
Light Emission Intensities of Luminescent Y2O3:Eu and Gd2O3:Eu Particles of Various Sizes

 ,
,
Abstract
:
1. Introduction
2. Experimental Procedure
2.1. Materials
- Y(NO3)3·6H2O, batch ‘YN1/13’, purity 99.9%, Treibacher Industrie AG, Althofen, Austria
- Gd(NO3)3·6H2O, batch ‘23989500’, purity 99.9%, Strem Chemicals, Kehl, Germany
- Eu(NO3)3·6H2O, batches ‘23118500’ and ‘21422100’, purity 99.9%, Strem Chemicals, Bischheim, France
- Na2O2, batch ‘MKBP0012V’, purity 97%, Sigma Aldrich Chemie GmbH, Steinheim, Germany
- Urea, CH4N2O, batch ‘SZBD2810V’, purity 99%, Sigma Aldrich Chemie GmbH, Steinheim, Germany
- Micron sized Y2O3:Eu (4 mol %), labeled ‘L581’, a commercial phosphor from Osram (a former Siemens subsidiary) for use in fluorescent lamps. It is used as a standard because it has optimized emission intensity in the orange-red range when excited by a mercury vapour lamp at 254 nm with a reported quantum yield of 85%
2.2. Peroxidic Precipitation (PP)
2.3. Urea Based Homogeneous Precipitation (UBHP)
2.4. Treatment of the PP and UBHP Precipitates
2.5. Physical Particle Properties—Morphology and Size Information
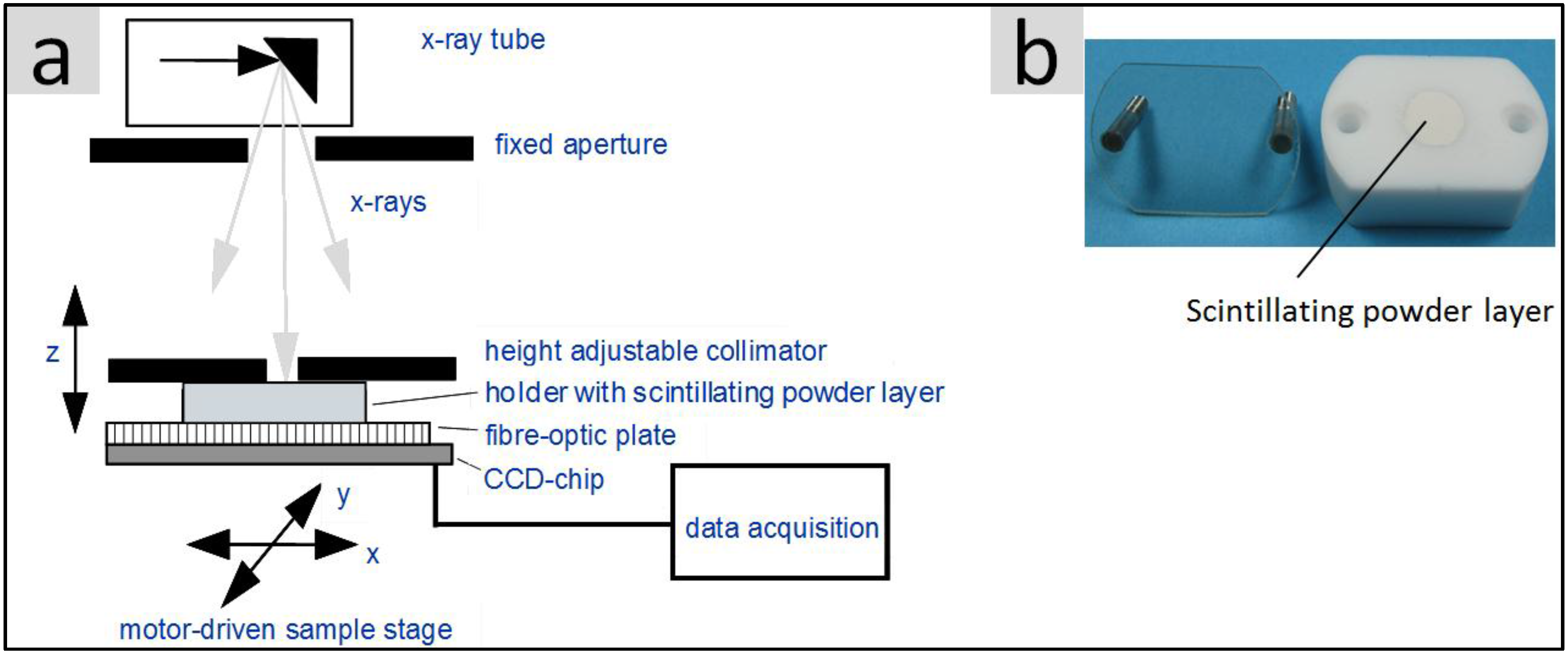
2.6. Luminescence Properties of the Particles
2.6.1. Fluorescence and Quantum Yield (QY) under UV Excitation
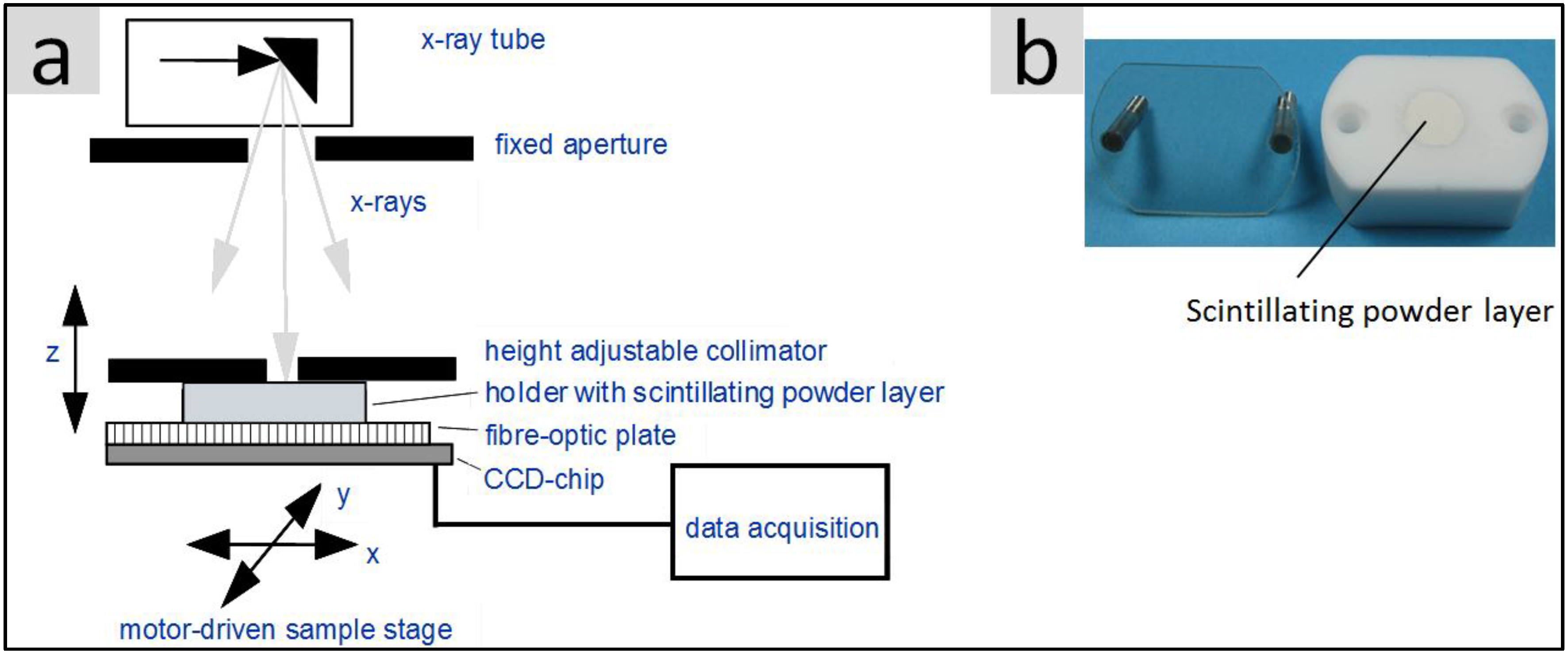
2.6.2. Light Yield under X-Ray Excitation (Scintillation)
2.6.3. Light Yield and Spectra Obtained under Electron Beam Irradiation (Cathodoluminescence/SEM)
3. Results and Discussion
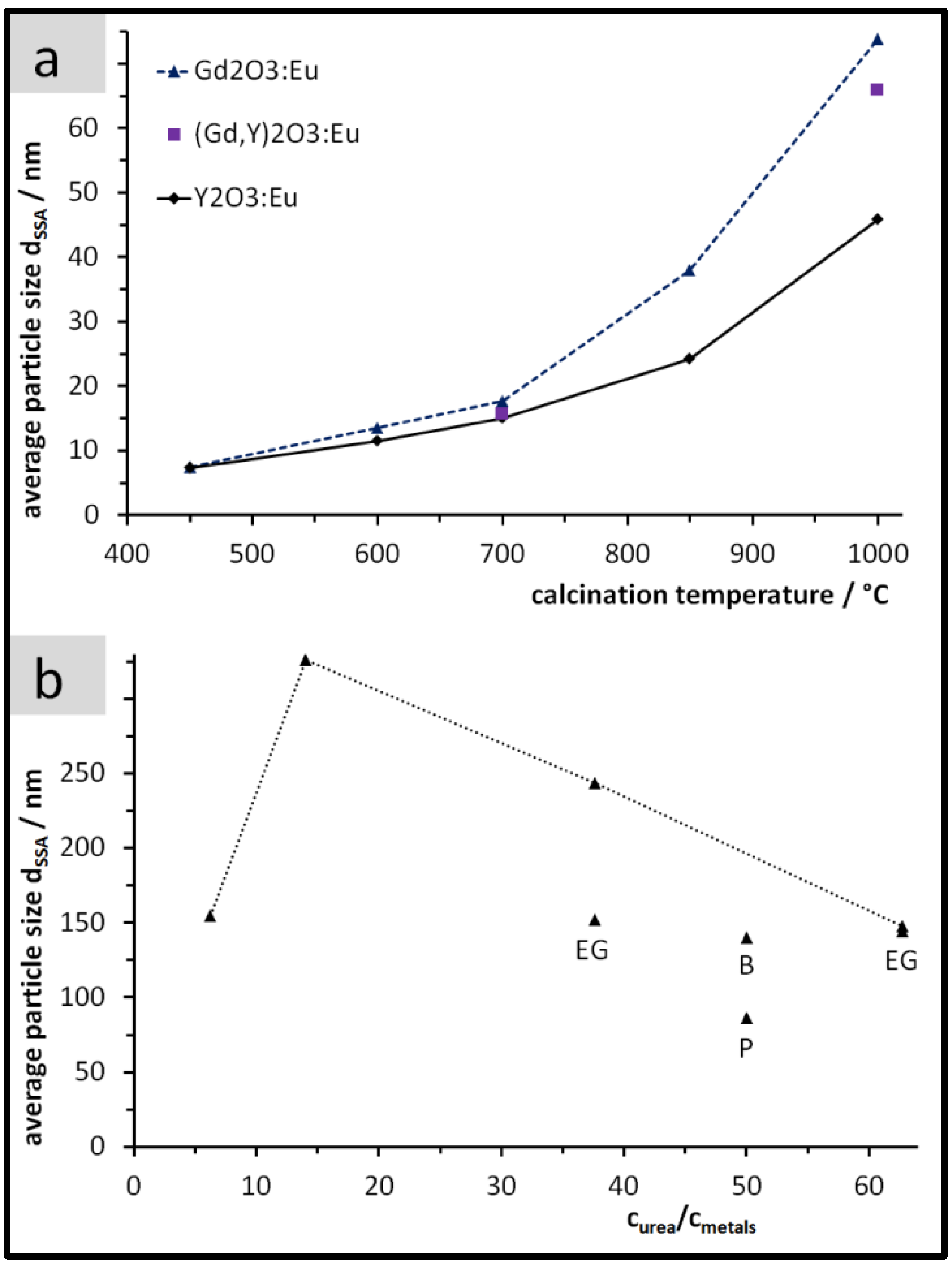
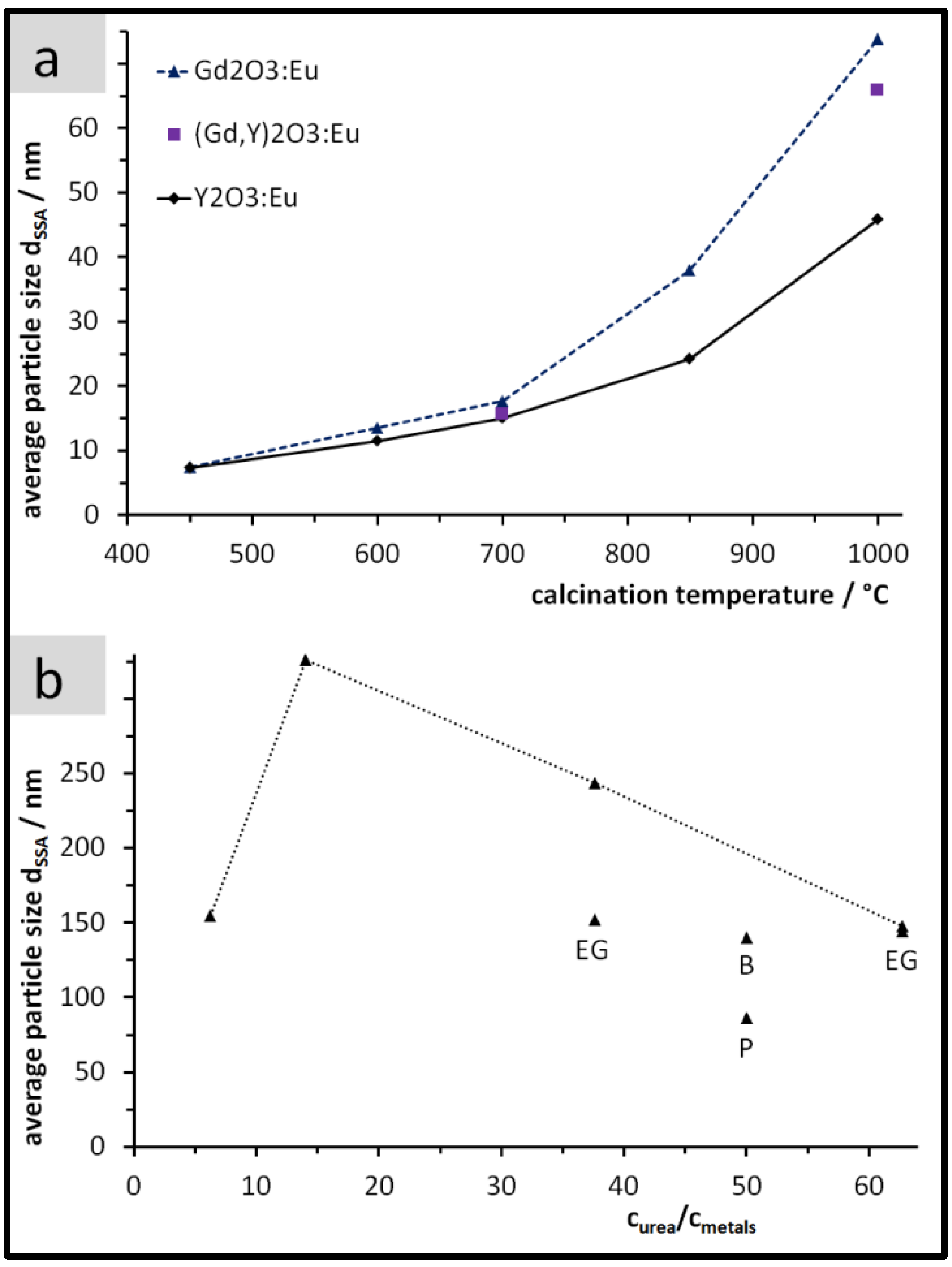
3.1. Yield, Composition, and Specific Surface Area of the Precipitates
- There were peroxide groups detected in the evolving O2 by thermogravimetry coupled with mass spectrometry (TG/MS) and by the test indicating the presence of peroxide anions by the yellow-brown colour of [TiO2 aq.]2+ upon addition of aqueous TiOSO4-solution. A peroxidic composition can also be expected from the reaction deployed.
- The carbonate fraction is concluded from emerging CO2 in TG/MS and from carbon found by C/H/N/S combustion analysis. Carbonate arises by the reaction of the peroxidic fraction with CO2 from the air during storage.
- Hydroxide can also be expected based on the precipitation reaction and provides charge neutrality. A precipitate composition without hydroxide (but with more associated water) can be excluded by comparison of the theoretical and measured weight losses and the ratio of released H2O and CO2 upon calcination.
- Associated/adsorbed water is usually present in and on precipitates with high surface areas, here as a result of the reaction conditions and by the adsorption of water from the air.
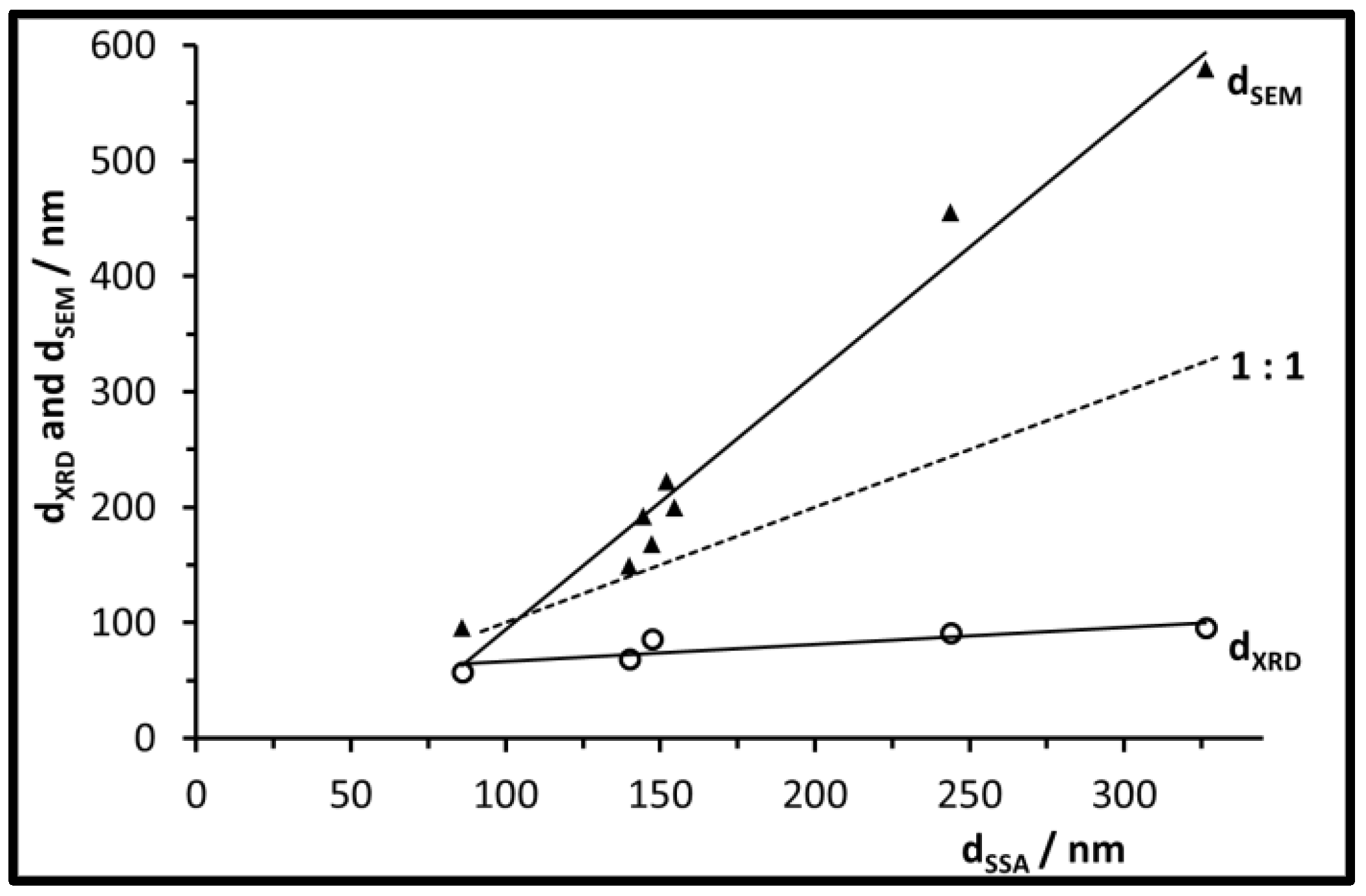
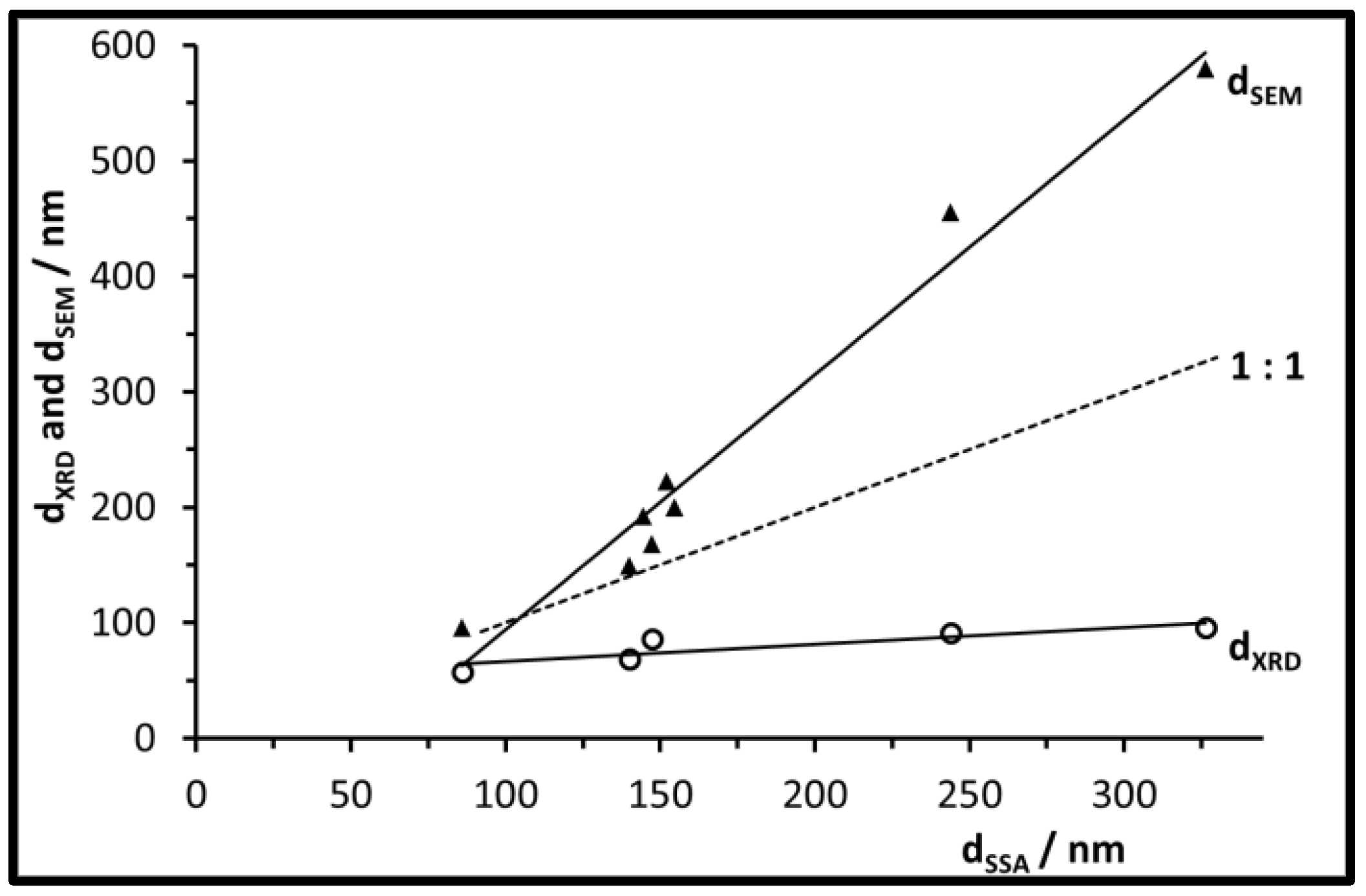
3.2. Size, Morphology, and Structure of the Calcined Particles
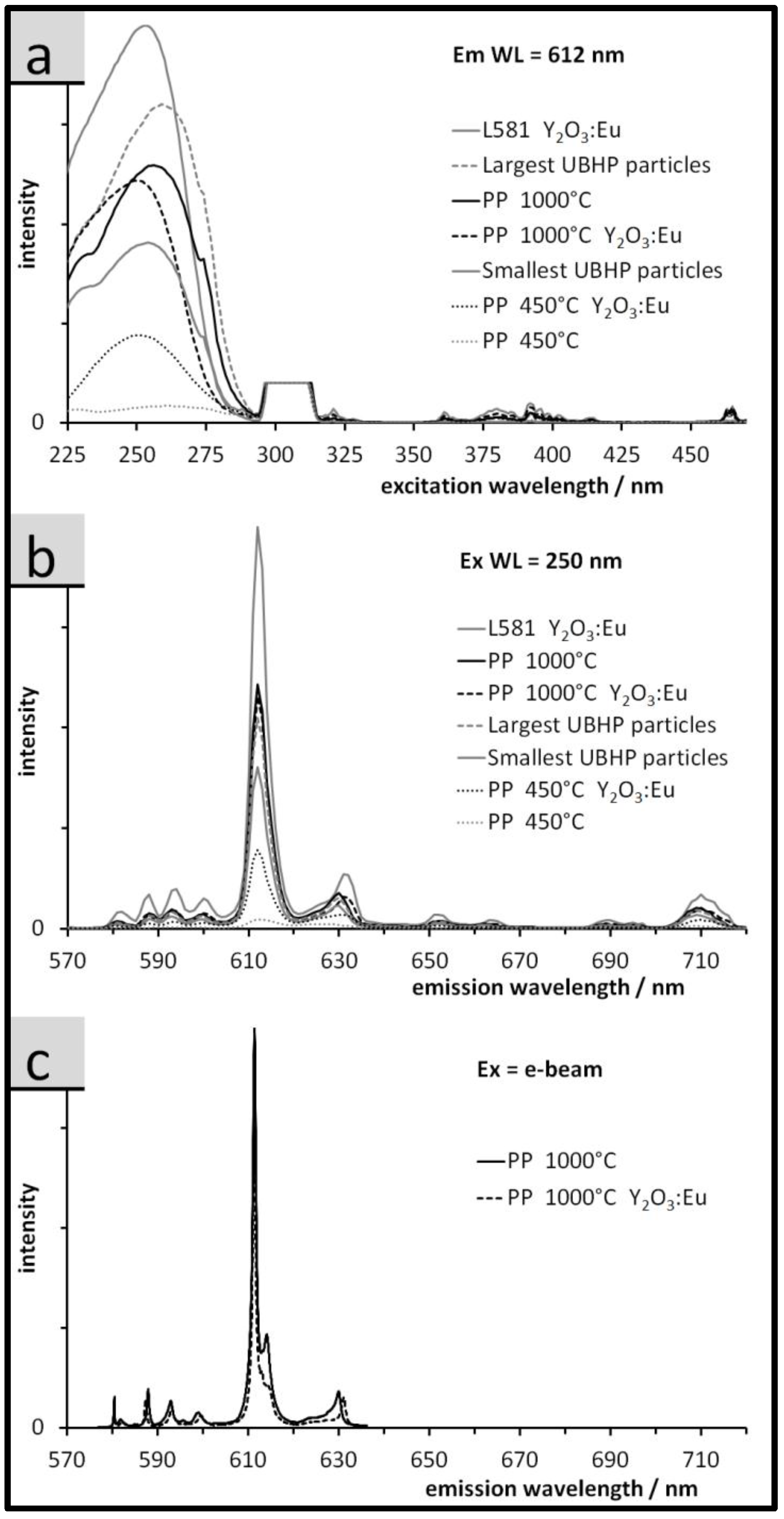
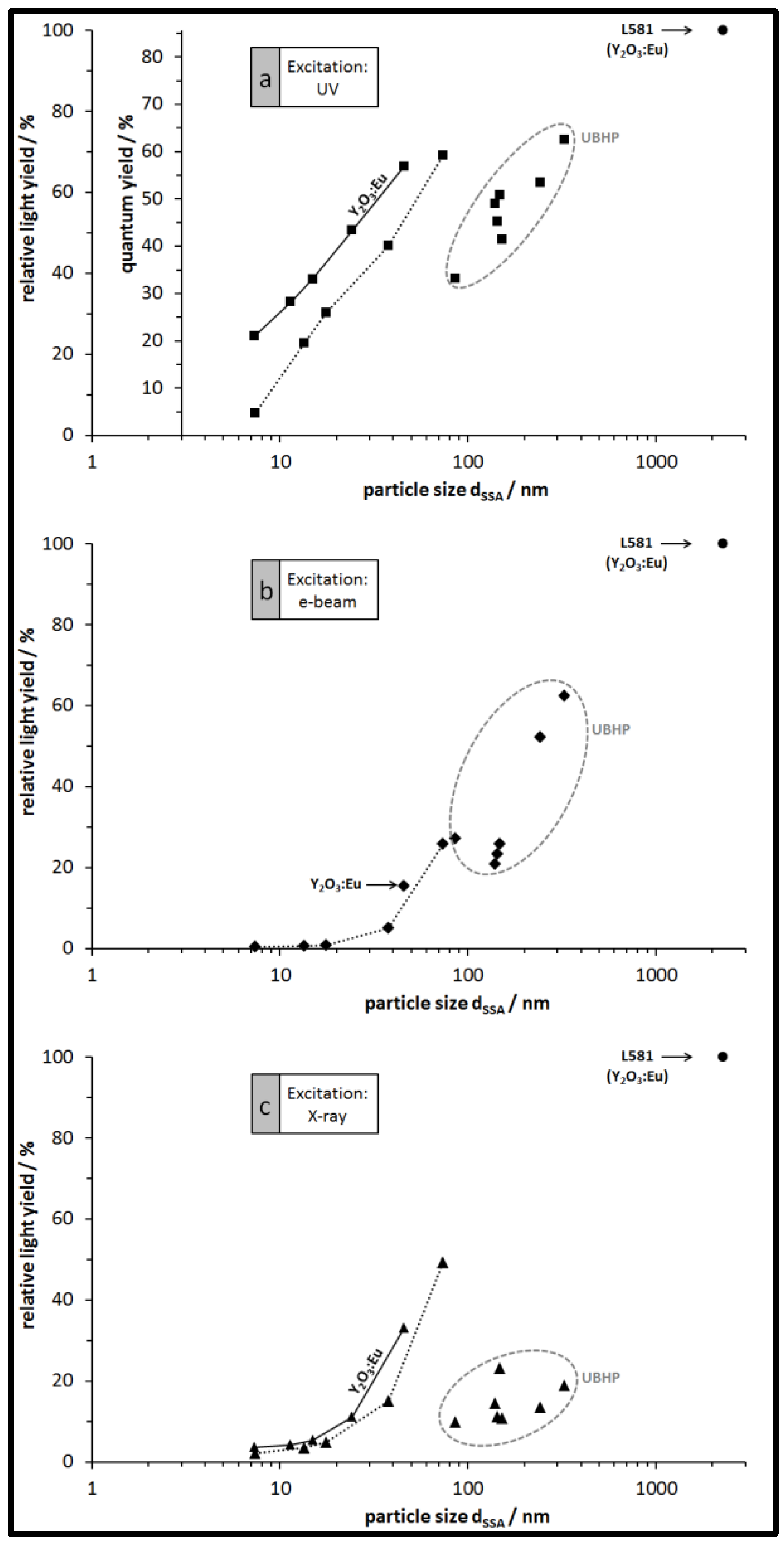
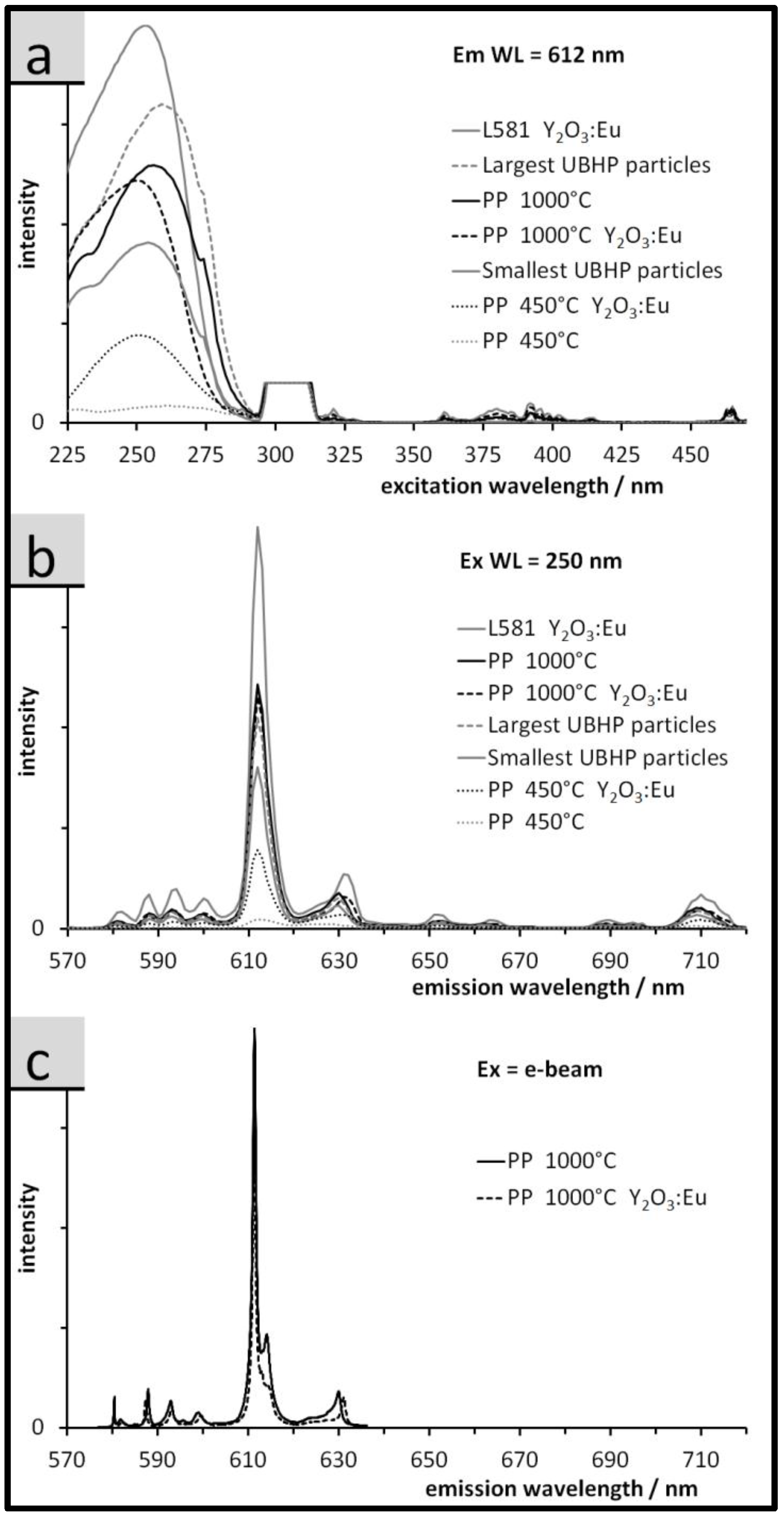
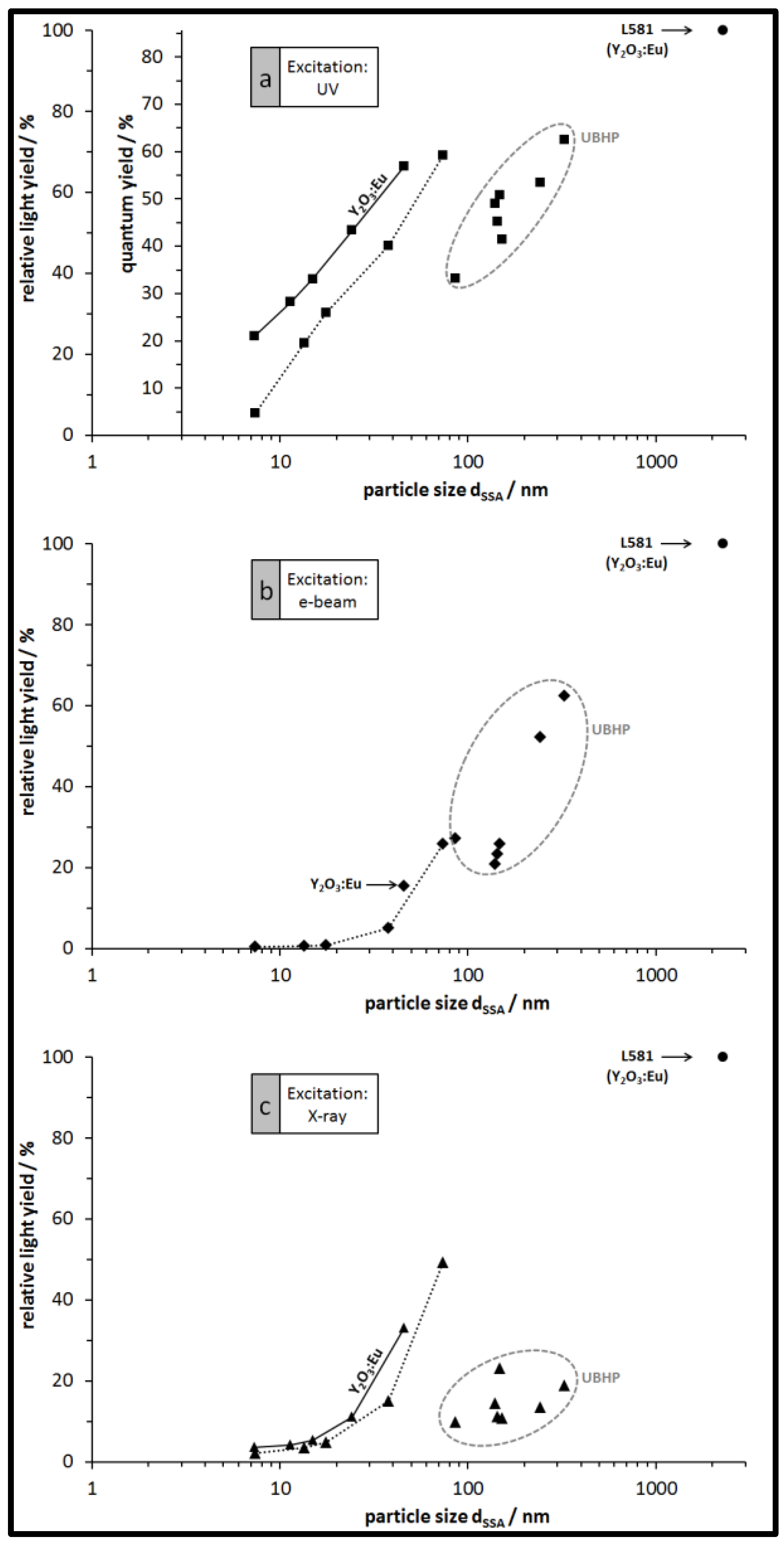
3.3. Luminescence of the Particles: Spectra, QY, and Relative Emission Light Intensities
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blasse, G.; Grabmaier, B.C. Luminescent Materials; Springer: Berlin Heidelberg, Germany, 1994. [Google Scholar]
- Den Engelsen, D.; Harris, P.; Ireland, T.; Silver, J. Cathodoluminescence of nanocrystalline Y2O3:Eu3+ with various Eu3+ concentrations. ECS J. Solid State Sci. Technol. 2015, 4, R1–R9. [Google Scholar] [CrossRef]
- Lindstrom, J.; Carlsson, G.A. A simple model for estimating the particle size dependence of absolute efficiency of fluorescent screens. Phys. Med. Biol. 1999, 44, 1353–1367. [Google Scholar] [CrossRef] [PubMed]
- Kubrin, R. Nanophosphor coatings: Technology and applications, opportunities and challenges. KONA Powder Part. J. 2014, 31, 22–52. [Google Scholar] [CrossRef]
- Yang, J.; Quan, Z.W.; Kong, D.Y.; Liu, X.M.; Lin, J. Y2O3:Eu3+ microspheres: Solvothermal synthesis and luminescence properties. Cryst. Growth Des. 2007, 7, 730–735. [Google Scholar] [CrossRef]
- Devaraju, M.K.; Yin, S.; Sato, T. Solvothermal synthesis and characterization of Eu3+ doped Y2O3 nanocrystals. J. Nanosci. Nanotechnol. 2010, 10, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, G.; Holland, E.; Dobson, P.J.; Hutchison, J.L. Luminescence properties of nanocrystalline Y2O3:Eu. Adv. Mater. 2001, 13, 1557–1560. [Google Scholar] [CrossRef]
- Yin, S.; Shinozaki, M.; Sato, T. Synthesis and characterization of wire-like and near-spherical Eu2O3-doped Y2O3 phosphors by solvothermal reaction. J. Lumin. 2007, 126, 427–433. [Google Scholar] [CrossRef]
- Zhu, Q.; Li, J.G.; Li, X.D.; Sun, X.D.; Sakka, Y. Monodisperse colloidal spheres for (Y,Eu)2O3 red-emitting phosphors: Establishment of processing window and size-dependent luminescence behavior. Sci. Technol. Adv. Mater. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, J.G.; Li, X.D.; Sun, X.D.; Ishigaki, T. Monodispersed colloidal spheres for uniform Y2O3:Eu3+ red-phosphor particles and greatly enhanced luminescence by simultaneous Gd3+ doping. J. Phys. Chem. C 2008, 112, 11707–11716. [Google Scholar] [CrossRef]
- Dujardin, C.; Amans, D.; Belsky, A.; Chaput, F.; Ledoux, G.; Pillonnet, A. Luminescence and scintillation properties at the nanoscale. IEEE Trans. Nucl. Sci. 2010, 57, 1348–1354. [Google Scholar] [CrossRef]
- Bazzi, R.; Flores-Gonzalez, M.A.; Louis, C.; Lebbou, K.; Dujardin, C.; Brenier, A.; Zhang, W.; Tillement, O.; Bernstein, E.; Perriat, P. Synthesis and luminescent properties of sub-5-nm lanthanide oxides nanoparticles. J. Lumin. 2003, 102, 445–450. [Google Scholar] [CrossRef]
- Bezkrovnyi, O.S.; Yermolayeva, Y.V.; Yanovskii, V.V.; Dulina, N.A.; Prymak, O.; Baumer, V.N.; Danilenko, N.I.; Tolmachev, A.V. Structure and morphology of spherical crystalline (Y1−xEux)2O3 particles. Inorg. Mater. 2015, 51, 51–56. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, H.C.; Zhang, W.S.; Lian, S.Y.; Kang, Z.H.; Liu, Y. Ultra-small sized Y2O3:Eu3+ nanocrystals: One-step polyoxometalate-assisted synthesis and their photoluminescence properties. J. Lumin. 2012, 132, 2155–2160. [Google Scholar] [CrossRef]
- Gupta, B.K.; Haranath, D.; Saini, S.; Singh, V.N.; Shanker, V. Synthesis and characterization of ultra-fine Y2O3:Eu3+ nanophosphors for luminescent security ink applications. Nanotechnology 2010, 21, 055607. [Google Scholar] [CrossRef] [PubMed]
- Gowd, G.S.; Patra, M.K.; Songara, S.; Shukla, A.; Mathew, M.; Vadera, S.R.; Kumar, N. Effect of doping concentration and annealing temperature on luminescence properties of Y2O3:Eu3+ nanophosphor prepared by colloidal precipitation method. J. Lumin. 2012, 132, 2023–2029. [Google Scholar] [CrossRef]
- Li, J.G.; Li, X.D.; Sun, X.D.; Ikegami, T.; Ishigaki, T. Uniform colloidal spheres for (Y1−xGdx)2O3 (x = 0–1): Formation mechanism, compositional impacts, and physicochemical properties of the oxides. Chem. Mater. 2008, 20, 2274–2281. [Google Scholar] [CrossRef]
- Jadhav, A.P.; Kim, C.W.; Cha, H.G.; Pawar, A.U.; Jadhav, N.A.; Pal, U.; Kang, Y.S. Effect of different surfactants on the size control and optical properties of Y2O3:Eu3+ nanoparticles prepared by coprecipitation method. J. Phys. Chem. C 2014, 113, 13600–13604, Erratum in 2014, 118, 14060. [Google Scholar] [CrossRef]
- Ledoux, G.; Mercier, B.; Louis, C.; Dujardin, C.; Tillement, O.; Perriat, P. Synthesis and optical characterization of Gd2O3:Eu3+ nanocrystals: Surface states and VUV excitation. Radiat. Meas. 2004, 38, 763–766. [Google Scholar] [CrossRef]
- He, C.A.; Guan, Y.F.; Yao, L.Z.; Cai, W.L.; Li, X.G.; Yao, Z. Synthesis and photoluminescence of nano-Y2O3:Eu3+ phosphors. Mater. Res. Bull. 2003, 38, 973–979. [Google Scholar] [CrossRef]
- Jing, X.; Ireland, T.; Gibbons, C.; Barber, D.J.; Silver, J.; Vecht, A.; Fern, G.; Trowga, P.; Morton, D.C. Control of Y2O3:Eu spherical particle phosphor size, assembly properties, and performance for FED and HDTV. J. Electrochem. Soc. 1999, 146, 4654–4658. [Google Scholar] [CrossRef]
- Li, J.G.; Zhu, Q.; Li, X.D.; Sun, X.D.; Sakka, Y. Colloidal processing of Gd2O3:Eu3+ red phosphor monospheres of tunable sizes: Solvent effects on precipitation kinetics and photoluminescence properties of the oxides. Acta Mater. 2011, 59, 3688–3696. [Google Scholar] [CrossRef]
- Mao, G.J.; Zhang, H.W.; Li, H.Y.; Jin, J.; Niu, S.Y. Selective synthesis of morphology and species controlled La2O3:Eu3+ and La2O2Co3:Eu3+ phosphors by hydrothermal method. J. Electrochem. Soc. 2012, 159, J48–J53. [Google Scholar] [CrossRef]
- Aiken, B.; Hsu, W.P.; Matijevic, E. Preparation and properties of monodispersed colloidal particles of lanthanide compounds: III, Yttrium(III) and mixed yttrium(III)/cerium(III) systems. J. Am. Ceram. Soc. 1988, 71, 845–853. [Google Scholar] [CrossRef]
- Sharma, P.K.; Nass, R.; Schmidt, H. Effect of solvent, host precursor, dopant concentration and crystallite size on the fluorescence properties of Eu(III) doped yttria. Opt. Mater. 1998, 10, 161–169. [Google Scholar] [CrossRef]
- Feldmann, C. Polyol-mediated synthesis of nanoscale functional materials. Adv. Funct. Mater. 2003, 13, 101–107. [Google Scholar] [CrossRef]
- Flores-Gonzalez, M.A.; Louis, C.; Bazzi, R.; Ledoux, G.; Lebbou, K.; Roux, S.; Perriat, P.; Tillement, O. Elaboration of nanostructured Eu3+-doped Gd2O3 phosphor fine spherical powders using polyol-mediated synthesis. Appl. Phys. Mater. Sci. Process. 2005, 81, 1385–1391. [Google Scholar] [CrossRef]
- Bazzi, R.; Flores, M.A.; Louis, C.; Lebbou, K.; Zhang, W.; Dujardin, C.; Roux, S.; Mercier, B.; Ledoux, G.; Bernstein, E.; et al. Synthesis and properties of europium-based phosphors on the manometer scale: Eu2O3, Gd2O3:Eu, and Y2O3:Eu. J. Coll. Interface Sci. 2004, 273, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Buechele, P.; Richter, M.; Tedde, S.F.; Matt, G.J.; Ankah, G.N.; Fischer, R.; Biele, M.; Metzger, W.; Lilliu, S.; Bikondoa, O.; et al. X-ray imaging with scintillator-sensitized hybrid organic photodetectors. Nat. Photonics 2015, 9, 843–848. [Google Scholar] [CrossRef]
- Coenen, T. Angle-Resolved Cathodoluminescence Nanoscopy. Ph.D. Thesis, University of Amsterdam, Amsterdam, The Netherlands, 2014. [Google Scholar]
- Coenen, T.; den Hoedt, S.V.; Polman, A. A new cathodoluminescence system for nanoscale optics, materials science, and geology. Microsc. Today 2016, 24, 12–18. [Google Scholar] [CrossRef]
- Vecht, A.; Gibbons, C.; Davies, D.; Jing, X.; Marsh, P.; Ireland, T.; Silver, J.; Newport, A.; Barber, D. Engineering phosphors for field emission displays. J. Vac. Sci. Technol. B 1999, 17, 750–757. [Google Scholar] [CrossRef]
- Susumu, S.; Masanobu, W. Relations between intramolecular energy transfer efficiencies and triplet state energies in rare earth β-diketone chelates. Bull. Chem. Soc. Jpn. 1970, 43, 1955–1962. [Google Scholar]
- Pang, M.L.; Lin, J.; Fu, J.; Xing, R.B.; Luo, C.X.; Han, Y.C. Preparation, patterning and luminescent properties of nanocrystalline Gd2O3:A (A = Eu3+, Dy3+, Sm3+, Er3+) phosphor films via pechini sol–gel soft lithography. Opt. Mater. 2003, 23, 547–558. [Google Scholar] [CrossRef]
- Schmechel, R.; Kennedy, M.; von Seggern, H.; Winkler, H.; Kolbe, M.; Fischer, R.A.; Xaomao, L.; Benker, A.; Winterer, M.; Hahn, H. Luminescence properties of nanocrystalline Y2O3:Eu3+ in different host materials. J. Appl. Phys. 2001, 89, 1679–1686. [Google Scholar] [CrossRef]
- Forest, H.; Ban, G. Evidence for Eu+3 emission from two symmetry sites in Y2O3:Eu+3. J. Electrochem. Soc. 1969, 116, 474–478. [Google Scholar] [CrossRef]
- Dai, Q.; Foley, M.E.; Breshike, C.J.; Lita, A.; Strouse, G.F. Ligand-passivated Eu:Y2O3 nanocrystals as a phosphor for white light emitting diodes. J. Am. Chem. Soc. 2011, 133, 15475–15486. [Google Scholar] [CrossRef] [PubMed]
- Konrad, A.; Herr, U.; Tidecks, R.; Kummer, F.; Samwer, K. Luminescence of bulk and nanocrystalline cubic yttria. J. Appl. Phys. 2001, 90, 3516–3523. [Google Scholar] [CrossRef]
- Buijs, M.; Meyerink, A.; Blasse, G. Energy transfer between Eu3+ ions in a lattice with two different crystallographic sites: Y2O3:Eu3+, Gd2O3:Eu3+ and Eu2O3. J. Lumin. 1987, 37, 9–20. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Batch | NS129 | NS133 | NS126 | NS130 | NS141 | NS134 | NS127 | NS132 |
|---|---|---|---|---|---|---|---|---|
| Solvent [vol.-ratio] | H2O | H2O | H2O | H2O | 2-propanol + H2O (6:4) | 1-butanol + H2O (6:4) | ethylene glycol + H2O (2:8) | ethylene glycol + H2O (2:8) |
| curea/cmetals | 6.3 | 14.1 | 37.6 | 62.7 | 50.1 | 50.1 | 37.6 | 62.7 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adam, J.; Metzger, W.; Koch, M.; Rogin, P.; Coenen, T.; Atchison, J.S.; König, P. Light Emission Intensities of Luminescent Y2O3:Eu and Gd2O3:Eu Particles of Various Sizes. Nanomaterials 2017, 7, 26. https://doi.org/10.3390/nano7020026
Adam J, Metzger W, Koch M, Rogin P, Coenen T, Atchison JS, König P. Light Emission Intensities of Luminescent Y2O3:Eu and Gd2O3:Eu Particles of Various Sizes. Nanomaterials. 2017; 7(2):26. https://doi.org/10.3390/nano7020026
Chicago/Turabian StyleAdam, Jens, Wilhelm Metzger, Marcus Koch, Peter Rogin, Toon Coenen, Jennifer S. Atchison, and Peter König. 2017. "Light Emission Intensities of Luminescent Y2O3:Eu and Gd2O3:Eu Particles of Various Sizes" Nanomaterials 7, no. 2: 26. https://doi.org/10.3390/nano7020026
APA StyleAdam, J., Metzger, W., Koch, M., Rogin, P., Coenen, T., Atchison, J. S., & König, P. (2017). Light Emission Intensities of Luminescent Y2O3:Eu and Gd2O3:Eu Particles of Various Sizes. Nanomaterials, 7(2), 26. https://doi.org/10.3390/nano7020026