

Molecularly Imprinted Nanozymes for Selective Hydrolysis of Aromatic Carbonates Under Mild Conditions


Abstract

1. Introduction
2. Materials and Methods
2.1. General Experimental Methods
2.2. Methods
2.2.1. General Procedure for the Synthesis of Diaryl Carbonate Substrates 1b–f
2.2.2. Procedure for the Preparation of NP-Zn (Scheme 1)
2.2.3. Kinetic Measurement for the Hydrolysis of Diaryl Carbonates
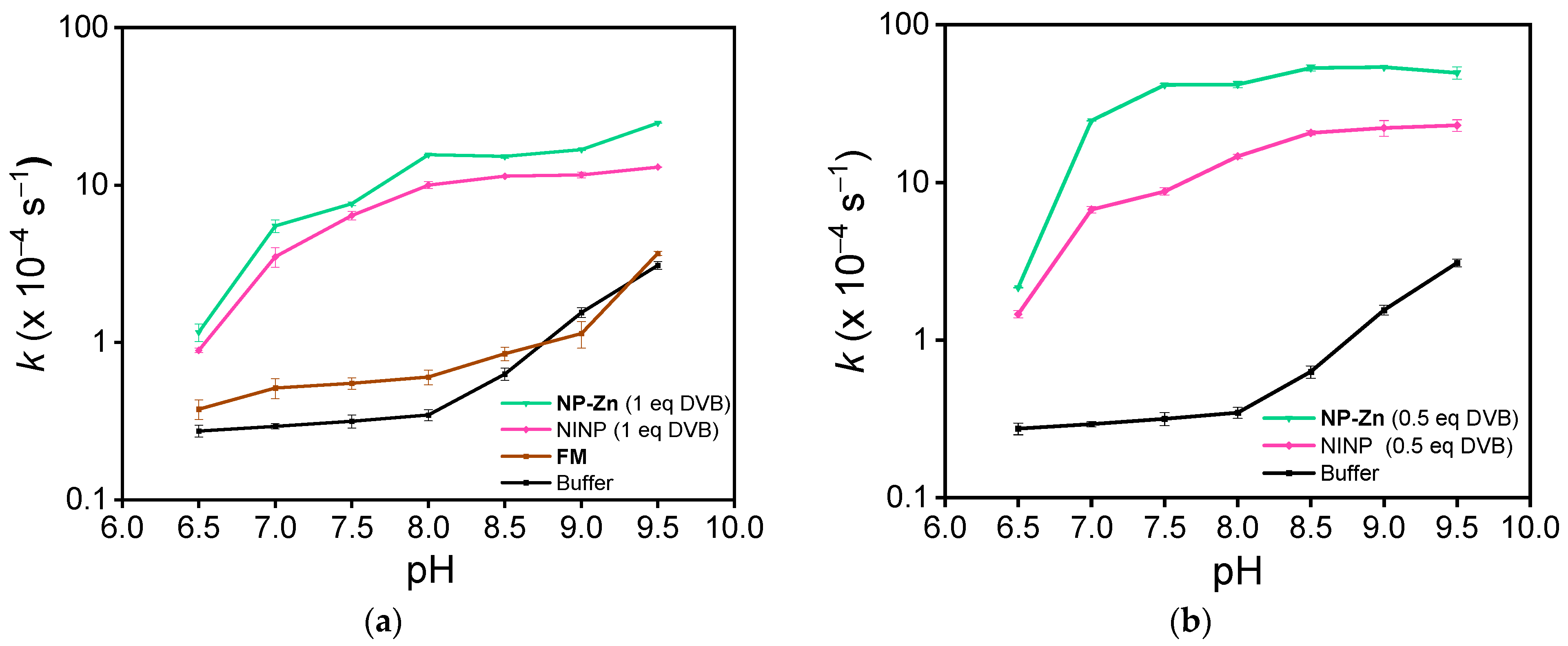
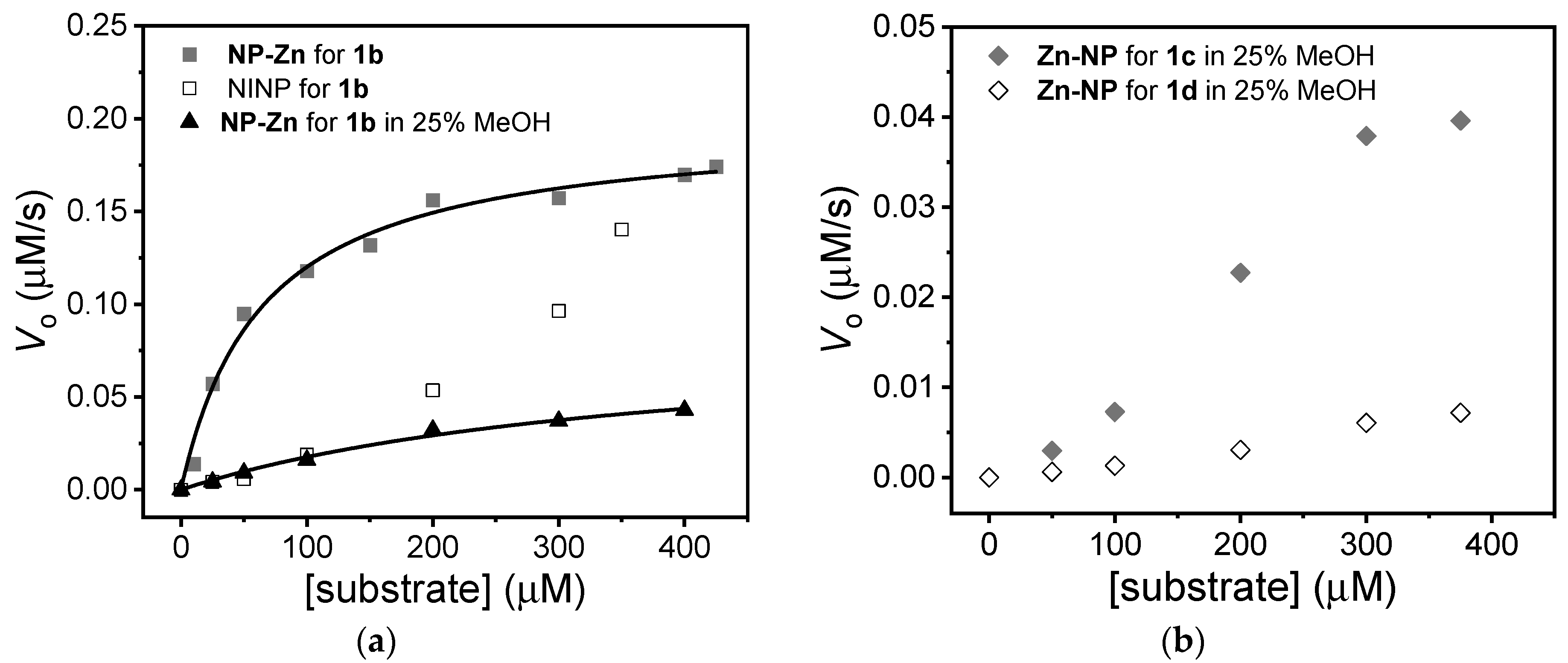
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tournier, V.; Duquesne, S.; Guillamot, F.; Cramail, H.; Taton, D.; Marty, A.; André, I. Enzymes’ Power for Plastics Degradation. Chem. Rev. 2023, 123, 5612–5701. [Google Scholar] [CrossRef] [PubMed]
- Britt, P.F.; Coates, G.W.; Winey, K.I.; Byers, J.; Chen, E.; Coughlin, B.; Ellison, C.; Garcia, J.; Goldman, A.; Guzman, J. Report of the Basic Energy Sciences Roundtable on Chemical Upcycling of Polymers; USDOE Office of Science (SC)(United States): Washington, DC, USA, 2019. [Google Scholar]
- Suyama, T.; Tokiwa, Y. Enzymatic degradation of an aliphatic polycarbonate, poly(tetramethylene carbonate). Enzyme Microb. Technol. 1997, 20, 122–126. [Google Scholar] [CrossRef]
- Thiyagarajan, S.; Maaskant-Reilink, E.; Ewing, T.A.; Julsing, M.K.; van Haveren, J. Back-to-monomer recycling of polycondensation polymers: Opportunities for chemicals and enzymes. RSC Adv. 2022, 12, 947–970. [Google Scholar] [CrossRef] [PubMed]
- Artham, T.; Mohanalakshmi, N.; Paragi-Vedanthi, P.P.; Doble, M. Mechanistic investigations of lipase-catalyzed degradation of polycarbonate in organic solvents. Enzyme Microb. Technol. 2011, 48, 71–79. [Google Scholar] [CrossRef]
- Artham, T.; Doble, M. Biodegradation of Physicochemically Treated Polycarbonate by Fungi. Biomacromolecules 2010, 11, 20–28. [Google Scholar] [CrossRef]
- Yue, W.; Yin, C.-F.; Sun, L.; Zhang, J.; Xu, Y.; Zhou, N.-Y. Biodegradation of bisphenol-A polycarbonate plastic by Pseudoxanthomonas sp. strain NyZ600. J. Hazard. Mater. 2021, 416, 125775. [Google Scholar] [CrossRef]
- Breslow, R. Artificial Enzymes; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Nothling, M.D.; Xiao, Z.; Bhaskaran, A.; Blyth, M.T.; Bennett, C.W.; Coote, M.L.; Connal, L.A. Synthetic Catalysts Inspired by Hydrolytic Enzymes. ACS Catal. 2019, 9, 168–187. [Google Scholar] [CrossRef]
- Kim, J.G. Chemical recycling of poly(bisphenol A carbonate). Polym. Chem. 2020, 11, 4830–4849. [Google Scholar] [CrossRef]
- Gilbert, E.A.; Polo, M.L.; Maffi, J.M.; Guastavino, J.F.; Vaillard, S.E.; Estenoz, D.A. The organic chemistry behind the recycling of poly(bisphenol-A carbonate) for the preparation of chemical precursors: A review. J. Polym. Sci. 2022, 60, 3284–3317. [Google Scholar] [CrossRef]
- Hata, S.; Goto, H.; Yamada, E.; Oku, A. Chemical conversion of poly(carbonate) to 1,3-dimethyl-2-imidazolidinone (DMI) and bisphenol A: A practical approach to the chemical recycling of plastic wastes. Polymer 2002, 43, 2109–2116. [Google Scholar] [CrossRef]
- Quaranta, E.; Sgherza, D.; Tartaro, G. Depolymerization of poly(bisphenol A carbonate) under mild conditions by solvent-free alcoholysis catalyzed by 1,8-diazabicyclo[5.4.0]undec-7-ene as a recyclable organocatalyst: A route to chemical recycling of waste polycarbonate. Green Chem. 2017, 19, 5422–5434. [Google Scholar] [CrossRef]
- Do, T.; Baral, E.R.; Kim, J.G. Chemical recycling of poly(bisphenol A carbonate): 1,5,7-Triazabicyclo [4.4.0]-dec-5-ene catalyzed alcoholysis for highly efficient bisphenol A and organic carbonate recovery. Polymer 2018, 143, 106–114. [Google Scholar] [CrossRef]
- Quaranta, E.; Minischetti, C.C.; Tartaro, G. Chemical Recycling of Poly(bisphenol A carbonate) by Glycolysis under 1,8-Diazabicyclo[5.4.0] undec-7-ene Catalysis. ACS Omega 2018, 3, 7261–7268. [Google Scholar] [CrossRef] [PubMed]
- Holst, L.H.; Madsen, N.G.; Toftgård, F.T.; Rønne, F.; Moise, I.-M.; Petersen, E.I.; Fojan, P. De novo design of a polycarbonate hydrolase. Protein Eng. Des. Sel. 2023, 36, gzad022. [Google Scholar] [CrossRef]
- Pasquato, L.; Rancan, F.; Scrimin, P.; Mancin, F.; Frigeri, C. N-methylimidazole-functionalized gold nanoparticles as catalysts for cleavage of a carboxylic acid ester. Chem. Commun. 2000, 2253–2254. [Google Scholar] [CrossRef]
- Manea, F.; Houillon, F.B.; Pasquato, L.; Scrimin, P. Nanozymes: Gold-nanoparticle-based transphosphorylation catalysts. Angew. Chem. Int. Ed. 2004, 43, 6165–6169. [Google Scholar] [CrossRef]
- Chen, J.L.-Y.; Pezzato, C.; Scrimin, P.; Prins, L.J. Chiral Nanozymes–Gold Nanoparticle-Based Transphosphorylation Catalysts Capable of Enantiomeric Discrimination. Chem. Eur. J. 2016, 22, 7028–7032. [Google Scholar] [CrossRef]
- Hu, L.; Zhao, Y. A Bait-and-Switch Method for the Construction of Artificial Esterases for Substrate-Selective Hydrolysis. Chem. Eur. J. 2019, 25, 7702–7710. [Google Scholar] [CrossRef]
- Arifuzzaman, M.D.; Bose, I.; Bahrami, F.; Zhao, Y. Imprinted polymeric nanoparticles as artificial enzymes for ester hydrolysis at room temperature and pH 7. Chem. Catal. 2022, 2, 2049–2065. [Google Scholar] [CrossRef]
- Zangiabadi, M.; Zhao, Y. Synergistic Hydrolysis of Cellulose by a Blend of Cellulase-Mimicking Polymeric Nanoparticle Catalysts. J. Am. Chem. Soc. 2022, 144, 17110–17119. [Google Scholar] [CrossRef]
- Wulff, G.; Liu, J. Design of biomimetic catalysts by molecular imprinting in synthetic polymers: The role of transition state stabilization. Acc. Chem. Res. 2012, 45, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Huang, C.; Shinde, S.; Jagadeesan, K.K.; Ekström, S.; Fritz, E.; Sellergren, B. Catalytic Formation of Disulfide Bonds in Peptides by Molecularly Imprinted Microgels at Oil/Water Interfaces. ACS Appl. Mater. Interfaces 2016, 8, 30484–30491. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Yang, Y.; Faheem, M.; Zou, X.; Ma, X.; Wang, Z.; Meng, Q.; Wang, L.; Zhao, S.; Zhu, G. Molecularly Imprinted Porous Aromatic Frameworks Serving as Porous Artificial Enzymes. Adv. Mater. 2018, 30, 1800069. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Chen, W.; Ma, Y.; Pan, G. Molecularly imprinted polymers as receptor mimics for selective cell recognition. Chem. Soc. Rev. 2018, 47, 5574–5587. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lieberzeit, P.A.; Piletsky, S.; Turner, A.P.F. Smart Polymer Catalysts and Tunable Catalysis; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Zhang, H. Molecularly Imprinted Nanoparticles for Biomedical Applications. Adv. Mater. 2020, 32, 1806328. [Google Scholar] [CrossRef]
- Haupt, K.; Medina Rangel, P.X.; Bui, B.T.S. Molecularly Imprinted Polymers: Antibody Mimics for Bioimaging and Therapy. Chem. Rev. 2020, 120, 9554–9582. [Google Scholar] [CrossRef]
- Muratsugu, S.; Shirai, S.; Tada, M. Recent progress in molecularly imprinted approach for catalysis. Tetrahedron Lett. 2020, 61, 151603. [Google Scholar] [CrossRef]
- Li, J.; Zhu, M.; Wang, M.; Qi, W.; Su, R.; He, Z. Molecularly imprinted peptide-based enzyme mimics with enhanced activity and specificity. Soft Matter 2020, 16, 7033–7039. [Google Scholar] [CrossRef]
- Wei, W.; Thakur, V.K.; Chew, Y.J.; Li, S. Towards next generation “smart” tandem catalysts with sandwiched mussel-inspired layer switch. Mater. Today Chem. 2020, 17, 100286. [Google Scholar] [CrossRef]
- Liu, J.-Q.; Wulff, G. Functional Mimicry of Carboxypeptidase A by a Combination of Transition State Stabilization and a Defined Orientation of Catalytic Moieties in Molecularly Imprinted Polymers. J. Am. Chem. Soc. 2008, 130, 8044–8054. [Google Scholar] [CrossRef]
- Robinson, D.K.; Mosbach, K. Molecular imprinting of a transition state analogue leads to a polymer exhibiting esterolytic activity. J. Chem. Soc. Chem. Commun. 1989, 969–970. [Google Scholar] [CrossRef]
- Ohkubo, K.; Urata, Y.; Hirota, S.; Honda, Y.; Fujishita, Y.-I.; Sagawa, T. Homogeneous esterolytic catalysis of a polymer prepared by molecular imprinting of a transition state analogue. J. Mol. Catal. 1994, 93, 189–193. [Google Scholar] [CrossRef]
- Sellergren, B.; Karmalkar, R.N.; Shea, K.J. Enantioselective Ester Hydrolysis Catalyzed by Imprinted Polymers. J. Org. Chem. 2000, 65, 4009–4027. [Google Scholar] [CrossRef]
- Awino, J.K.; Zhao, Y. Protein-Mimetic, Molecularly Imprinted Nanoparticles for Selective Binding of Bile Salt Derivatives in Water. J. Am. Chem. Soc. 2013, 135, 12552–12555. [Google Scholar] [CrossRef]
- Schaufelberger, F.; Seigel, K.; Ramström, O. Hydrogen-Bond Catalysis of Imine Exchange in Dynamic Covalent Systems. Chem. Eur. J. 2020, 26, 15581–15588. [Google Scholar] [CrossRef]
- Arifuzzaman, M.; Zhao, Y. Artificial Zinc Enzymes with Fine-Tuned Catalytic Active Sites for Highly Selective Hydrolysis of Activated Esters. ACS Catal. 2018, 8, 8154–8161. [Google Scholar] [CrossRef]
- Vallee, B.L.; Auld, D.S. Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry 1990, 29, 5647–5659. [Google Scholar] [CrossRef]
- Coleman, J.E. Zinc enzymes. Curr. Opin. Chem. Biol. 1998, 2, 222–234. [Google Scholar] [CrossRef]
- Zastrow, M.L.; Peacock, A.F.A.; Stuckey, J.A.; Pecoraro, V.L. Hydrolytic catalysis and structural stabilization in a designed metalloprotein. Nat. Chem. 2012, 4, 118–123. [Google Scholar] [CrossRef]
- Zastrow, M.L.; Pecoraro, V.L. Influence of Active Site Location on Catalytic Activity in de Novo-Designed Zinc Metalloenzymes. J. Am. Chem. Soc. 2013, 135, 5895–5903. [Google Scholar] [CrossRef]
- Rufo, C.M.; Moroz, Y.S.; Moroz, O.V.; Stohr, J.; Smith, T.A.; Hu, X.Z.; DeGrado, W.F.; Korendovych, I.V. Short peptides self-assemble to produce catalytic amyloids. Nat. Chem. 2014, 6, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Tezcan, F.A. A designed supramolecular protein assembly with in vivo enzymatic activity. Science 2014, 346, 1525–1528. [Google Scholar] [CrossRef] [PubMed]
- Burton, A.J.; Thomson, A.R.; Dawson, W.M.; Brady, R.L.; Woolfson, D.N. Installing hydrolytic activity into a completely de novo protein framework. Nat. Chem. 2016, 8, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Studer, S.; Hansen, D.A.; Pianowski, Z.L.; Mittl, P.R.E.; Debon, A.; Guffy, S.L.; Der, B.S.; Kuhlman, B.; Hilvert, D. Evolution of a highly active and enantiospecific metalloenzyme from short peptides. Science 2018, 362, 1285–1288. [Google Scholar] [CrossRef]
- Cooper, G.D.; Johnson, H.T.; Williams, B. Substituent Effects in Hydrolysis of Diaryl Carbonates. J. Org. Chem. 1965, 30, 3989–3991. [Google Scholar] [CrossRef]
- Roy, D.; Karmakar, R.; Mondal, S.K.; Sahu, K.; Bhattacharyya, K. Excited state proton transfer from pyranine to acetate in a CTAB micelle. Chem. Phys. Lett. 2004, 399, 147–151. [Google Scholar] [CrossRef]
- Chadha, G.; Zhao, Y. Properties of surface-cross-linked micelles probed by fluorescence spectroscopy and their catalysis of phosphate ester hydrolysis. J. Colloid Interface Sci. 2013, 390, 151–157. [Google Scholar] [CrossRef]
- Hammett, L.P. Physical Organic Chemistry; Reaction Rates, Equilibria, and Mechanisms, 1st ed.; McGraw-Hill Book Company, Inc.: New York, NY, USA; London, UK, 1940. [Google Scholar]
- Williams, A. Free Energy Relationships in Organic and Bio-Organic Chemistry; RSC: Cambridge, UK, 2003. [Google Scholar]
- Parkin, G. Synthetic analogues relevant to the structure and function of zinc enzymes. Chem. Rev. 2004, 104, 699–767. [Google Scholar] [CrossRef]
- Henao, J.D.; Suh, Y.-W.; Lee, J.-K.; Kung, M.C.; Kung, H.H. Striking Confinement Effect: AuCl4− Binding to Amines in a Nanocage Cavity. J. Am. Chem. Soc. 2008, 130, 16142–16143. [Google Scholar] [CrossRef]
- Bender, M.L.; Pollock, E.J.; Neveu, M.C. Deuterium Oxide Solvent Isotope Effects in the Nucleophilic Reactions of Phenyl Esters. J. Am. Chem. Soc. 1962, 84, 595–599. [Google Scholar] [CrossRef]
- Gibbs, R.A.; Benkovic, P.A.; Janda, K.D.; Lerner, R.A.; Benkovic, S.J. Substituent effects of an antibody-catalyzed hydrolysis of phenyl esters: Further evidence for an acyl-antibody intermediate. J. Am. Chem. Soc. 1992, 114, 3528–3534. [Google Scholar] [CrossRef]
- Jencks, W.P.; Carriuolo, J. General Base Catalysis of Ester Hydrolysis1. J. Am. Chem. Soc. 1961, 83, 1743–1750. [Google Scholar] [CrossRef]
- Stefanidis, D.; Jencks, W.P. General Base Catalysis of Ester Hydrolysis. J. Am. Chem. Soc. 1993, 115, 6045–6050. [Google Scholar] [CrossRef]
- Li, X.; Zangiabadi, M.; Zhao, Y. Molecularly Imprinted Synthetic Glucosidase for the Hydrolysis of Cellulose in Aqueous and Nonaqueous Solutions. J. Am. Chem. Soc. 2021, 143, 5172–5181. [Google Scholar] [CrossRef]
- Xing, X.; Zhao, Y. Fluorescent nanoparticle sensors with tailor-made recognition units and proximate fluorescent reporter groups. New J. Chem. 2018, 42, 9377–9380. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Catalyst | kcat (×10−3 s−1) | KM (μM) | kcat/KM (M−1s−1) | kcat/kuncat |
|---|---|---|---|---|---|---|
| 1 | 1b a | NP-Zn | 39.5 ± 0.6 | 64 ± 3 | 610 | - |
| 2 | 1b a | NINP | 7.2 ± 1.3 | 350 ± 15 | 20 | - |
| 3 | 1b b | NP-Zn | 17.2 ± 0.2 | 387 ± 19 | 44 | - |
| 4 | 1c b | NP-Zn | 5.4 ± 0.3 | 496 ± 3 | 11 | - |
| 5 | 1e a,c | NP-Zn | 11.9 ± 0.3 | 425 ± 1 | 28 | 1.0 × 106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bui, T.T.; Zhao, Y. Molecularly Imprinted Nanozymes for Selective Hydrolysis of Aromatic Carbonates Under Mild Conditions. Nanomaterials 2025, 15, 169. https://doi.org/10.3390/nano15030169
Bui TT, Zhao Y. Molecularly Imprinted Nanozymes for Selective Hydrolysis of Aromatic Carbonates Under Mild Conditions. Nanomaterials. 2025; 15(3):169. https://doi.org/10.3390/nano15030169
Chicago/Turabian StyleBui, Tien Tan, and Yan Zhao. 2025. "Molecularly Imprinted Nanozymes for Selective Hydrolysis of Aromatic Carbonates Under Mild Conditions" Nanomaterials 15, no. 3: 169. https://doi.org/10.3390/nano15030169
APA StyleBui, T. T., & Zhao, Y. (2025). Molecularly Imprinted Nanozymes for Selective Hydrolysis of Aromatic Carbonates Under Mild Conditions. Nanomaterials, 15(3), 169. https://doi.org/10.3390/nano15030169