
Atomic Pathways of Crystal-to-Crystal Transitions and Electronic Origins of Resistive Switching in MnTe for Ultralow-Power Memory



Abstract
1. Introduction
2. Computational Methods
3. Results and Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ovshinsky, S.R. Reversible electrical switching phenomena in disordered structures. Phys. Rev. Lett. 1968, 21, 1450. [Google Scholar] [CrossRef]
- Burr, G.W.; Franzon, P. Storage Class Memory. In Emerging Nanoelectronic Devices; Chen, A., Hutchby, J., Zhirnov, V., Bourianoff, G., Eds.; John Wiley and Sons Ltd.: Hoboken, NJ, USA, 2014; pp. 498–510. [Google Scholar]
- Quintino Palhares, J.H.; Garg, N.; Mouny, P.-A.; Beilliard, Y.; Sandrini, J.; Arnaud, F.; Anghel, L.; Alibart, F.; Drouin, D.; Galy, P. 28 nm FDSOI embedded PCM exhibiting near zero drift at 12 K for cryogenic SNNs. npj Unconv. Comput. 2024, 1, 8. [Google Scholar] [CrossRef]
- Kim, T.; Lee, S. Evolution of phase-change memory for the storage-class memory and beyond. IEEE Trans. Electron Devices 2020, 67, 1394–1406. [Google Scholar] [CrossRef]
- Freitas, R.F.; Wilcke, W.W. Storage-class memory: The next storage system technology. IBM J. Res. Dev. 2008, 52, 439–447. [Google Scholar] [CrossRef]
- Fong, S.W.; Neumann, C.M.; Wong, H.-S.P. Phase-change memory—Towards a storage-class memory. IEEE Trans. Electron Devices 2017, 64, 4374–4385. [Google Scholar] [CrossRef]
- Wei, S.-T.; Gao, B.; Wu, D.; Tang, J.-S.; Qian, H.; Wu, H.-Q. Trends and challenges in the circuit and macro of RRAM-based computing-in-memory systems. Chip 2022, 1, 100004. [Google Scholar] [CrossRef]
- Peng, H.; Gan, L.; Guo, X. Memristor based Spiking Neural Networks: Cooperative Development of Neural Network Architecture/Algorithms and Memristors. Chip 2024, 8, 100093. [Google Scholar] [CrossRef]
- Wuttig, M.; Bhaskaran, H.; Taubner, T. Phase-change materials for non-volatile photonic applications. Nat. Photonics 2017, 11, 465–476. [Google Scholar] [CrossRef]
- Burr, G.W.; Brightsky, M.J.; Sebastian, A.; Cheng, H.-Y.; Wu, J.-Y.; Kim, S.; Sosa, N.E.; Papandreou, N.; Lung, H.-L.; Pozidis, H. Recent progress in phase-change memory technology. IEEE J. Emerg. Sel. Top. Circuits Syst. 2016, 6, 146–162. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Z.; Li, C.; Xu, R.; Yang, D.; Sun, L. Van der Waals materials-based floating gate memory for neuromorphic computing. Chip 2023, 2, 100059. [Google Scholar] [CrossRef]
- Zhou, K.-J.; Mu, C.; Wen, B.; Zhang, X.-M.; Wu, G.-J.; Li, C.; Jiang, H.; Xue, X.-Y.; Tang, S.; Chen, C.-X. The trend of emerging non-volatile TCAM for parallel search and AI applications. Chip 2022, 1, 100012. [Google Scholar] [CrossRef]
- Li, X.-B.; Chen, N.-K.; Wang, X.-P.; Sun, H.-B. Phase-Change Superlattice Materials toward Low Power Consumption and High Density Data Storage: Microscopic Picture, Working Principles, and Optimization. Adv. Funct. Mater. 2018, 28, 1803380. [Google Scholar] [CrossRef]
- Zhang, W.; Mazzarello, R.; Wuttig, M.; Ma, E. Designing crystallization in phase-change materials for universal memory and neuro-inspired computing. Nat. Rev. Mater. 2019, 4, 150–168. [Google Scholar] [CrossRef]
- Su, Z.-J.; Xuan, Z.-H.; Liu, J.; Kang, Y.; Liu, C.-S.; Zuo, C.-J. Sub-femto-Joule energy consumption memory device based on van der Waals heterostructure for in-memory computing. Chip 2022, 1, 100014. [Google Scholar] [CrossRef]
- Chen, N.-K.; Li, X.-B. Unconventional phase transition of phase-change-memory materials for optical data storage. Chin. Phys. B 2019, 28, 104202. [Google Scholar] [CrossRef]
- Chen, N.-K.; Wang, B.-Q.; Wang, X.-P.; Li, X.-B. Orbital-selective electronic excitation in phase-change memory materials: A brief review. Z. Naturforsch. B 2021, 76, 537–541. [Google Scholar] [CrossRef]
- Simpson, R.E.; Fons, P.; Kolobov, A.V.; Fukaya, T.; Krbal, M.; Yagi, T.; Tominaga, J. Interfacial phase-change memory. Nat. Nanotechnol. 2011, 6, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Dumcenco, D.O.; Huang, Y.-S.; Suenaga, K. Atomic mechanism of the semiconducting-to-metallic phase transition in single-layered MoS2. Nat. Nanotechnol. 2014, 9, 391–396. [Google Scholar] [CrossRef]
- Guan, M.-X.; Liu, X.-B.; Chen, D.-Q.; Li, X.-Y.; Qi, Y.-P.; Yang, Q.; You, P.-W.; Meng, S. Optical control of multistage phase transition via phonon coupling in MoTe2. Phys. Rev. Lett. 2022, 128, 015702. [Google Scholar] [CrossRef]
- Cho, S.; Kim, S.; Kim, J.H.; Zhao, J.; Seok, J.; Keum, D.H.; Baik, J.; Choe, D.-H.; Chang, K.J.; Suenaga, K. Phase patterning for ohmic homojunction contact in MoTe2. Science 2015, 349, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Vanacore, G.M.; Yang, Z.; Miao, X.; Zewail, A.H. Transient structures and possible limits of data recording in phase-change materials. ACS Nano 2015, 9, 6728–6737. [Google Scholar] [CrossRef]
- Matsubara, E.; Okada, S.; Ichitsubo, T.; Kawaguchi, T.; Hirata, A.; Guan, P.; Tokuda, K.; Tanimura, K.; Matsunaga, T.; Chen, M. Initial atomic motion immediately following femtosecond-laser excitation in phase-change materials. Phys. Rev. Lett. 2016, 117, 135501. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.-K.; Li, X.-B.; Bang, J.; Wang, X.-P.; Han, D.; West, D.; Zhang, S.; Sun, H.-B. Directional forces by momentumless excitation and order-to-order transition in peierls-distorted solids: The case of GeTe. Phys. Rev. Lett. 2018, 120, 185701. [Google Scholar] [CrossRef]
- Tao, X.; Gu, Y. Crystalline–crystalline phase transformation in two-dimensional In2Se3 thin layers. Nano Lett. 2013, 13, 3501–3505. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-T.; Chen, N.-K.; Li, Z.-Z.; Wang, X.-P.; Sun, H.-B.; Zhang, S.; Li, X.-B. Two-dimensional In2Se3: A rising advanced material for ferroelectric data storage. InfoMat 2022, 4, e12341. [Google Scholar] [CrossRef]
- Choi, M.S.; Cheong, B.k.; Ra, C.H.; Lee, S.; Bae, J.H.; Lee, S.; Lee, G.D.; Yang, C.W.; Hone, J.; Yoo, W.J. Electrically driven reversible phase changes in layered In2Se3 crystalline film. Adv. Mater. 2017, 29, 1703568. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Hatayama, S.; Shuang, Y.; Ando, D.; Sutou, Y. Reversible displacive transformation in MnTe polymorphic semiconductor. Nat. Commun. 2020, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Ando, D.; Sutou, Y. Sequential two-stage displacive transformation from β to α via β’ phase in polymorphic MnTe film. Mater. Des. 2020, 196, 109141. [Google Scholar] [CrossRef]
- Mori, S.; Sutou, Y. Polymorphic transformation from metastable β to stable α phase in MnTe flake. Jpn. J. Appl. Phys. 2024, 63, 045502. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Bechstedt, F. Structural and magnetic properties of MnTe phases from ab initio calculations. J. Supercond. Nov. Magn. 2013, 26, 1963–1972. [Google Scholar] [CrossRef]
- Giannozzi, P.; De Gironcoli, S.; Pavone, P.; Baroni, S. Ab initio calculation of phonon dispersions in semiconductors. Phys. Rev. B 1991, 43, 7231. [Google Scholar] [CrossRef]
- Fonari, A.; Sutton, C. Effective Mass Calculator. 2012. Available online: https://github.com/afonari/ (accessed on 1 November 2024).
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Ong, S.P.; Richards, W.D.; Jain, A.; Hautier, G.; Kocher, M.; Cholia, S.; Gunter, D.; Chevrier, V.L.; Persson, K.A.; Ceder, G. Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis. Comput. Mater. Sci. 2013, 68, 314–319. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Kriegner, D.; Výborný, K.; Olejník, K.; Reichlová, H.; Novák, V.; Marti, X.; Gazquez, J.; Saidl, V.; Němec, P.; Volobuev, V. Multiple-stable anisotropic magnetoresistance memory in antiferromagnetic MnTe. Nat. Commun. 2016, 7, 11623. [Google Scholar] [CrossRef]
- Yan, J.-Q.; Liu, Y.; Parker, D.; Wu, Y.; Aczel, A.; Matsuda, M.; McGuire, M.; Sales, B. A-type antiferromagnetic order in MnBi4Te7 and MnBi6 Te10 single crystals. Phys. Rev. Mater. 2020, 4, 054202. [Google Scholar] [CrossRef]
- Mori, S.; Sutou, Y.; Ando, D.; Koike, J. Optical and electrical properties of α-MnTe thin films deposited using RF magnetron sputtering. Mater. Trans. 2018, 59, 1506–1512. [Google Scholar] [CrossRef]
- Siol, S.; Han, Y.; Mangum, J.; Schulz, P.; Holder, A.M.; Klein, T.R.; van Hest, M.F.; Gorman, B.; Zakutayev, A. Stabilization of wide band-gap p-type wurtzite MnTe thin films on amorphous substrates. J. Mater. Chem. C 2018, 6, 6297–6304. [Google Scholar] [CrossRef]
- Li, S.; Wu, J.; Liang, B.; Liu, L.; Zhang, W.; Wazir, N.; Zhou, J.; Liu, Y.; Nie, Y.; Hao, Y. Antiferromagnetic α-MnTe: Molten-salt-assisted chemical vapor deposition growth and magneto-transport properties. Chem. Mat. 2022, 34, 873–880. [Google Scholar] [CrossRef]
- Mori, S.; Hatayama, S.; Ando, D.; Sutou, Y. Thermal stability and polymorphic transformation kinetics in β-MnTe films deposited via radiofrequency magnetron sputtering. Jpn. J. Appl. Phys. 2021, 60, 045504. [Google Scholar] [CrossRef]
- Shuang, Y.; Mori, S.; Yamamoto, T.; Hatayama, S.; Saito, Y.; Fons, P.J.; Song, Y.-H.; Hong, J.-P.; Ando, D.; Sutou, Y. Soret-Effect Induced Phase-Change in a Chromium Nitride Semiconductor Film. ACS Nano 2024, 18, 21135–21143. [Google Scholar] [CrossRef]
- Kittel, C.; McEuen, P. Introduction to Solid State Physics; John Wiley & Sons: Hoboken, NJ, USA, 2018. [Google Scholar]
- Mori, S.; Tanimura, H.; Ichitsubo, T.; Sutou, Y. Photoinduced Nonvolatile Displacive Transformation and Optical Switching in MnTe Semiconductors. ACS Appl. Mater. Interfaces 2023, 15, 42730–42736. [Google Scholar] [CrossRef]
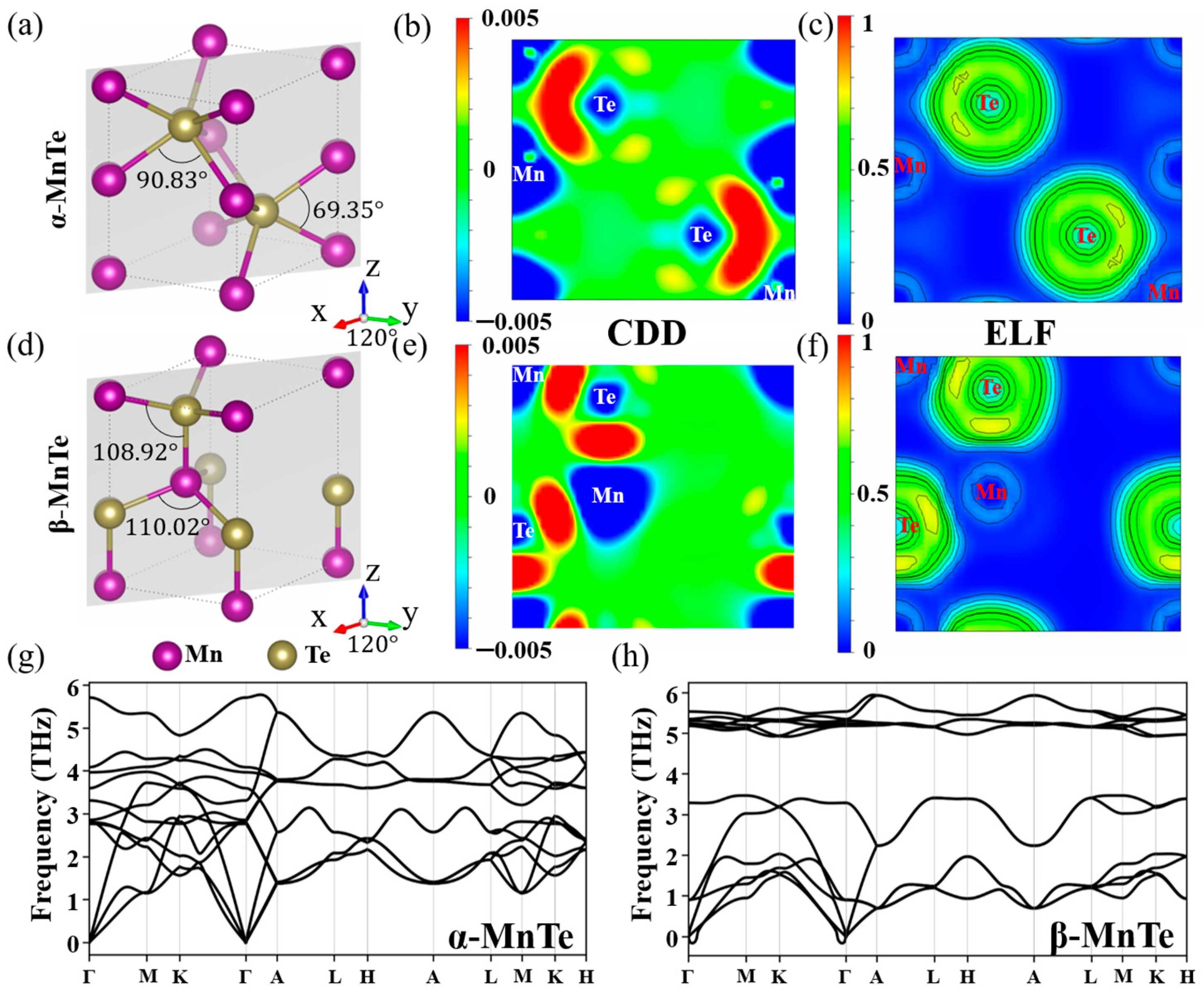
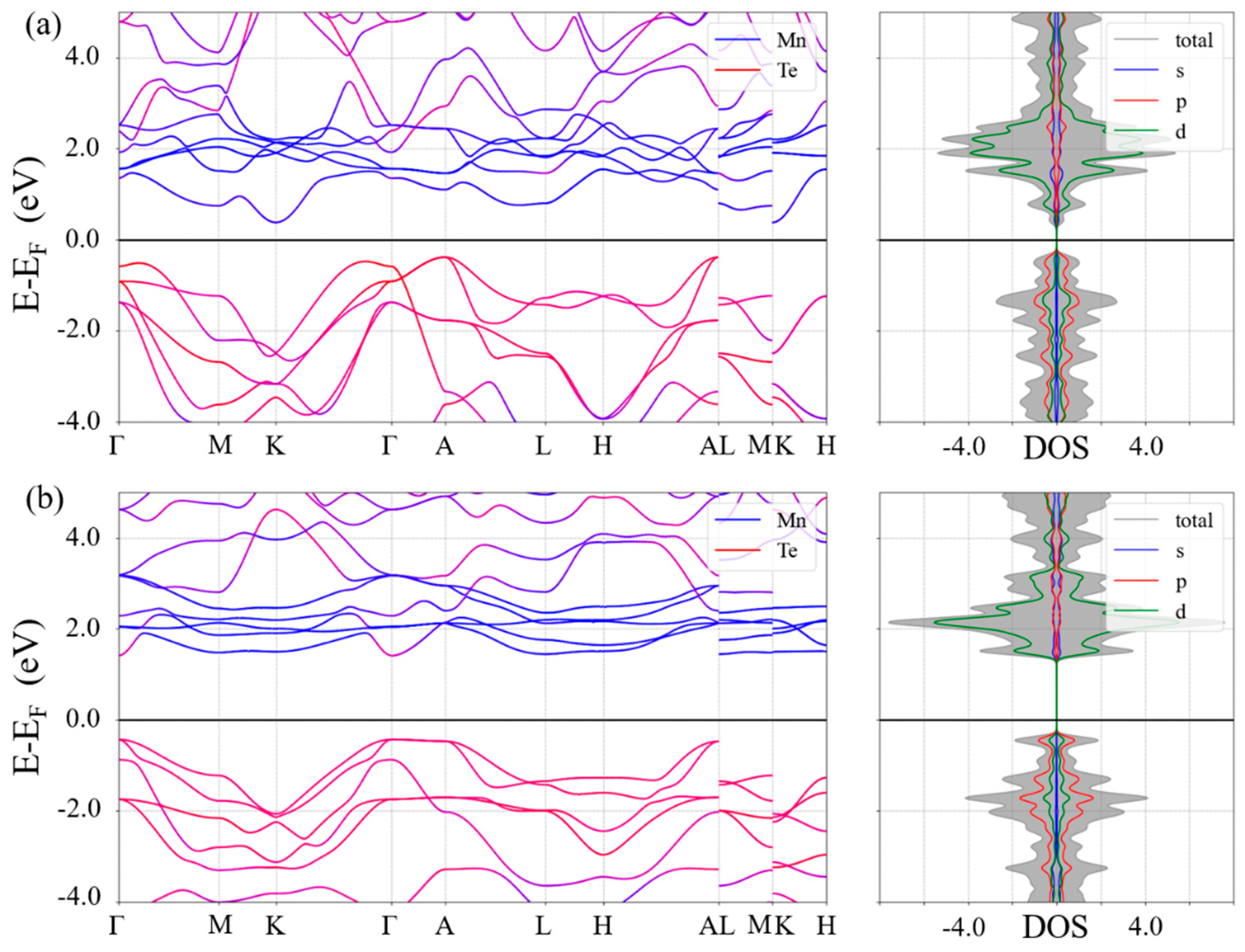
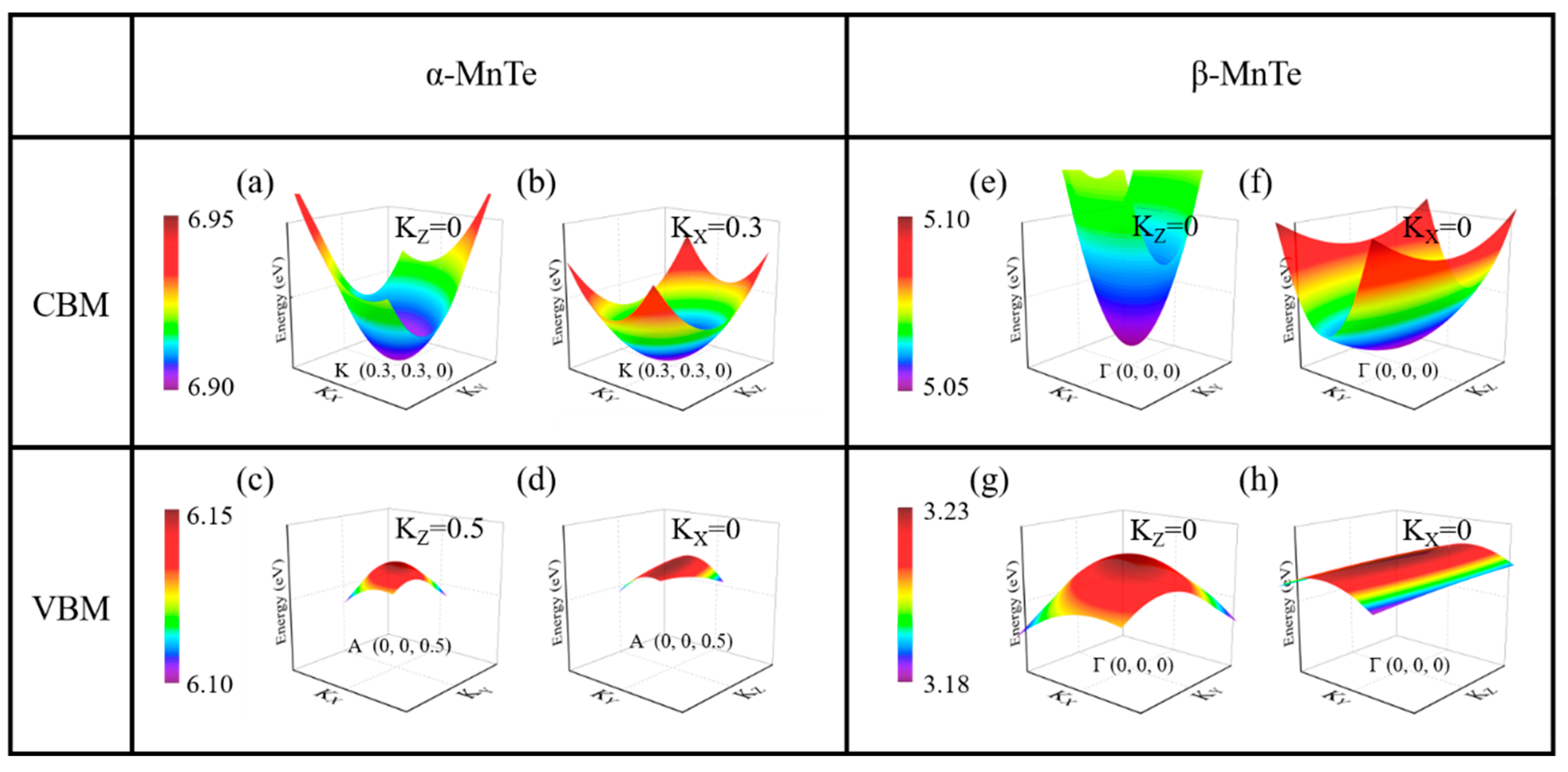
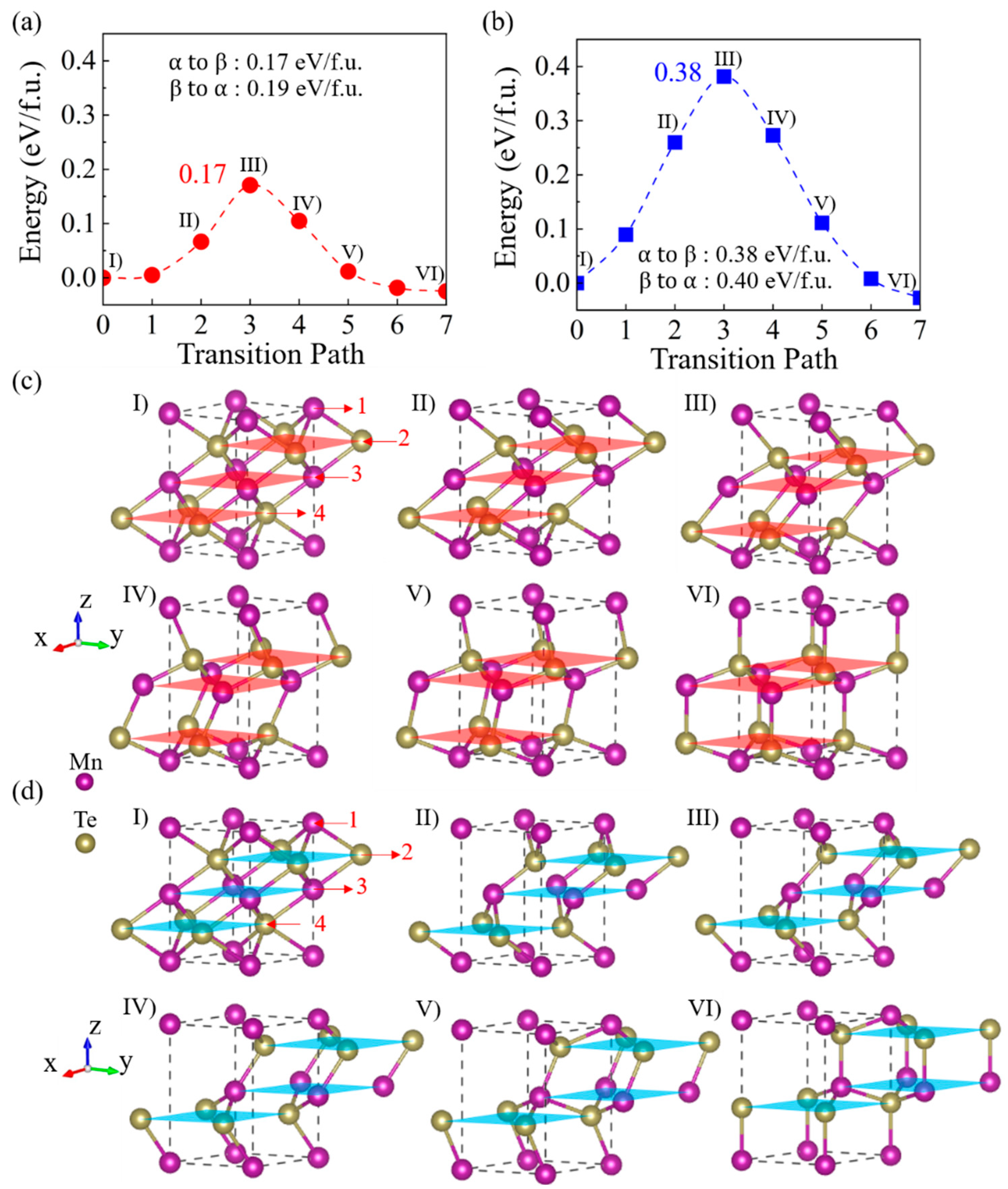

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| α-CBM | α-VBM | β-CBM | β-VBM | |
|---|---|---|---|---|
| 0.352 | −0.326 | 0.209 | −0.915 | |
| 0.352 | −0.459 | 0.058 | −0.918 | |
| 0.259 | −3.120 | 0.042 | −7.550 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, R.; Chen, N.-K.; Ma, M.-Y.; Wang, B.-Q.; Huang, Y.-T.; Zhang, B.; Li, X.-B. Atomic Pathways of Crystal-to-Crystal Transitions and Electronic Origins of Resistive Switching in MnTe for Ultralow-Power Memory. Nanomaterials 2025, 15, 231. https://doi.org/10.3390/nano15030231
Wu R, Chen N-K, Ma M-Y, Wang B-Q, Huang Y-T, Zhang B, Li X-B. Atomic Pathways of Crystal-to-Crystal Transitions and Electronic Origins of Resistive Switching in MnTe for Ultralow-Power Memory. Nanomaterials. 2025; 15(3):231. https://doi.org/10.3390/nano15030231
Chicago/Turabian StyleWu, Rui, Nian-Ke Chen, Ming-Yu Ma, Bai-Qian Wang, Yu-Ting Huang, Bin Zhang, and Xian-Bin Li. 2025. "Atomic Pathways of Crystal-to-Crystal Transitions and Electronic Origins of Resistive Switching in MnTe for Ultralow-Power Memory" Nanomaterials 15, no. 3: 231. https://doi.org/10.3390/nano15030231
APA StyleWu, R., Chen, N.-K., Ma, M.-Y., Wang, B.-Q., Huang, Y.-T., Zhang, B., & Li, X.-B. (2025). Atomic Pathways of Crystal-to-Crystal Transitions and Electronic Origins of Resistive Switching in MnTe for Ultralow-Power Memory. Nanomaterials, 15(3), 231. https://doi.org/10.3390/nano15030231