; Sumpter, B.G.; Yoon, M. Strain-Engineered Optoelectronic Properties of 2D Transition Metal Dichalcogenide Lateral Heterostructures. 2D Mater. 2017, 4, 021016. [Google Scholar] [CrossRef]
- Shockley, W.; Queisser, H.J. Detailed Balance Limit of Efficiency of P-N Junction Solar Cells. J. Appl. Phys. 1961, 32, 510–519. [Google Scholar] [CrossRef]
- Hanna, M.C.; Nozik, A.J. Solar Conversion Efficiency of Photovoltaic and Photoelectrolysis Cells with Carrier Multiplication Absorbers. J. Appl. Phys. 2006, 100, 074510. [Google Scholar] [CrossRef]
- Wadia, C.; Alivisatos, A.P.; Kammen, D.M. Materials Availability Expands the Opportunity for Large-Scale Photovoltaics Deployment. Environ. Sci. Technol. 2009, 43, 2072–2077. [Google Scholar] [CrossRef] [PubMed]
- Motta, C.; El-Mellouhi, F.; Kais, S.; Tabet, N.; Alharbi, F.; Sanvito, S. Revealing the Role of Organic Cations in Hybrid Halide Perovskite CH3NH3PbI3. Nat. Commun. 2015, 6, 7026. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Tan, L.Z.; Liu, S.; Rappe, A.M. Rashba Spin–Orbit Coupling Enhanced Carrier Lifetime in CH3NH3PbI3. Nano Lett. 2015, 15, 7794–7800. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Sun, Q. A Honeycomb BeN2 Sheet with a Desirable Direct Bandgap and High Carrier Mobility. J. Phys. Chem. Lett. 2016, 7, 2664–2670. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier Type | E1 (eV) | C (N/m) | m* (me) | (cm2V−1s−1) | |
|---|---|---|---|---|---|
| Pd2Se3 | electron (x) | 3.756 | 33.02 | 0.762 | 101.9 |
| electron (y) | 3.785 | 32.93 | 0.543 | 140.4 | |
| hole (x) | 2.870 | 33.02 | 9.029 | 7.3 | |
| hole (y) | 12.082 | 32.93 | 0.187 | 19.9 | |
| PdSe2 | electron (x) | 9.542 | 32.45 | 0.429 | 43.36 |
| electron (y) | 9.982 | 55.05 | 0.390 | 73.99 | |
| hole (x) | 3.352 | 32.45 | 0.656 | 97.94 | |
| hole (y) | 3.074 | 55.05 | 1.401 | 92.54 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhang, S.; Guo, Y.; Wang, F.Q.; Wang, Q. Physical Properties and Photovoltaic Application of Semiconducting Pd2Se3 Monolayer. Nanomaterials 2018, 8, 832. https://doi.org/10.3390/nano8100832
Li X, Zhang S, Guo Y, Wang FQ, Wang Q. Physical Properties and Photovoltaic Application of Semiconducting Pd2Se3 Monolayer. Nanomaterials. 2018; 8(10):832. https://doi.org/10.3390/nano8100832
Chicago/Turabian StyleLi, Xiaoyin, Shunhong Zhang, Yaguang Guo, Fancy Qian Wang, and Qian Wang. 2018. "Physical Properties and Photovoltaic Application of Semiconducting Pd2Se3 Monolayer" Nanomaterials 8, no. 10: 832. https://doi.org/10.3390/nano8100832
APA StyleLi, X., Zhang, S., Guo, Y., Wang, F. Q., & Wang, Q. (2018). Physical Properties and Photovoltaic Application of Semiconducting Pd2Se3 Monolayer. Nanomaterials, 8(10), 832. https://doi.org/10.3390/nano8100832