1. Introduction
Heterofullerenes, which involve the incorporation of heteroatoms into the cage structures of fullerenes, can be classified into three categories: exo-doping, substitutional doping, and endo-doping. In comparison to pristine carbon fullerenes, these doped nanocages exhibit distinct electronic, magnetic, and optical properties, thereby expanding their potential applications in various fields such as catalysis [
1], hydrogen storage materials [
2], lithium battery cathodes [
3], nonlinear optical materials [
4,
5], and molecular electronics [
6,
7,
8]. Among all types of heterofullerenes, nitrogen- and boron-doped fullerenes have attracted significant attention due to the trivalent nature of nitrogen and boron atoms along with their comparable size and electronegativity to carbon. Notably, the chemical inertness of BCN materials surpasses that of diamond. Consequently, they are suitable for use in high-temperature semiconductor devices as well as short-wavelength optoelectronic devices [
9,
10].
Since the synthesis of carbon fullerene cages [
11,
12], there has been considerable interest in exploring their novel structures alongside their electronic and optical properties. In a pioneering experiment conducted in 1991 by Guo et al., boron-substituted fullerenes C
60−nB
n (
n = 1–6) were reported [
13]. Chen et al. performed semiempirical and ab initio calculations on C
60 substituted with two to eight B or N atoms [
14] as well as on C
70 substituted with two to ten B or N atoms [
15]. They found that fullerene structures doped with fewer heteroatoms tend to exhibit greater stability.
In 2011, Garg et al. empirically identified several structures of C
60−nB
n (where
n = 1–12) and discovered potential lower-energy configurations [
16]. They noted that boron-doped fullerenes contain no more than one B atom on a pentagon, while two B atoms can coexist in either para or meta positions on the same hexagon. Xie investigated a lower isomer of C
48B
12, which demonstrates enhanced third-order optical non-linearity, thereby suggesting its promising application in photonic devices [
8]. Cheng conducted a systematic study of the C
60−nB
n systems for
n = 1–6 and uncovered new low-energy structures [
17].
Comparatively, C
60−nN
n azafullerenes have been studied more extensively than boron-doped fullerenes. Several azafullerenes, including C
59N
+ and C
69N
+ [
18] and C
57N
3 [
19], have been successfully synthesized in laboratory settings. Hultman et al. [
20] discovered a cage structure of C
48N
12 exhibiting S
6 symmetry and synthesized carbon-nitrogen nano-onions with a nitrogen content of 20%. A more stable configuration of C
48N
12 was theoretically proposed by Manna et al. [
21], which retains the same S
6 symmetry as reported by Hultman et al. [
20]. Chen found that nitrogen substitution in host carbon fullerenes is generally more stable than boron substitution [
14,
15]. Sharma et al. [
22] investigated the structural, electronic, and vibrational properties of C
60−nN
n (
n = 1–12). Srinivasu et al. [
23] calculated the structure, stability, and nonlinear optical properties of C
60−2nN
2n (for
n = 1–12). In 2019, Cheng et al. [
24] conducted a systematic investigation into the structures of C
60−nN
n up to
n = 12. They calculated all possible isomers for
n = 1–4, estimated isomer energies for
n = 5–9, and proposed a classification method to filter out unstable isomers for
n = 10–12.
Despite the fact that some groups have investigated the substitution of B or N for carbon in C
60 and other sized carbon fullerenes, the ground state structures of these heterofullerenes remain uncertain. This uncertainty primarily stems from the exponential increase in the number of isomers of C
60−nX
n with respect to
n [
25], rendering it nearly impossible to calculate all possible isomers using first-principles methods. Generally speaking, previous studies have employed semiempirical approaches or applied strict filtering conditions to compute a limited set of isomers. However, excessive filtering may result in the exclusion of critical ground state structures [
24].
In the past decade, machine learning (ML) techniques have been successfully employed to accelerate the structural prediction of clusters, including main-group clusters [
26,
27,
28], coinage metal clusters [
29], and specific cage clusters [
30]. A ML model for atomistic simulations of boron and carbon, constructed using Gaussian approximation potential, can expedite the global minimum search for B
n (
n = 36, 40, 84) and C
n (
n up to 720) clusters. The global minimum structures of small clusters such as Pt
n (
n = 8–14), Ta
n (
n = 9–13), and Ag
n (
n = 14–26) have been successfully obtained using various deep neural networks. Furthermore, global optimization efforts for larger Ag
n (
n = 30–60) and Al
n clusters (
n = 21–55) have demonstrated the scalability and transferability of machine learning methods.
Combining optimization techniques with machine learning could significantly enhance the current state-of-the-art in structural search methodologies. However, since these models rely on atomic coordinates as inputs, their accuracy is contingent upon a reasonable initial structure that typically necessitates DFT calculations. To further improve efficiency, it is desirable to develop effective methods capable of directly predicting energy based on topological connections within cage structures. A notable attempt in this direction was made by Liu et al., who utilized a neural network potential based on SchNet [
31] to investigate the exohedral functionalization of fullerenes [
30], achieving a mean absolute error (MAE) of just 0.37 eV. Moreover, in our previous work, we developed a graph neural network (GNN) model that directly predicts the binding energy of boron-nitride fullerene cages solely from their topological connections [
32].
In this paper, we generated all possible isomers of C60−nBn and C60−nNn (n = 2–12) utilizing a recursive algorithm combined with isomorphic judgment techniques. Subsequently, we developed a modified GNN model to predict their binding energies both rapidly and accurately. This model employs ring type as initial features and performs convolution on both the source graph and dual graph to aggregate vertex and ring characteristics effectively. As a result, we confirmed several previous findings while also identifying some lower-energy structures.
2. Data Generation
In this study, we began with the well-known C60 buckyball with Ih symmetry and subsequently replace a portion of the C atoms with B and N atoms. Taking azafullerenes as an example, we first substituted a specified number (n) of N atoms into C60. The B-substituted fullerenes can be readily obtained by replacing all N atoms in the azafullerenes with B atoms. Given that all 60 C atoms are equivalent, we will consistently replace the first C atom with an N atom. Following this substitution, we will generate combinations represented by C(59, n − 1). Due to the high symmetry inherent in the structure of C60, some substitutions may yield equivalent configurations.
The symmetry group of C60 has been determined to be a group of order 120, consisting of permutations of the 60 atoms. For each permutation, a hash value is computed by encoding the replacement position using sexagesimal notation. The minimal hash among all 120 equivalent replacements is utilized to uniquely identify each substitution. During the generation of each combination, we verified whether it already exists in the hash set. If it does not exist, a new substitution is identified and its minimal hash is added to the set. Additionally, the replacement position is recorded as a valid structure. Each generated structure retains the minimum replacement position among its equivalent replacement isomers.
The theoretical number of isomers can be determined by Pólya’s enumeration theorem based on the symmetry group of the C
60 structure. We generated the atom permutation group of C
60 and then deduced that the number of replacement of
k B or N atoms corresponds to coefficient of
xk in following polynomial:
The number of isomers for C
60 substituted by N (or B) atoms is summarized in
Table 1. As noted in previous studies [
14,
16], structures containing adjacent N or B atoms are energetically unfavorable. Consequently, we restrict our analysis to configurations where N and B atoms are not adjacent. When introducing a new N atom, its position and neighboring positions are excluded from the possible positions already in the set. The subsequent N atom is then placed in one of the remaining available positions. However, as the number of N atoms increases, the number of isomers without adjacent N atoms grows excessively large. For instance, when there are 10 N atoms, the number of isomers exceeds 10 million (see
Table 1). Garg et al. also observed that a pentagon cannot accommodate more than one boron atom [
16]. Furthermore, Srinivasu et al. [
23] and Cheng et al. [
24] excluded isomers with meta-position substitutions in pentagons and introduced additional filtering criteria to further reduce the number of isomers. In this study, we applied a rule prohibiting meta-position substitutions on pentagons for
n = 10–12, which results in a tenfold reduction in the number of isomers (see
Table 1). This filtering significantly enhances computational efficiency, allowing GNN to predict all isomer energies within an acceptable time frame.
Some structures of C
60−nB
n and C
60−nN
n were selected, with the number
n ranging from 3 to 12. All isomers of C
57B
3 and C
56N
4 were included, while a subset of isomers for
n > 4 was randomly selected. These structures were optimized with spin unrestricted for both open and close shell clusters using DFT as implemented in the DMol
3 package [
33], forming our dataset. The double numerical basis set along with the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) [
34] was employed for self-consistent field calculations. The relaxed C
60-I
h structure yielded two distinct types of C-C bond lengths: 1.401 Å and 1.460 Å, which align reasonably well with experimental values of 1.401 Å and 1.458 Å [
35,
36]. Our dataset encompasses six compositions of C-B and C-N cluster systems: C
57B
3, C
54B
6, C
56N
4, C
54N
6, C
52N
8, C
51N
9, C
49N
11, and C
48N
12, and two test datasets—C
49N
11 and C
48N
12—while the remaining are designated as training data (
Table 2). In total, the training and testing datasets comprise 11,594 and 1916 structures, respectively.
3. Train and Test
GNN is employed to predict the energies of C
60−nB
n and C
60−nN
n heterofullerenes. Based on our previous research, we found that utilizing the dual graph simplifies handling compared to its original graph [
32]. Therefore, in this study, we adopted the dual graph as input. The first critical step is to the initial features for the vertices. We should emphasize that only topology information can be used as input because using coordinates would require optimizing structures first. However, optimizing millions of isomers using DFT calculations is impractical. Our task is to predict binding energies without relying on DFT calculations. In a simple graph, the degree or element type serves as a natural feature of a vertex; however, degree alone cannot differentiate between various isomers. Additionally, there is no element type present in the dual graph since each vertex corresponds to a ring in the original graph. In our prior investigation of (BN)
n fullerenes [
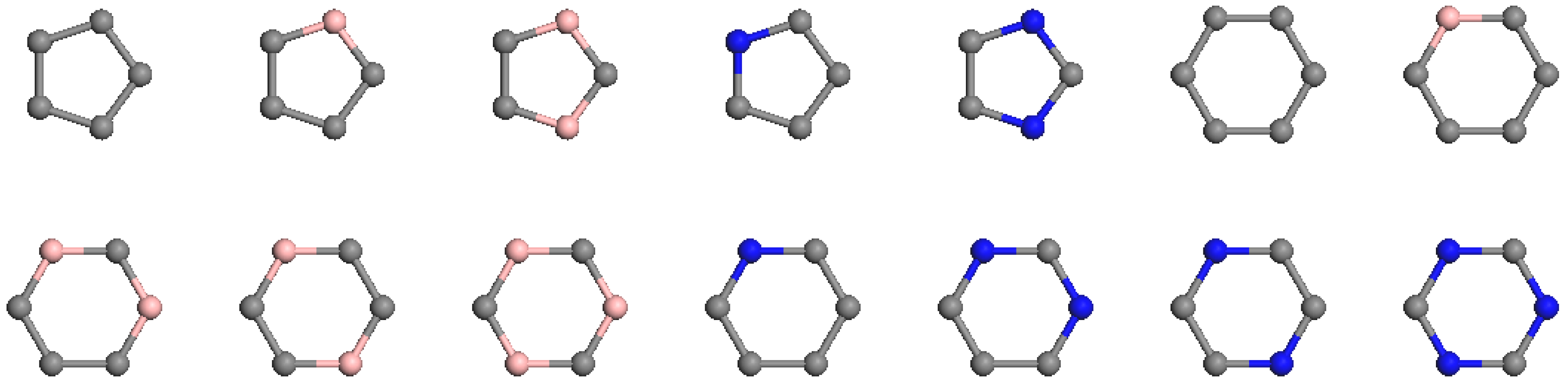
32], we established that initial vertex features could be represented by permutations of neighboring vertices for each vertex, effectively capturing different rings. For C-B and C-N cage clusters, there are only 14 distinct types of pentagons and hexagons, as illustrated in
Figure 1. Consequently, an integer ranging from 0 to 13 can be utilized to represent a vertex within the dual graph. This integer is then mapped to a vector through an embedding layer to form the feature vector corresponding to that specific vertex. Following this process, vertex features are updated by aggregating features from neighboring vertices into central vertices using various methods. Below are two approaches: one method involves summing all neighbor features [
32,
37] according to the following formula:
Here,
W represents a weight matrix used for transferring feature dimensions;
Hl denotes
l-th layer of vertices’ features; σ is the active function; and
I is identity matrix with the same order of
A.
signifies a matrix obtained from adjacent matrix
A.
An enhanced approach entails aggregating neighboring features with weighted coefficients, a technique commonly referred to as the attention mechanism [
38]:
where
hi represents feature of node
i. α
ij is attention coefficients of node
i. Softmax, LeakyReLU, and Elu are activation functions.
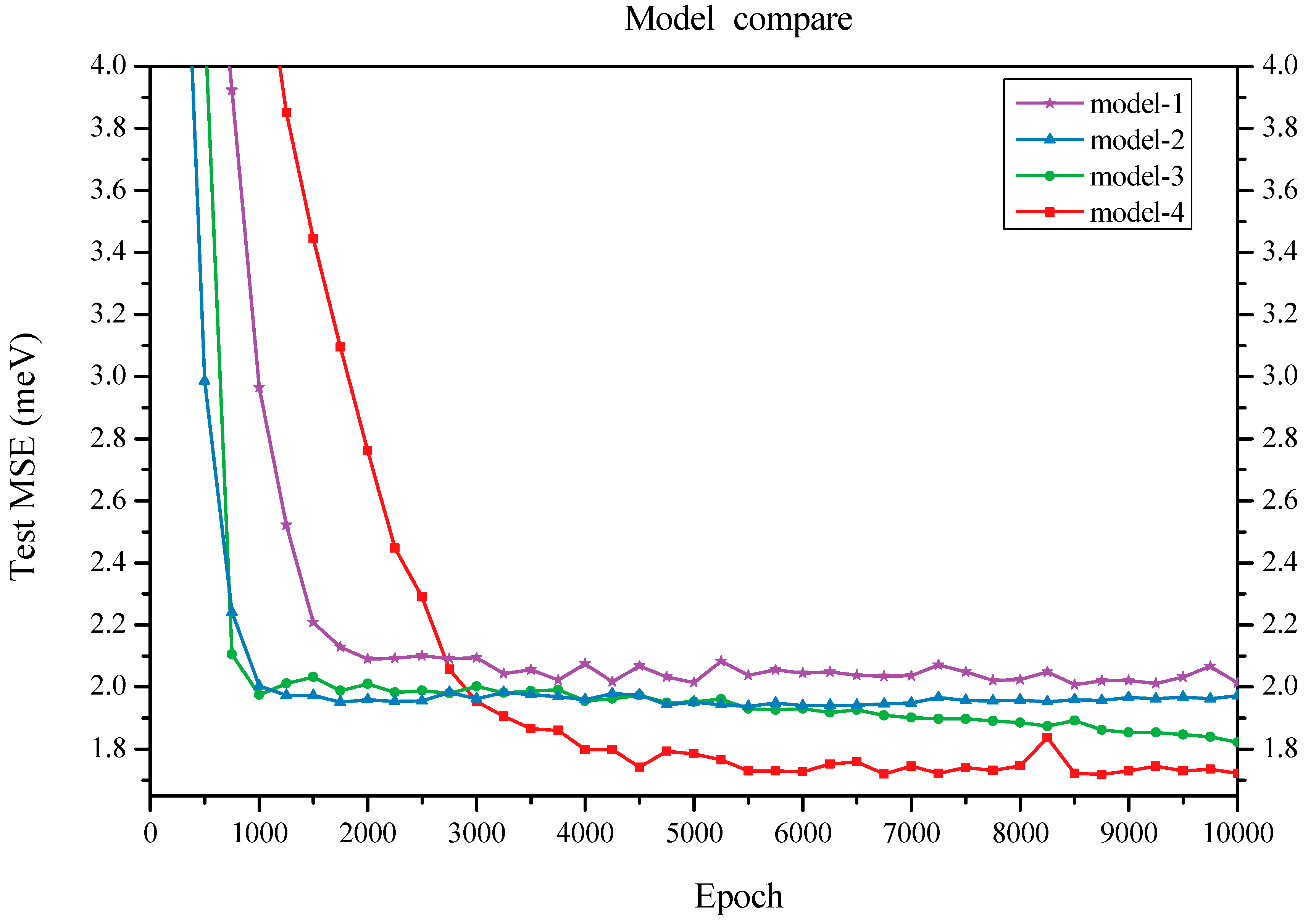
We initially constructed a model that comprises a feature embedding layer, three graph convolution layers, a fully connected layer, and a final readout function. The feature embedding layer transforms the input ring type into vector features. The graph convolution layers capture local structural information. The fully connected layer maps each vertex feature to one dimension and subsequently sums all atom values into a single value that serves as the prediction for average binding energy. This model (referred to as Model-1) achieved a test mean square error (MSE) of 1.974 meV for average binding energy (see
Table 3). Model-2 introduces an additional graph convolution layer following the three graph convolution layers from Model-1; this new layer aggregates dual-graph features onto the original graph vertices. As a result, this model attained a lower MSE of 1.932 meV (
Figure 2). A more effective model (Model-3) was developed by incorporating element information: the elemental data from all 60 sites were embedded into vectors, followed by application of a
tanh activation function and integration with another fully connected layer. Elemental information was added to vertex features prior to the readout function. Consequently, Model-3 achieved an even lower MSE of 1.822 meV.
When varying the number of graph convolution layers to two or four, we observed an increase in test MSEs to 1.862 and 1.872 meV, respectively; both results exceeded the test MSE obtained with three graph convolution layers. Furthermore, we compared the performance of graph attention layers against traditional graph convolution layers. By substituting two graph attention layers for the original ones in our architecture, we recorded a minimum test error of 1.713 meV for the resulting Model-4; however, employing three graph attention layers led to an increased test error of 1.868 meV (as shown in
Figure 2), suggesting that two attention layers are sufficient. Overall, these findings indicate that networks utilizing ring type as input topology information alongside graph convolutional mechanisms for substructure feature extraction demonstrate robustness and exhibit minimal sensitivity to specific architectural details.
4. Prediction
Our GNN model has successfully and rapidly predicted all isomers of C60−nBn and C60−nNn for n = 4–12. The prediction speed reaches 6000 isomers per second on a PC equipped with an 8-core Xeon Gold 6139 CPU, allowing for the prediction of up to 2 million isomers within just 5 min. For any given system, the subsequent step involves selecting stable structures from the top list generated by the GNN predictions for further examination using DFT calculations, aimed at identifying the true ground state structure. Taking C52B8 as an example, we identified a total of 4,158,712 isomers, with the highest predicted binding energy being 8.5348 eV according to our GNN model. It is essential to determine a cutoff value Ec such that those isomers exhibiting predicted binding energies greater than 8.5348–Ec may potentially include the true ground state structure. Hence, only structures with predicted energies exceeding 8.5348–Ec will be considered for further analysis via DFT calculations.
It should be noted that both the distribution of predicted energies and the number of isomers can vary across different systems. To address this variability, we conducted 1000 simulations for each composition in order to ascertain its corresponding Ec value. In each simulation, we introduced random errors into the predicted energies of all isomers based on a distribution derived from sample statistics representing their true binding energy. Subsequently, we establish an Ec value such that in 5 out of 1000 simulations, the difference between the lowest predicted energy and simulated true energy remains less than Ec.
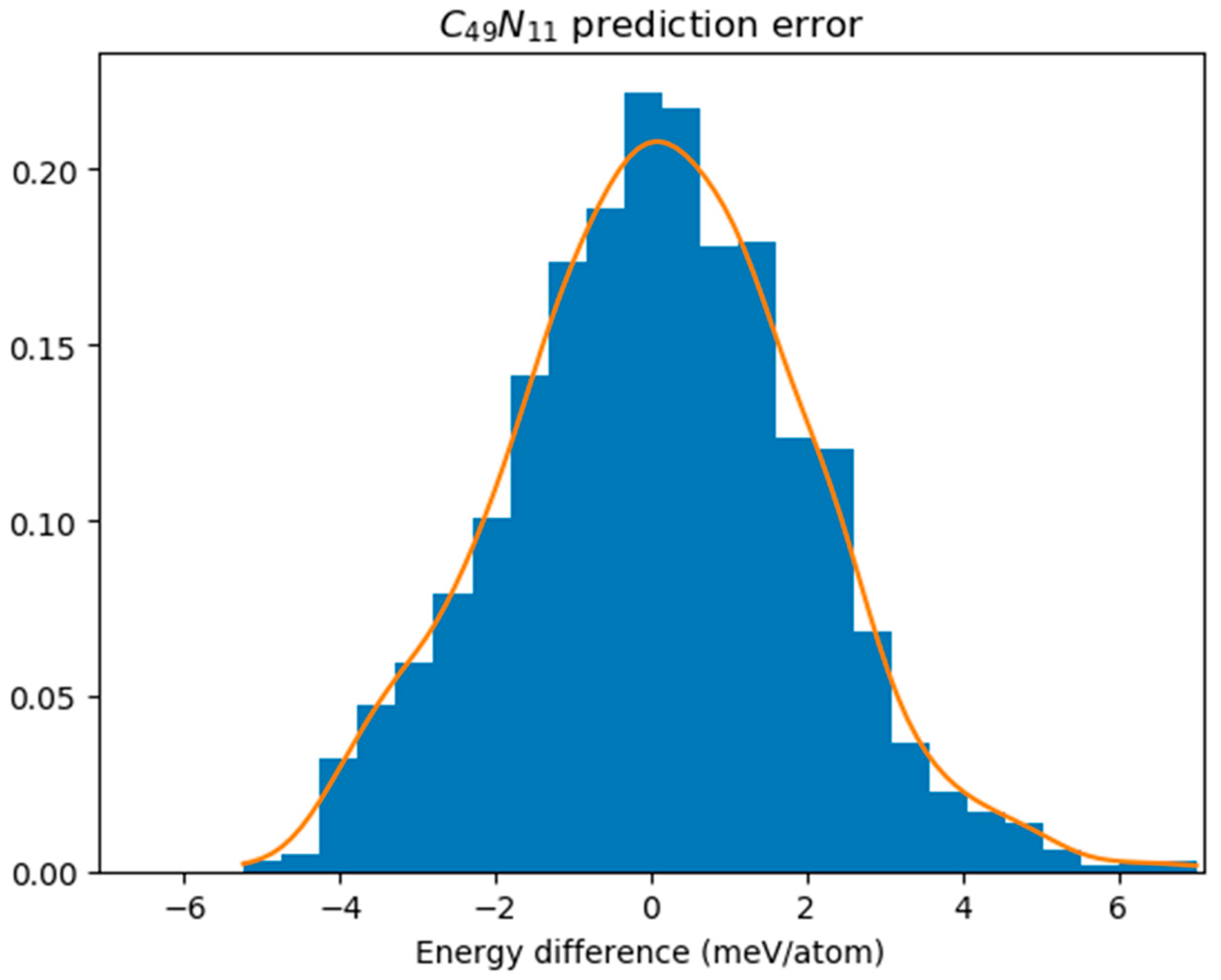
In
Figure 3, we illustrated the energy differences between GNN predictions and DFT results specifically for C
49N
11 obtained from these simulations; this reveals a sample standard deviation (σ) of energy difference equal to 0.002147 eV. The prediction error was assessed using the Kolmogorov–Smirnov test, which yielded a
p-value of 0.9802, significantly higher than our significance level set at 0.05. This indicates that we cannot reject the hypothesis asserting conformity to a normal distribution. We assume that discrepancies between GNN predictions and DFT results follow a normal distribution denoted as
N(0, σ
2). Accordingly, each GNN-predicted energy receives an associated random error ε~
N(0, σ
2). By selecting E
c = 3.5σ, only 91 isomers are predicted to have energies exceeding 8.5348–E
c, leading to just four instances where the lowest DFT energy does not fall within this cutoff range. As a consequence, we can assert with 99.6% confidence that one of these 91 isomers corresponds to the ground state structure.
Table 4 presents all cutoff values and the number of isomers to be examined through DFT calculations for C
60−nB
n and C
60−nN
n with
n = 4–12. In most cases, less than 1% of all possible isomers require evaluation. The selected combinations of cutoff values and isomers were optimized using DMol
3 program with high precision. For
n ≤ 6, our results align with previously reported ground state structures; however, for
n > 7, new lower-energy structures were identified except in the case of C
48N
12.
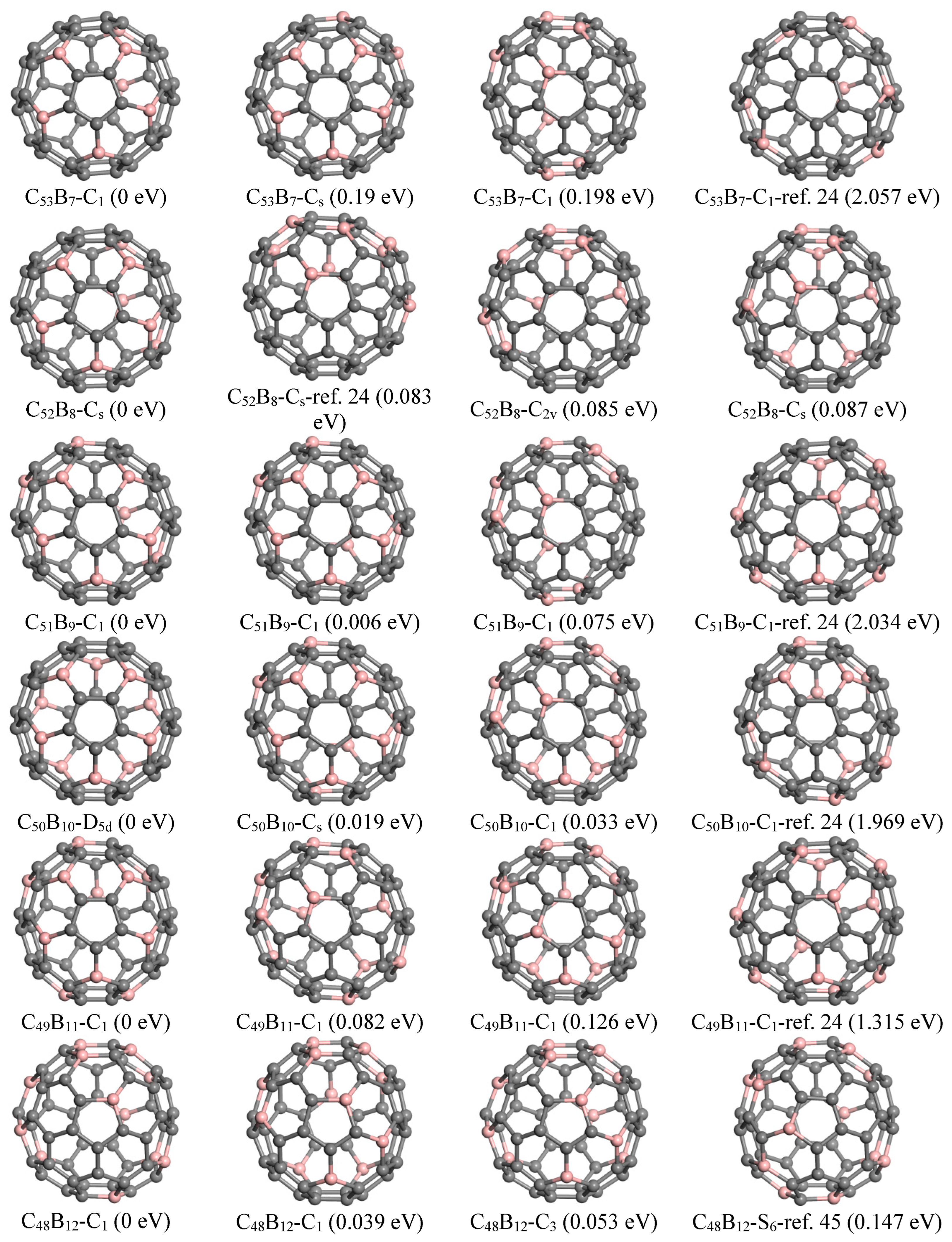
The previously reported ground state structures and the top three lowest energy configurations identified by GNN for C
60−nB
n and C
60−nN
n with
n = 7–12 are shown in
Figure 4. The ground state structure of C
53B
7, as determined in this study, features C atom indices (1, 7, 11, 16, 24, 27, 36) that have been substituted with boron atoms. This configuration exhibits a flower-like substructure composed of five petal-shaped hexagons surrounding a pentagon; specifically, five B atoms are symmetrically arranged on the petals of these hexagons. Additionally, two other B atoms occupy para-positions within another hexagon, resulting in the formation of five pairs of para-positioned B-B bonds. Notably, this structure is energetically favored by 2.057 eV compared to that reported by Garg et al. [
16]. The second lowest-energy configuration for C
53B
7 identified here has B positions at (1, 7, 11, 14, 24, 27, 31), which also maintains a similar arrangement involving five B positions.
Regarding the fullerene C
60−nB
n series for
n = 7, 9, 10, and 11, we have discovered several additional isomers exhibiting greater stability than those previously reported. The top five stable isomers are detailed in
Table S1. For instance, the lowest-energy structure found for C
52B
8 corresponds to B positions (1, 7, 11, 15, 24, 27, 36, 39). It shares a comparable arrangement of five B atoms akin to that observed in C
53B
7 7 while incorporating three additional boron atoms organized into two pairs located at para-positions within their respective hexagons. This particular configuration demonstrates an energy reduction of approximately 0.083 eV relative to isomer(b), which contains two B atoms situated within a pentagon as reported by Chen et al. [
14], and it is also lower in energy by 1.014 eV compared to the structure presented by Garget al. [
16].
The lowest-energy structure of C
51B
9 identified by GNN features B atoms located at positions (1, 7, 11, 14, 24, 27, 31, 36, and 39), exhibiting an energy reduction of 2.034 eV compared to the previously reported configuration [
16]. This structure also includes a flower-like sub-structure akin to that found in C
53B
7; additionally, the remaining four B atoms are arranged in two para-positioned hexagons.
For C
50B
10, the ground state structure reveals a substitution pattern for B atoms at positions (1, 7, 11, 24, 27, 34, 37, and 50). It comprises two groups of five B atom substructures similar to those observed in C
53B
7 and demonstrates D
5d symmetry. Notably, this specific structure is energetically more favorable by approximately 1.969 eV when compared with earlier reported structure [
16].
In the case of
n = 11, the lowest-energy structure involves substituting B atoms at positions (1, 7, 11, 14, 17, 24, 27, 31, 35, 41, 57). This configuration showcases five B atoms positioned analogously to petal arrangements seen in C
53B
7. The remaining six B atoms are organized into three pairs situated in para positions. This particular geometry exhibits greater stability than the isomeric structure documented by Garg et al. [
16], which contains a pair of meta-positioned B atoms within a pentagon and is less stable by approximately 1.315 eV.
For C
48B
12, we found that the most energetically favorable configuration consists of B atoms placed at locations (1, 6, 8, 11, 16, 18, 23, 28, 31, 36, 54, 60). In contrast to previous structures examined, it incorporates a hexagon containing three B atoms. Furthermore, the S
6 symmetric structure reported by Manna et al. [
39] is 0.147 eV higher in energy than our current ground state configuration and ranks fifth on our list.
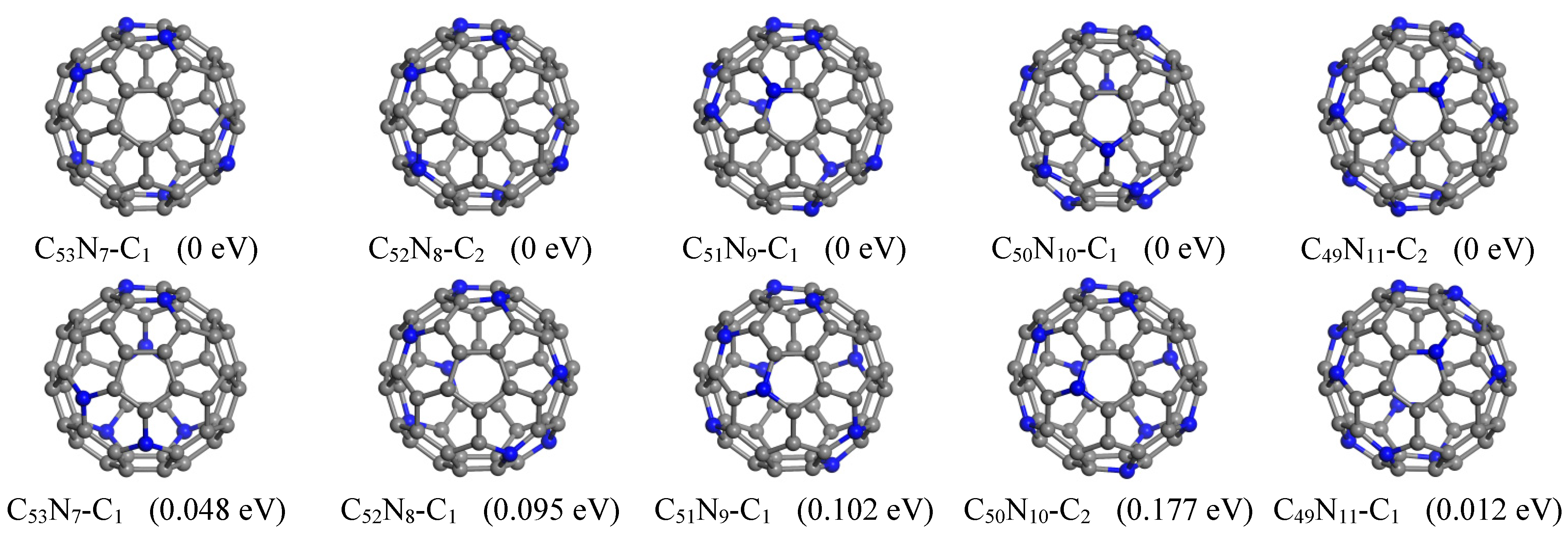
In this study, we found lower energy structures for all C
60−nN
n systems with
n = 7–11, while the previously reported ground state structure for
n = 12 has been confirmed (
Figure 5). Our predicted lowest energy configuration of C
53N
7 features nitrogen atoms located at locations (1, 7, 26, 31, 37, 51, 54). This structure includes three pairs of para-positioned N and is energetically favored by 0.048 eV compared to the structure with two pairs of para-positioned N [
24].
For C
52N
8, we identified eight structures (
Table S1) that possess lower energies than those previously reported [
24]. Among these configurations, the lowest-energy structure has N positioned at (1, 7, 26, 31, 37, 46, 51, 54), which is found to be energetically more favorable by 0.095 eV relative to that in literature [
24]. The current ground state structure of C
51N
9 contains N atoms situated at positions (1, 7, 11, 14, 24, 27, 35, 54, 60). This configuration comprises four pairs of para-positioned N; this is in contrast to the previously reported one that contained only three pairs [
24]. Our proposed structure is lower in energy by 0.102 eV in comparison to the previously reported structure featuring just three pairs of para-positioned N [
24].
Furthermore, we have identified four additional structures with even lower energies. For C
50N
10, we discovered a total of sixteen structures that exhibit greater energetic favorability compared to those previously reported. Among these, the lowest-energy structure is favored by as much as 0.177 eV. This specific configuration comprises three pairs each of meta-positioned and para-positioned N atoms, which are substituted at positions (1, 6, 11, 15, 18, 43, 46, 49, 52, 56). In the case of C
49N
11, we found six isomers with lower energy. Notably, the ground state structure is 0.012 eV lower than the previously reported structure [
24]. In this configuration, N atoms are positioned at (1, 6, 11, 18, 23, 27, 33, 40, 48, 51, 59), consisting of two pairs of meta-positioned N and six pairs of para-positioned N atoms.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}