First-principles Investigations of Magnetic Semiconductors: An example of Transition Metal Decorated Two-dimensional SnS Monolayer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
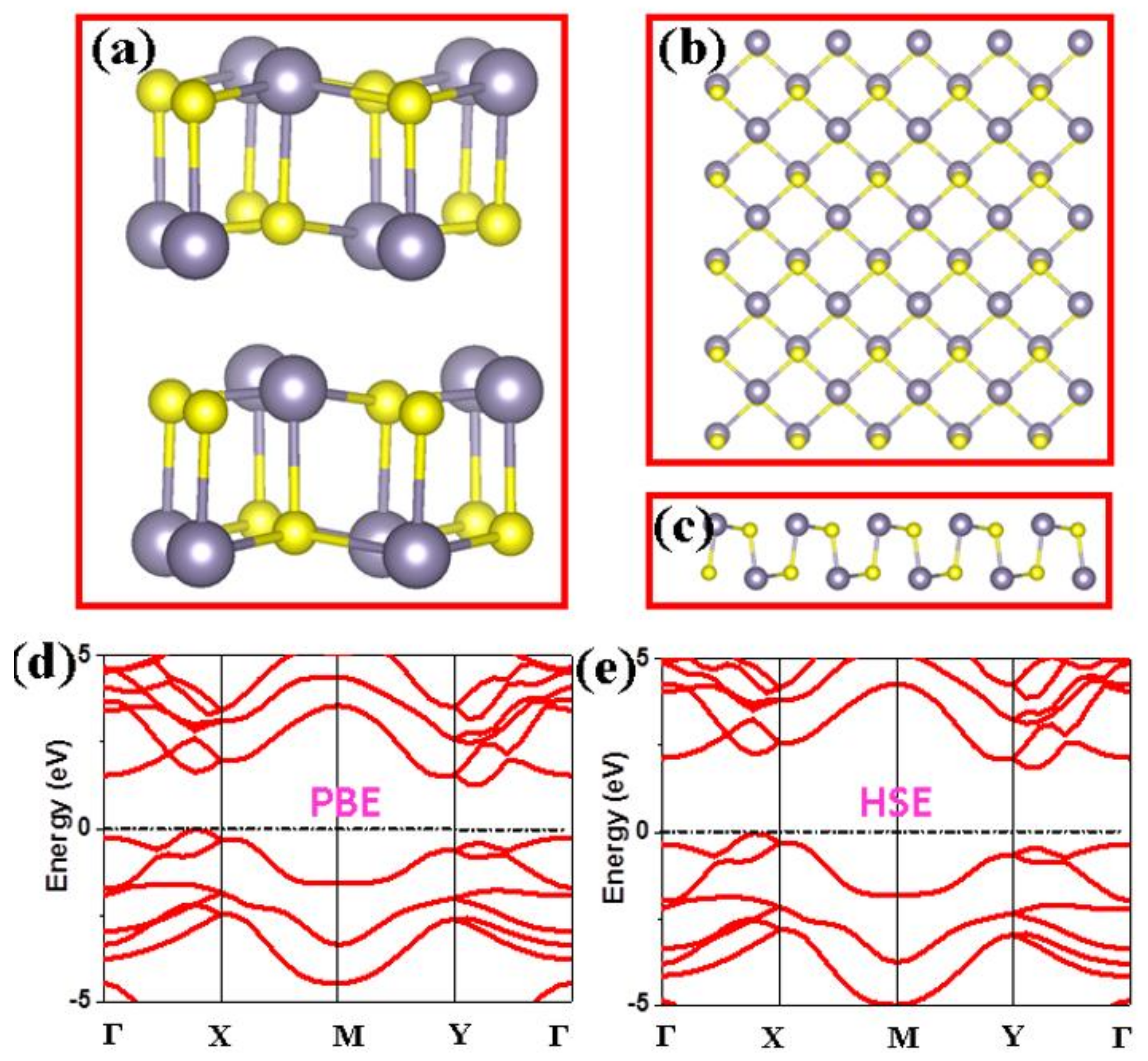
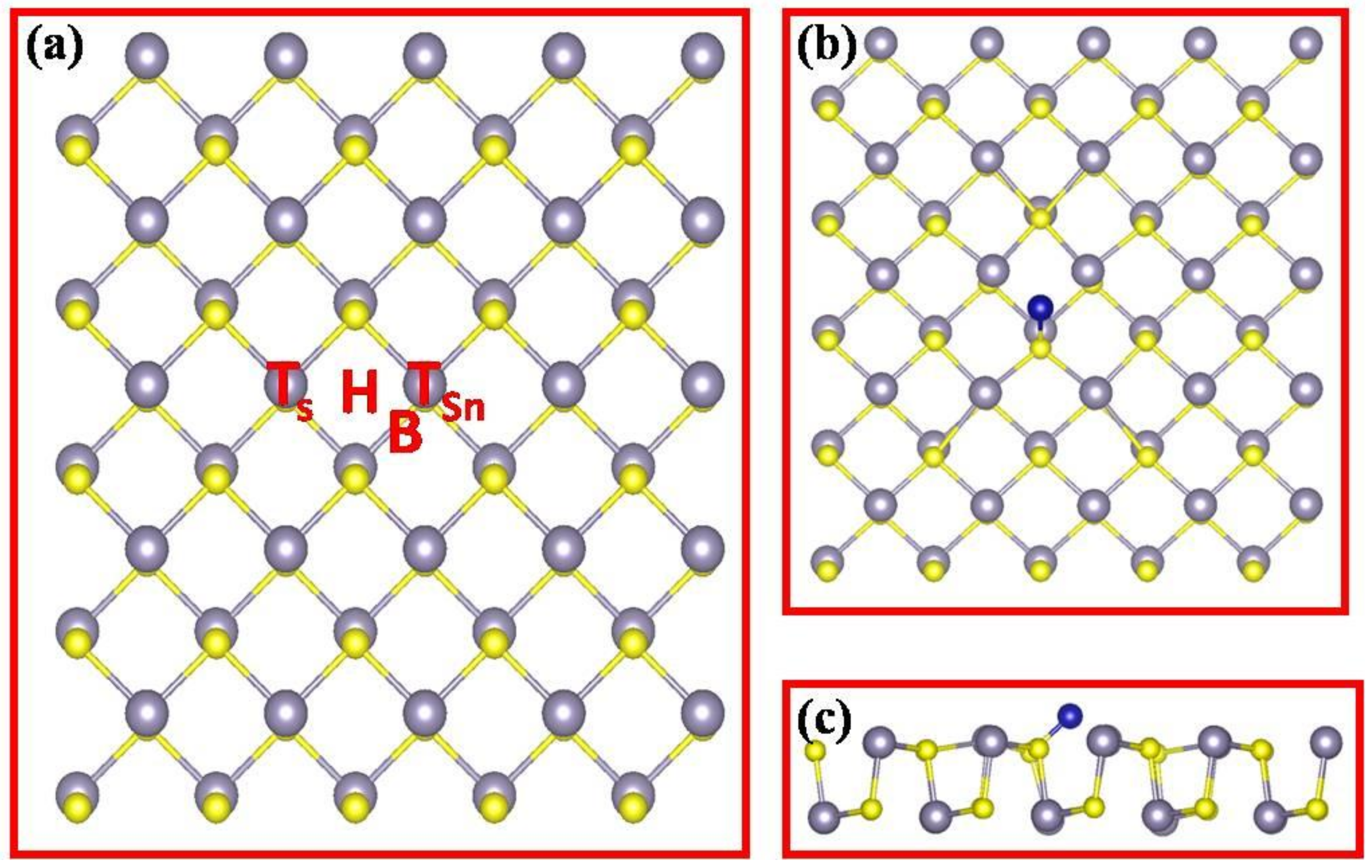
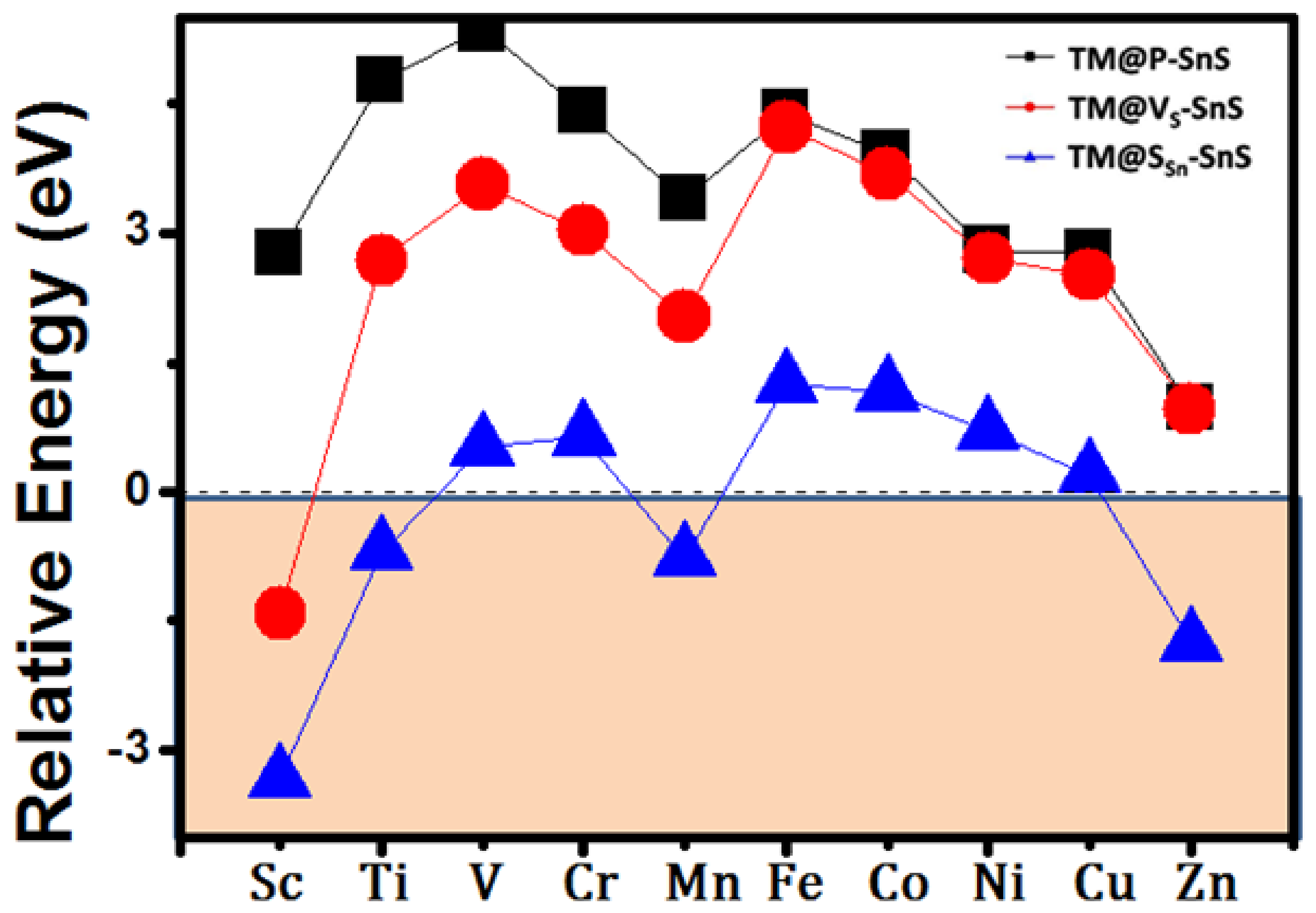
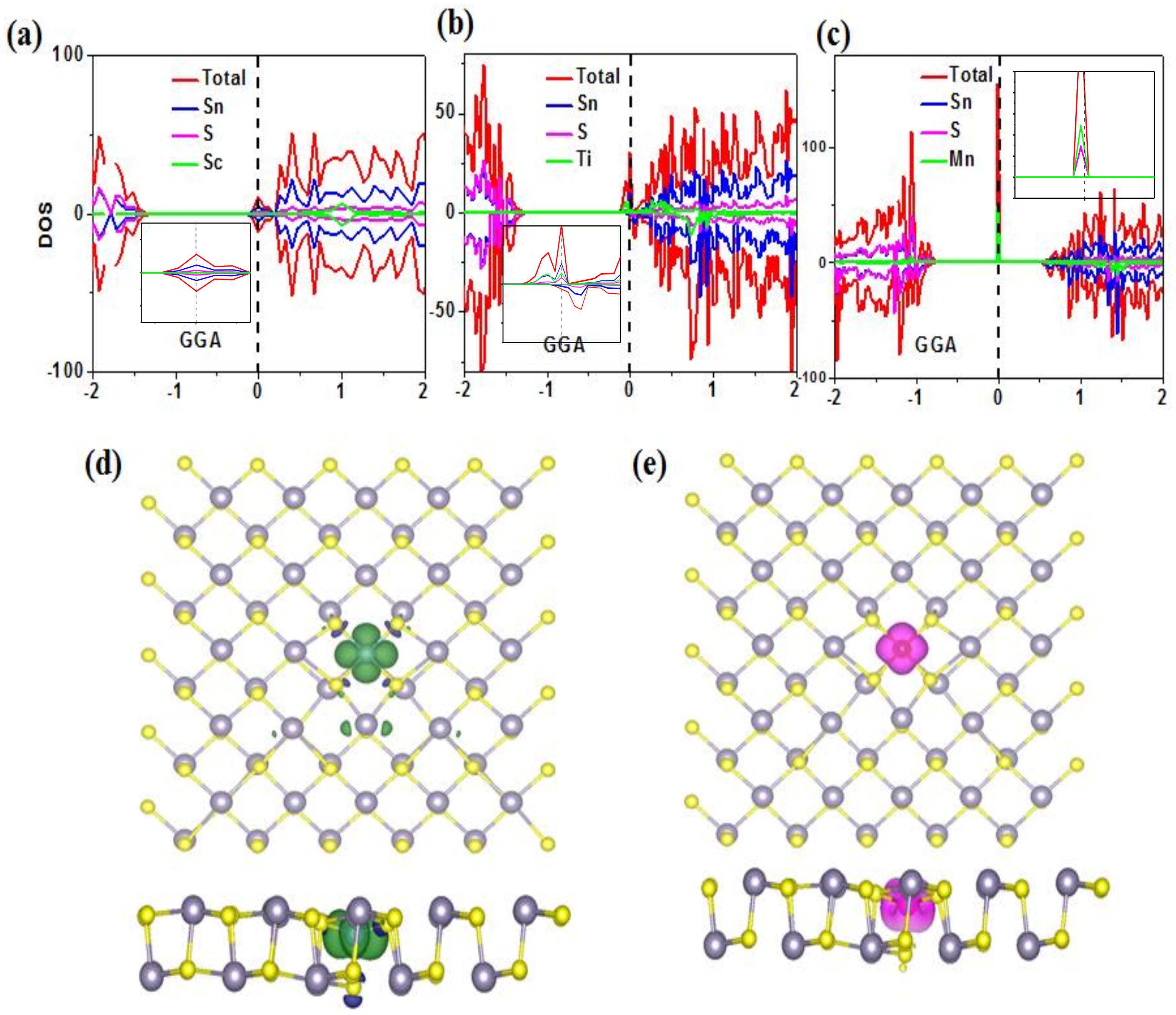
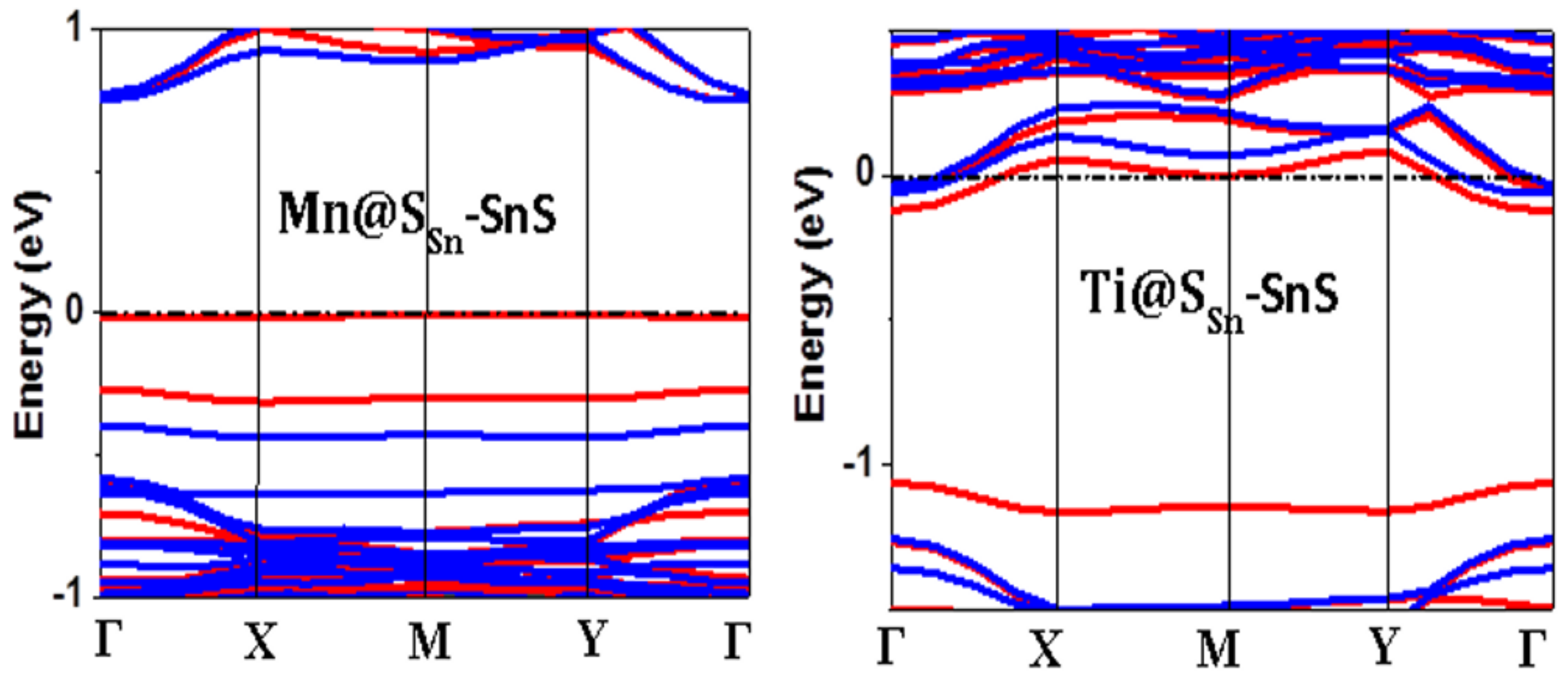
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V. Firsov A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Song, Z.; Li, X.; Wu, X.; Brown, N.; Naud, C.; Mayou, D.; Li, T.; Hass, J.; Marchenkov, A. Electronic confinement and coherence in patterned epitaxial graphene. Science 2006, 312, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Cinquanta, E.; Chiappe, D.; Grazianetti, C.; Fanciulli, M.; Dubey, M.; Molle, A.; Akinwande, D. Silicene field-effect transistors operating at room temperature. Nat. Nanotechnol. 2015, 10, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, Y.; Ye, G.; Ge, Q.; Ou, X.; Wu, H.; Feng, D.; Chen, X.H.; Zhang, Y. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Kan, E.; Hu, W.; Xiao, C.; Lu, R.; Deng, K.; Yang, J.; Su, H. Half-Metallicity in Organic Single Porous Sheets. J. Am. Chem. Soc. 2012, 134, 5718–5721. [Google Scholar] [CrossRef] [PubMed]
- Splendiani, A.; Sun, L.; Zhang, Y.; Li, T.; Kim, J.; Chim, C.Y.; Galli, G.; Wang, F. Emerging Photoluminescence in Monolayer MoS2. Nano Lett. 2010, 10, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Xiang, H.; Yu, J.; Wei, S. Effective control of the charge and magnetic states of transition-metal atoms on single-layer boron nitride. Phys. Rev. Lett. 2012, 108, 206802. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS₂ transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pan, Y.; Shen, Y.; Wang, Z.; Ong, Z.; Xu, T.; Xin, R.; Pan, L.; Wang, B.; Sun, L.; et al. Towards intrinsic charge transport in monolayer molybdenum disulfide by defect and interface engineering. Nat. Commun. 2014, 5, 5290. [Google Scholar] [CrossRef] [PubMed]
- Naguib, M.; Mashtalir, O.; Carle, J.; Presser, V.; Lu, J.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-dimensional transition metal carbides. ACS Nano 2012, 6, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.; Norman, C. What don’t we know? Science 2005, 309, 75. [Google Scholar] [CrossRef] [PubMed]
- Ugeda, M.M.; Brihuega, I.; Guinea, F.; Gómez-Rodríguez, J.M. Missing atom as a source of carbon magnetism. Phys. Rev. Lett. 2010, 104, 096804. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Montanari, B.; Harrison, N. A Defective Graphene Phase Predicted to be a Room Temperature Ferromagnetic Semiconductor. New. J. Phys. 2008, 10, 033002. [Google Scholar] [CrossRef]
- Kan, E.; Xiang, H.; Wu, F.; Chuan, T.; Lee, C.; Yang, J.; Whangbo, M.H. Prediction for room-temperature half-metallic ferromagnetism in the half-fluorinated single layers of BN and ZnO. Appl. Phys. Lett. 2010, 97. [Google Scholar] [CrossRef]
- Krasheninnikov, A.; Lehtinen, P.O.; Foster, A.; Pyykkö, P.; Nieminen, R.M. Embedding transition-metal atoms in graphene: Structure, bonding, and magnetism. Phys. Rev. Lett. 2009, 102, 126807. [Google Scholar] [CrossRef] [PubMed]
- Brent, J.R.; Lewis, D.J.; Lorenz, T.; Lewis, E.A.; Savjani, N.; Haigh, S.J.; Seifert, G.; Derby, B.; O’Brien, P. Tin(II) Sulfide (SnS) Nanosheets by Liquid-Phase Exfoliation of Herzenbergite: IV–VI Main Group Two-Dimensional Atomic Crystals. J. Am. Chem. Soc. 2015, 137, 12689–12696. [Google Scholar] [CrossRef] [PubMed]
- Buscema, M.; Groenendijk, D.J.; Blanter, S.I.; Steele, G.A.; vander Zant, H.S.J.; Castellanos-Gomez, A. Fast and broadband photoresponse of few-layer black phosphorus field-effect transistors. Nano Lett. 2014, 14, 3347–3352. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Kong, X.; Hu, Z.; Yang, F.; Ji, W. High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus. Nat. Commun. 2014, 5, 4475. [Google Scholar] [CrossRef] [PubMed]
- Reich, E.S. Phosphorene excites materials scientists. Nature 2014, 506, 19. [Google Scholar] [CrossRef] [PubMed]
- Biacchi, A.J.; Vaughn, D.D.; Schaak, R.E. Synthesis and Crystallographic Analysis of Shape-Controlled SnS Nanocrystal Photocatalysts: Evidence for a Pseudotetragonal Structural Modification. J. Am. Chem. Soc. 2013, 135, 11634–11644. [Google Scholar] [CrossRef] [PubMed]
- Malone, B.D.; Gali, A.; Kaxiras, E. First principles study of point defects in SnS. Phys. Chem. Chem. Phys. 2014, 16, 26176–26183. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Yang, D.; Ji, Y.; Zhang, H.; Shen, X. Two-dimensional SnS nanosheets fabricated by a novel hydrothermal method. J. Mater. Science. 2005, 40, 591–595. [Google Scholar] [CrossRef]
- Ning, J.; Men, K.; Xiao, G.; Wang, L.; Dai, Q.; Zou, B.; Liu, B.; Zou, G. Facile synthesis of IV-VI SnS nanocrystals with shape and size control: Nanoparticles, nanoflowers and amorphous nanosheets. Nanoscale 2010, 2, 1699–1703. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, J.; Shen, S.; Xu, H.; Wang, Q. Ultralarge single crystal SnS rectangular nanosheets. Chem. Commun 2011, 47, 5226–5228. [Google Scholar] [CrossRef] [PubMed]
- Xin, C.; Zheng, J.; Su, Y.; Li, S.; Zhang, B.; Feng, Y.; Pan, F. Few-layer Tin Sulfide: A new black-phosphorus-analogue 2D material with a sizeable band gap, odd-even quantum confinement effect, and high carrier mobility. J. Phys. Chem. C 2016, 120, 22663–22669. [Google Scholar] [CrossRef]
- Tian, Z.; Guo, C.; Zhao, M.; Li, R.; Xue, J. Two-dimensional SnS: A phosphorene analogue with strong in-plane electronic anisotropy. Acs Nano 2017, 11, 2219–2226. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, M.; Huang, Y.; Chen, Q.; Cao, C.; He, Y. Tuning the electronic properties of bilayer group-IV monochalcogenides by stacking order, strain and an electric field: a computational study. Phys. Chem. Chem. Phys. 2017, 20, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Cao, H.; Dong, J. Electronic properties of group-IV monochalcogenides nanoribbons: studies from first-principles. Phys. Lett. A 2017, 381, 3747–3753. [Google Scholar] [CrossRef]
- Zhou, Y. MX ( M = Ge, Sn; X = S, Se) sheets: Theoretical prediction of new promising electrode materials for Li ion batteries. J. Mater. Chem. A. 2016, 4, 10906–10913. [Google Scholar] [CrossRef]
- Zhang, Y.; Shang, B.; Li, L.; Lei, J. Coupling effects of strain on structural transformation and bandgap engineering in SnS monolayer. Rsc. Adv. 2017, 7, 30327–30333. [Google Scholar] [CrossRef]
- Xiong, W.; Xia, C.; Zhao, X.; Wang, T.; Jia, Y. Effects of strain and electric field on electronic structures and Schottky barrier in graphene and SnS hybrid heterostructures. Carbon 2016, 109, 737–746. [Google Scholar]
- Ma, Z.; Wang, B.; Ou, L.; Zhang, Y.; Zhang, X.; Zhou, Z. Structure and properties of phosphorene-like IV-VI 2D materials. Nanotechnology 2016, 27, 415203. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Guo, Y.; Han, N.; Su, Y.; Zhang, J.; Zhao, J. Lateral heterostructures of monolayer group-IV monochalcogenides: band alignment and electronic properties. J. Mater. Chem. C. 2017, 5, 3788–3795. [Google Scholar] [CrossRef]
- Guo, R.; Wang, X.; Kuang, Y.; Huang, B. First-principles study of anisotropic thermoelectric transport properties of IV-VI semiconductor compounds SnSe and SnS. Phys. Rev. B. 2015, 92, 115202. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics of Liquid Metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. ERRATA:Generalized gradient approximation made simple. Phys. Rev. Lett. 1998, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E. Efficient hybrid density functional calculations in solids: assessment of the Heyd-Scuseria-Ernzerhof screened Coulomb hybrid functional. J. Chem. Phys. 2004, 121, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Dudarev, S.; Botton, G.; Savrasov, Y.; Humphreys, C.; Sutton, A. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Wang, F.; Yao, Q.; Zhou, L.; Ma, Z.; He, M.; Wu, F. Theoretical understanding of SnS monolayer as Li ion battery anode material. J. Phys. Chem. Solid. 2018, 121, 261–265. [Google Scholar] [CrossRef]
- Chowdhury, C.; Karmakar, S.; Datta, A. Monolyaer Group IV-VI monochalcogenides: Low-dimensional materials for photocatalytic water splitting. J. Phys. Chem. C. 2017, 121, 7615–7624. [Google Scholar] [CrossRef]
- Kamal, C.; Chakrabarti, A.; Ezawa, M. Direct band gaps in group IV-VI monolayer materials: Binary counterparts of phosphorene. Phys. Rev. B. 2016, 93, 125428. [Google Scholar] [CrossRef]
- Dietl, T.; Ohno, H. Dilute ferromagnetic semiconductors: Physics and spintronic structures. Rev. Mod. Phys. 2014, 86, 187. [Google Scholar] [CrossRef]
- Sui, X.; Si, C.; Shao, B.; Zou, X.; Wu, J.; Gu, B.; Duan, W. Tunable Magnetism in Transition-Metal-Decorated Phosphorene. J. Phys. Chem. C. 2015, 119, 10059–10063. [Google Scholar] [CrossRef]
- Hashmi, A.; Hong, J. Transition Metal Doped Phosphorene: First-Principles Study. J. Phys. Chem. C. 2015, 119, 9198–9204. [Google Scholar] [CrossRef]
- Lany, S.; Zunger, A. Accurate prediction of defect properties in density functional supercell calculations. Modell. Simul. Mater. Sci. Eng. 2009, 17, 084002. [Google Scholar] [CrossRef]
- Kumagai, Y.; Oba, F. Electrostatics-based finite-size corrections for first-principles point defect calculations. Phys. Rev. B. 2014, 89, 195205. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Zhou, L.; Ma, Z.; He, M.; Wu, F.; Liu, Y. First-principles Investigations of Magnetic Semiconductors: An example of Transition Metal Decorated Two-dimensional SnS Monolayer. Nanomaterials 2018, 8, 789. https://doi.org/10.3390/nano8100789
Wang F, Zhou L, Ma Z, He M, Wu F, Liu Y. First-principles Investigations of Magnetic Semiconductors: An example of Transition Metal Decorated Two-dimensional SnS Monolayer. Nanomaterials. 2018; 8(10):789. https://doi.org/10.3390/nano8100789
Chicago/Turabian StyleWang, Fangfang, Liyu Zhou, Zhen Ma, Mingxue He, Fang Wu, and Yunfei Liu. 2018. "First-principles Investigations of Magnetic Semiconductors: An example of Transition Metal Decorated Two-dimensional SnS Monolayer" Nanomaterials 8, no. 10: 789. https://doi.org/10.3390/nano8100789
APA StyleWang, F., Zhou, L., Ma, Z., He, M., Wu, F., & Liu, Y. (2018). First-principles Investigations of Magnetic Semiconductors: An example of Transition Metal Decorated Two-dimensional SnS Monolayer. Nanomaterials, 8(10), 789. https://doi.org/10.3390/nano8100789